

SUMMARY
The cholesterol-dependent cytolysins (CDCs) punch holes in target cell membranes through a highly regulated process. Streptococcus mitis lectinolysin (LLY) exhibits another layer of regulation with a lectin domain that enhances the pore-forming activity of the toxin. We have determined the crystal structures of the lectin domain by itself and in complex with various glycans that reveal the molecular basis for the Lewis antigen specificity of LLY. A small-angle X-ray scattering study of intact LLY reveals the molecule is flat and elongated with the lectin domain oriented so that the Lewis antigen-binding site is exposed. We suggest that the lectin domain enhances the pore-forming activity of LLY by concentrating toxin molecules at fucose-rich sites on membranes, thus promoting the formation of pre-pore oligomers on the surface of susceptible cells.
INTRODUCTION
The cholesterol-dependent cytolysins (CDCs) are one of the most widely distributed toxins known, having been identified in five different genera of Gram-positive bacteria (Tweten, 2005). The CDCs exhibit a number of unique features among pore-forming toxins, including an absolute dependence on the presence of cholesterol-rich membranes for their activity and the formation of oligomeric transmembrane pores greater than 150 Å in diameter. There are more than 20 members of the CDC family identified so far, and there exists a high degree of sequence homology (40%–70%), suggesting they all have similar activities and three-dimensional structures. The latter has been confirmed with crystal structures determined for perfringolysin O (PFO) (Rossjohn et al., 1997, 2007), intermedilysin (ILY) (Polekhina et al., 2005), anthrolysin O (ALO) (Bourdeau et al., 2009), and suilysin (SLY) (Xu et al., 2010). Functional studies have revealed that CDCs undergo a highly regulated stepwise process in assembling as a large membrane pore consisting of more than 30 monomers (Tweten, 2005). Not only is the conversion from water-soluble monomer to pore highly complex, but it is also essential that the pore does not form prematurely, otherwise the target cell will not be successfully breached.
Streptococcus mitis is a member of the viridans streptococci and usually found in the normal flora of the mouth and throat. Together with other members of the viridans family, it can cause a numberof diseases such as infective endocarditis, bacteremia, and septicemia (Hall and Baddour, 2002; Huang et al., 2002; Gowda et al., 2003; Kennedy et al., 2004). S. mitis was a causative agent for a large outbreak of toxic shock-like syndrome in China (Lu et al., 2003) and has also been associated with Kawasaki disease (Ohkuni et al., 1997). A possible pathogenesis factor for these diseases is a protein secreted by the bacterium that was isolated from serum of patients who suffered from Kawasaki disease. The protein was suggested to have the ability to aggregate human platelets on the basis of an observed change in light-scattering properties and, therefore, was called platelet aggregation factor (PAF). Ohkuni et al. (2006) showed that antibody titers to a PAF-derived peptide were significantly elevated in children with Kawasaki disease, a disease often associated with platelet aggregation and coronary artery thrombosis. Amino acid sequence analysis of PAF (Sm-hPAF-NM-65, GenBank accession number AB051299.1) revealed that the DNA-derived sequence was related to ILY, a CDC produced by Streptococcus intermedicus (Nagamune et al., 2000).
Farrand et al. (2008) performed an extensive study of PAF and found that it shared a number of characteristics typical of CDCs. Of note, their studies showed that PAF did not appear to aggregate platelets. The changes in light-scattering properties of the platelets observed by Ohkuni et al. (1997) were apparently due to changes of the shape of the platelets induced by the formation of pores, not their aggregation. A special feature of the toxin is the presence of an additional amino-terminal domain of 162 amino acids that is not present in other CDCs. This domain was found to share significant sequence identity with proteins that bind glycans-containing fucosylated structures. These observations led Farrand et al. to rename PAF as lectinolysin (LLY).
Farrand et al. (2008) showed that the presence of the lectin domain (LLYlec) enhanced the formation of pores on platelets compared to LLYCDC (where LLYCDC is a mutant molecule that lacks the lectin domain), presumably because the domain interacted with one or more glycans on the cell surface of platelets. Glycan array analysis revealed that LLYlec had a preference for binding to the difucosylated glycans Lewis y (Ley) antigen and Lewis b (Leb) antigen. These Lewis carbohydrate antigens are blood group antigens, which are classified as either type 1 or type 2 antigens. Leb is one of the type 1 antigens that are important histo-blood group antigens, whereas Ley belongs to type 2 and is only expressed in a few cell types (Yuriev et al., 2005). The latter has been associated with tumor cells due to elevated expression levels in a number of cancerous cells and, as such, is a promising target for cancer therapy (Yuriev et al., 2005).
Opportunistic bacteria often utilize proteins bearing lectin domains to help target host cell glycoconjugates for recognition (Imberty and Varrot, 2008). However, LLY is the only reported CDC to date that utilizes this strategy as a part of its host cell recognition mechanism. Here we describe the crystal structure of LLYlec complexed to glycerol and three saccharides: fucose and the two difucosylated glycans Leb and Ley. These structures show that LLYlec adopts a relatively rare fold in the lectin super-family and reveals the molecular basis for the lectin’s glycan specificity. Small-angle X-ray scattering (SAXS) was used to determine how the lectin domain interacts with the rest of the toxin molecule. These studies show that the fucose-binding site is fully accessible to the environment, in contradiction to earlier reports (Farrand et al., 2008), and thus already primed for interaction with host cell glycoreceptors.
RESULTS
Structure of LLYlec
The final structure of LLYlec consists of residues 44 to 185 (Table 1), which adopt an eight-stranded β sandwich fold made up by a five-stranded antiparallel β sheet (β1, β2, β4, β5, and β7) on one side and a three-stranded antiparallel β sheet (β3, β6, and β8) on the other side (Figure 1). (A cartoon diagram of a model of full-length LLY indicating where LLYlec is located in comparison with the rest of the molecule is shown in Figure S1, available online.) β strands 1 and 2 are separated by a long loop (residues 59 to 91) containing two short α helices (α1 and α2), and another short α helix (α3) is positioned between β strands 3 and 4. There are numerous salt bridges in the structure (Glu 45 to Arg 182, Glu 47 to Arg 182, Asp 75 to His 80, Asp 90 to Lys 169, Asp 97 to Arg 160, Glu 151 to Lys 106, Glu 149 to Lys 108, Asp 114 to Arg 112, Asp 125 to Arg 138, Asp 142 to His 140, Glu 165 to Lys 163, and Glu 179 to Lys 108). An intrasheet salt bridge from Asp 97 to Arg 160 forms a link between the N terminus of β strand 2 with the C terminus of β strand 7.
Table 1.
Crystallographic Data Processing and Refinement Statistics
| Variable | LLYlec | LLYlec-Fucose | LLYlec-Ley | LLYlec-Leb |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength (Å) | 0.95 | 1.54 | 1.54 | 1.54 |
| Temperature (K) | 100 | 100 | 100 | 100 |
| Maximum resolution (Å) | 1.9 | 1.9 | 2.0 | 2.2 |
| Space group | P43212 | P43212 | P43212 | P43212 |
| Unit cell dimensions (Å) | 67.2, 67.2, 98.4 | 67.0, 67.0, 97.8 | 67.1, 67.1, 98.6 | 67.0, 67.0, 97.7 |
| No. of observations | 268,267 | 154,987 | 184,195 | 134,919 |
| No. of unique reflections | 16,856 | 18,008 | 15,622 | 11,851 |
| Redundancy | 15.9 | 8.6 | 11.8 | 11.4 |
| Data completeness (%) | 91.5 (72.3) | 98.6 (87.7) | 99.8 (99.8) | 99.7 (99.7) |
| I/σI | 28.0 (4.1) | 9.6 (4.0) | 9.7 (4.1) | 14.0 (6.9) |
| Rmerge (%)a | 9.1 (55.6) | 12.4 (42.9) | 14.3 (49.2) | 9.6 (27.0) |
| Refinement | ||||
| Nonhydrogen atoms | ||||
| Protein | 1,112 | 1,114 | 1,115 | 1,100 |
| Water | 164 | 144 | 121 | 135 |
| Ni2+ | 1 | 1 | 1 | 1 |
| Ca2+ | 1 | 1 | 1 | 1 |
| Ligands | 6 | 11 | 46 | 46 |
| Resolution (Å) | 1.9 | 1.9 | 2.0 | 2.2 |
| Rwork (%)b | 17.5 | 19.9 | 19.3 | 20.2 |
| Rfree (%)c | 20.6 | 24.7 | 23.4 | 23.7 |
| Rms deviations from ideal geometry | ||||
| Bond lengths (Å) | 0.015 | 0.017 | 0.018 | 0.017 |
| Bond angles (degrees) | 1.5 | 1.4 | 1.6 | 1.5 |
| Bonded Bs | 1.7 | 1.7 | 2.0 | 1.6 |
| Mean B (Å2) | ||||
| Main chain | 23.5 | 22.3 | 20.5 | 17.8 |
| Side chain | 25.7 | 23.7 | 22.2 | 19.6 |
| Water | 34.5 | 30.1 | 27.3 | 25.5 |
| Residues in most favored regions of the Ramachandran plot (%) | 89.1 | 85.4 | 86.0 | 85.9 |
| Residues in the disallowed regions of the Ramachandran plot (%) | 0 | 0 | 0 | 0 |
The values in parentheses are for the highest resolution bin (approximately 0.1 Å width).
Rmerge = ΣhklΣi|Ii − < I > |/| < I > |, where Ii is the intensity for the ith measurement of a symmetry-related reflection with indices h,k,l.
Rwork = Σ||Fobs| − |Fcalc||/Σ|Fobs|, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively.
Rfree was calculated with 5% of the diffraction data that were selected randomly and not used throughout refinement.
Figure 1. Structure of LLYlec.
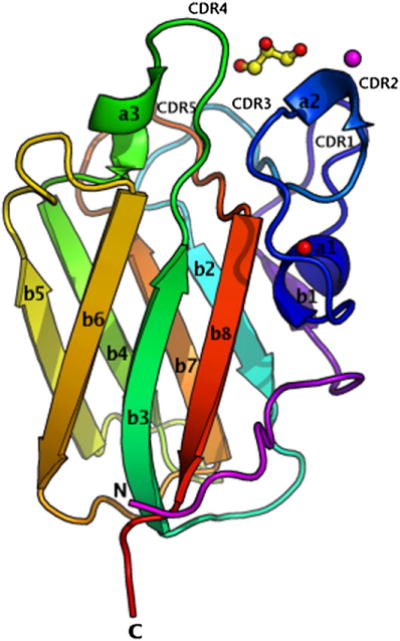
(See also Figure S1, which shows how LLYlec is oriented with respect to the rest of the toxin molecule.) The structure is represented in colors, from the N terminus in magenta to the C terminus in red. Glycerol is shown as ball and stick, the calcium ion is shown as a red sphere, and the Ni ion is shown as a magenta sphere. Secondary structural elements and CDR loops are labeled as described in the text. This and all following structural figures were drawn with PyMol (Delano, 2002).
A calcium ion is positioned between backbone carbonyls of Arg 68, Asp 73, Ser 82, and Ala 176 and side-chain residues of Asp 71, Ser 82, and Glu 177. The position of this ion is conserved in other fucolectins domains (Bianchet et al., 2002; Boraston et al., 2006) and in a number of carbohydrate-binding module (CBM) families (Boraston et al., 2004). It plays an important role in the interaction of many lectins with their target ligands, either by maintaining the binding site in the correct conformation or through direct coordination with the carbohydrate itself.
The electron density map revealed that one glycerol molecule was embedded in a hydrophobic pocket created by residues Tyr 62, Tyr 78, Phe 88, and Val 117 and the basic residues His 85, Arg 112, and Arg 120 (Figures 1 and 2A). The glycerol molecule forms hydrogen bonds with His 85, Arg 112, and Arg 120. The equivalent residues are conserved in other lectin domains (Bianchet et al., 2002; Boraston et al., 2006). In addition to the direct hydrogen bonds, the glycerol molecule also takes part in 66 van der Waals interactions with various residues in the pocket. More than two thirds (69% or 151 Å2) of the surface of the glycerol molecule is buried in the complex. The origin of this glycerol molecule was the cryosolution that was used to protect the crystal from radiation damage.
Figure 2. Close-up Stereo Views of the Ligand Binding Pocket of LLYlec.
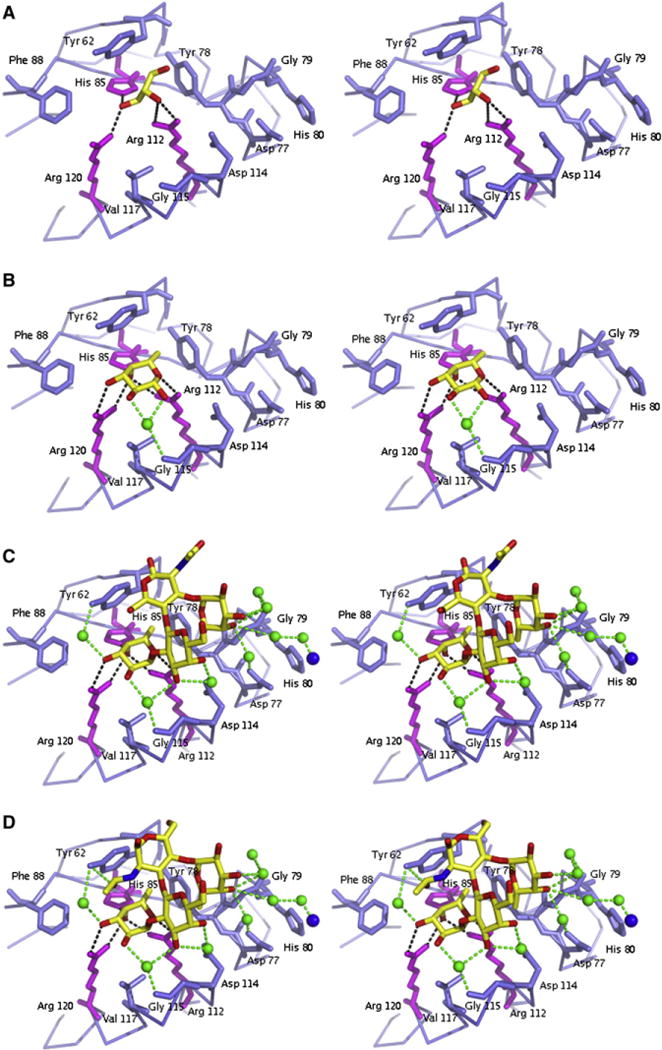
LLYlec is complexed to (A) glycerol, (B) α-L-fucose, (C) Ley antigen, and (D) Leb antigen. Residues that create the binding pocket are shown as blue and magenta sticks and the ligands as yellow sticks. Water molecules are shown as green spheres, and hydrogen bonds by dashed lines.
Glycan Interactions with LLYlec
LLYlec-Fucose Complex
The electron density for the α-L-fucose was well defined, located in the same pocket as the glycerol molecule described above, with an occupancy of 0.6 (Figure 2B). The fucose molecule is directly bound to the NE atom of His 85 and the NH1 and NH2 atoms of Arg 112 and Arg 120. Arg 112 is an important residue for binding of sugars by LLYlec as it was shown by Farrand et al. (2008) that the binding activity of LLYlec was eliminated when this residue was mutated to an alanine residue. There is one water-mediated hydrogen bond between fucose and Gly 115. The fucose molecule also takes part in hydrophobic interactions with Tyr 62, Tyr 78, Phe 88, and Val 117. In total, there are 94 van der Waals interactions between fucose and protein. The formation of the complex results in a buried surface area of more than two thirds (66% or 192 Å2) of the fucose molecule. The structure of the monomer is identical to that found in the LLYlec – glycerol complex, comprising the same number of residues and no significant changes in temperature factors.
LLYlec-Lewisy Antigen Complex
The overall protein structure of the Ley complex is identical to the LLYlec structures described earlier, with the same number of residues and no significant changes in temperature factors. The electron density for the Ley antigen Fuc(α1–2)Gal(β1–4) [Fuc(α1–3)]GlcNAc was readily modeled in the initial electron density maps. Henceforth, we refer to Fuc(α1–2) as the fucose 1 moiety and Fuc(α1–3) as the fucose 2 moiety. The Ley antigen is fully occupied in its binding site (Figure 2C). Fucose 1 of the Ley antigen was located in the same place and binds in the same manner as the fucose molecule seen in the structure of the LLYlec – fucose complex. In addition, there are eight water molecules that are involved in water-mediated hydrogen bonds between ligand and protein (including interactions with the Ni ion; see Experimental Procedures). Two of these water molecules mediate interactions between one fucose moiety and Gly 115 and Tyr 62, and another mediates an interaction between the galactose moiety and Asp 114. The remaining water molecules mediate interactions between the fucose 2 and Asp 77, Gly 79, and the Ni ion. In contrast, the GlcNAc moiety is not involved in any direct or indirect hydrogen bonds. In total, there are 19 van der Waals interactions between the ligand and the protein. Of these interactions, 15 are with fucose 1, 1 with galactose, 1 with GlcNAc, and 2 with fucose2. About one third (38% or 283 Å2) of the Ley antigen is buried because of the formation of the complex. Fucose 1, which is hydrogen bonded to residues His 85, Arg 112, and Arg 120, is embedded most in the shallow binding pocket. Galactose and fucose 2 are located further away from the pocket and increase the complementarity by forming water-mediated hydrogen bonds.
We wondered whether the Ley antigen could also bind into the fucose 1-binding site (described earlier) through the fucose 2 moiety instead. However, model building showed that, with fucose 2 deep in the pocket, the galactose moiety would likely clash with Tyr 62 and the fucose 1 moiety would have no interactions with the protein. Thus, fucose with an α1–2 linkage may be responsible for determining the specificity of LLY for Ley and Leb over Lex and Lea.
LLYlec-Lewisb Antigen Complex
The model of the Leb complex consists of residues 44 to 184. The electron density for the Leb antigen (Fuc(α1–2)Gal(β1–3) [Fuc(α1–3)]GlcNac) was clearly visible and located in the same binding pocket as found for the Ley antigen (Figure 2D). The site is fully occupied by the ligand in the crystal. The Ley and Leb antigens of the LLYlec complex structures superimpose well and there are no structural differences in the binding site of LLYlec. The only structural difference between Ley antigen and the Leb antigen is the opposing projections of the N-acetyl and CH2OH groups of the N-acetylglucosamine (GlcNAc). The hydrogen bonding interactions for Leb antigen are the same as for Ley antigen with the exception of the O atom in the N-acetyl group that forms a hydrogen bond to Tyr 62. Overall, there are 23 van der Waals interactions. The protein forms 15 van der Waals contacts with fucose 1, 1 with galactose, 3 with GlcNac and 4 with fucose 2. The complementarity of Leb to the protein is enhanced by water-mediated hydrogen bonds between galactose and fucose 2 with the protein, as was observed in the Ley complex structure. More than one third (40% or 292 Å2) of the total surface area of the Leb antigen and 221 Å2 of the total surface area of the protein is buried due to the formation of the complex.
LLYlec Dimers
All four crystal structures revealed a head-to-head dimer, generated by crystallographic symmetry (Figure 3). Superposition of all four complex structures showed that the structures are almost identical with a root-mean-square (rms) deviation of the alpha carbon atoms of ~0.15 Å (Figure 3). The total surface area of the dimer interfaces is approximately 318 Å2 per monomer. The residues of the monomers that are involved in dimer contacts are all located on a long loop opposite the β sandwich and on β strand 1. There are four direct hydrogen bonds—Gln 54A (OE1) to Gln 54B (NE2), Gln 54B (OE1) to Gln 54A (NE2), Gly 63A (O) to Arg 68B (NH1), and Gly 63B (O) to Arg 68A (NH1)—as well as four water-mediated hydrogen bonds at the interface. The recombinant forms of LLYlec and full-length LLY used in this study exist as mixtures of monomers and dimers in solution but were readily dissociated to monomers in the presence of DTT (data not shown). Thus, the LLYlec dimers seen in the crystal do not appear relevant to the intact toxin in its secreted state, although they may bear some relevance to how the toxin molecules pack in the prepore/pore forms of the toxin.
Figure 3. Superposition of the Dimers of LLYlec.
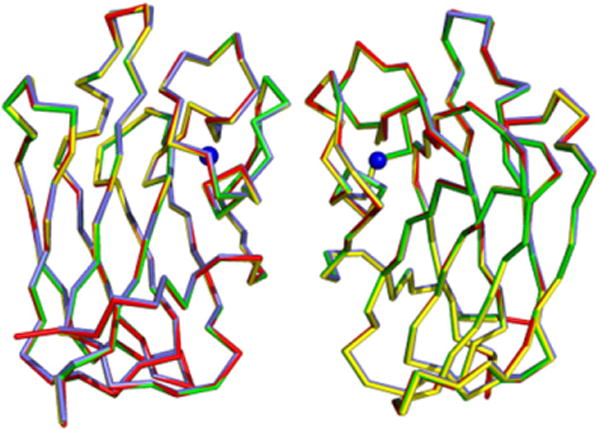
The dimers are created by crystallographic symmetry. The dimers of LLYlec, LLYlec-fucose, LLYlec-Ley, and LLYlec-Leb are shown in red, yellow, blue, and green, respectively. The Ca2+ ion is shown in blue.
Comparison with Other Lectin Domains
A search for similar structures to LLYlec in the Protein Data Bank using the DALI server (Holm and Sander, 1993) revealed two structures with scores greater than 20: Anguilla anguilla (eel) agglutinin (AAA) and the CBM family 98 glycoside hydrolase of Streptococcus pneumonia (SpX-1). LLYlec shares 33% sequence identity with AAA and 47% sequence identity with SpX-1. The crystal structure of the AAA fucolectin domain was shown to be a novel fold within the lectin family (Bianchet et al., 2002). Superposition of this structure and LLYlec shows similarities in their cores. The domains superimpose with rms deviations of the alpha carbon atoms of 1.5 Å (Figure 4A). In contrast to LLYlec, the fucolectin domain of AAA forms a tight trimer in the crystal. The AAA domain has additional residues at the N terminus and the C terminus, which form an antiparallel two-stranded β sheet. The loop between strands 5 and 6 in LLYlec has moved away from the core structure. Like the AAA domain, LLYlec contains five loops that form the boundary for the carbohydrate-binding site and connect the two β sheets at one end of the barrel. These loops were named complementarity-determining regions (CDRs) by Bianchet et al. (2002) in analogy with the nomenclature for immunoglobulins. In immunoglobulins, the CDRs are hypervariable regions and serve to recognize and bind specifically to antigens. The CDRs in LLYlec are CDR1 (Thr 60 to Gly 64), CDR2 (Val 74 to Val 83), CDR3 (Thr 86 to Asn 91), CDR4 (Arg 112 to Phe 124), and CDR5 (Leu 167 to Pro 172). CDR1 in AAA is five residues longer and not as tightly packed as in LLYlec. This loop creates a deeper pocket for the fucose compared to LLYlec. CDR4 and CDR5 are one residue longer in LLYlec compared to the AAA domain. As in AAA, CDR5 is the loop farthest away from the binding site. The loop between strands 4 and 5 is four residues longer than in LLY. In the AAA domain, there is an additional helix between β strands 1 and 2 (definition according to LLYlec). Among different fucolectins of Anguilla japonica (Japanese eel), the CDR loops around the binding site contain high sequence variability, suggesting that these loops modulate the fine specificity of lectins for different host cell receptors.
Figure 4. Structural Similarities of LLYlecto Other Proteins.
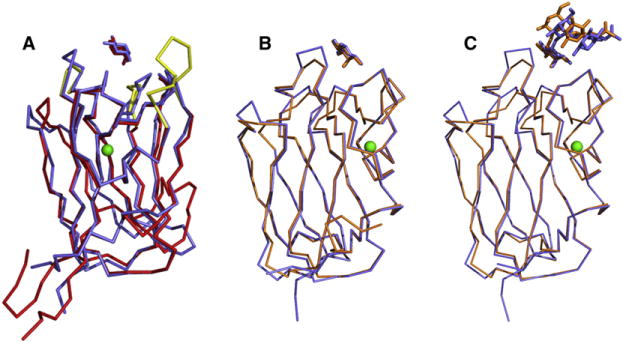
(A) Superposition of the LLYlec domain (in blue) on to the structure of the fucolection of AAA (in red). Both structures are complexed with fucose. The Ca2+ atom, which is located in the same positionin both structures, is shown in green. The CDR loops that are different in AAA, compared to the LLYLec domain, are shown in yellow.
(B) Superposition of the fucose complex structures of LLYlec domain (in blue) and the fucolectin module of SpX-1 (in orange).
(C) Superposition of the Ley antigen complex structures of LLYlec domain (in blue) and the fucolectin module of SpX-1 (orange).
Fucose is positioned in the same pocket in AAA, as observed in the LLYlec structures, and makes contact with the conserved residues His 52, Arg 79, and Arg 86 (His 85, Arg 112, and Arg 120 in LLYlec). However, AAA lacks affinity for Ley antigens or any other antigens belonging to type 2, but it can bind the fucosylated terminals of H and Lewis (a) blood groups. Modeling by Bianchet et al. (2002) suggested that the CDR1 loop of AAA must undergo a large structural disruption for binding of type 2 antigens to occur, but the loop is very rigid with low B factors and hence unlikely to be able to move. A Ca2+ ion is located in the same position in the AAA and LLYlec structures. It was found that the addition of calcium to AAA and related proteins (Saito et al., 1997) increased the affinity for carbohydrates despite the fact that the Ca2+ ion was not directly located near the residues involved in sugar binding. Thus, the ion is considered to play an important structural role in carbohydrate recognition.
The structure of SpX-1 (Boraston et al., 2006) was determined uncomplexed (PDB code 2J1R) and in complex with a number of carbohydrates including fucose (PDB code 2J1S), the type 2 H-trisaccharide (PDB code 2J1V), the blood group A-tetrasaccharide analog (PDB code 2J1U), and the Ley antigen (PDB code 2J1T). SpX-1 forms a head-to-tail dimer in the asymmetric unit in contrast with the head-to-head dimer found for LLYlec. It is not clear whether the SpX-1 dimer has any physiological relevance. Superposition of the alpha carbons of the LLYlec on to SpX-1 gives rms deviations of 0.6 Å (Figure 4B). Thus, the structures superimpose closely, but there are differences in the loop regions. The loop connecting β strands 4 and 5 is one residue longer in SpX-1 and points outward in SpX-1 rather than upward (because of crystal contacts). CDR4 is one amino acid longer in LLYlec compared to SpX-1. The tips of CDR3 and CDR5 are slightly shifted outward compared to the LLYlec structure as the residues form crystal contacts to the other monomer of the dimer.
Fucose, in complex with SpX-1, as well as in the complex structure of SpX-1 and Ley antigen, is located in the same pocket and makes the same contacts as those seen for fucose in the LLYlec complex structure. Again, a Ca2+ ion is positioned in SpX-1 in the same binding pocket as that found in the LLYlec and AAA structures. Analysis of the complex structure of SpX-1 and Ley antigen shows that the Gal-GlcNAc-Fuc groups of the Ley antigen have shifted approximately 2.9 Å compared to the same groups in the LLYlec-LeY complex (Figure 4C). This is due to differences in the amino acid sequence of the CDR2 and CDR4 loops and crystal contacts in the complex structure of SpX-1 and Ley antigen compared to LLYlec. It is not surprising that the water network around the Ley antigen is strikingly different between LLYlec and SpX-1. The water-mediated hydrogen bonds between fucose 2 and protein and between galactose and Asp 114 pulls the Ley antigen further into the binding pocket of LLYlec compared to SpX-1.
LLYlec Recognition of Lewis Antigens and Comparison with Other Lewis Antigen-Binding Proteins
Lea of type 1 and Lex of type 2 structures contain one fucose unit, whereas Leb of type 1 and Ley of type 2 have two fucose units each. The difference between type 1 and 2 antigens is the core disaccharide linkage with an opposing projection of the N-acetyl and –CH2OH groups of the N-acetylglucosamine monosaccharide. The overall Lewis determinants maintain the same conformation in both free and bound states (Yuriev et al., 2005). The rings of the two constant moieties, fucose and galactose, are usually tightly stacked in a rigid conformation. The addition of the fucose residue to the GlcNAc residue (forming Ley from type 2 and Leb from type 1 chains) does not cause a significant conformational change. To date, only a few structures complexed to Lewis antigens have been described. Apart from SpX-1 (described earlier), lectin structures complexed with Ley and Leb antigens have been reported for the legume lectin from Griffonia simplifolia (Delbaere et al., 1993) (PDB codes: 1LED and 1GSL). The structures of both Ley and Leb antigen as observed bound to the legume lectins and to LLYlec are very similar despite the binding pockets adopting very different shapes and nonsuperimposable CDR loops (Figure 5A).
Figure 5. Protein Recognition of Lewis Antigens.
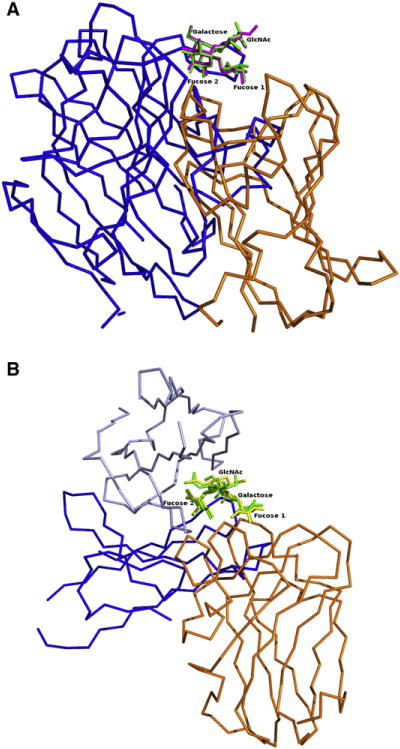
All superpositions are based on the Lewis antigen, and LLYlec is shown in orange.
(A) Griffonia simplifolia legume lectin (PDB code: 1LED) is shown in blue with Lewis b antigens superimposed.
(B) Monoclonal antibody hu3S193 (PDB code: 1SK3) heavy and light chains are shown in different shades of blue, with Lewis y antigens superimposed.
The structure of a humanized antibody with high specificity toward type 2 antigens (hu3S193) has been reported as a complex with Ley (Ramsland et al., 2004; Farrugia et al., 2009). The Ley antigen was positioned in a pocket between heavy and light chains of the antibody, and each of the hexose rings was involved in hydrogen bonding (Figure 5B). The structure also revealed an extensive network of water molecules, which was thought to play a role in recognition of the Ley antigen. Specificity of Ley over Leb antigen was determined by chemical restraints. The N-acetyl oxygen atoms of Ley are bound by a conserved hydrogen bond to the main-chain amide group of residue Tyr 33H. Additionally, the side chain of Tyr 33H was stacked against the hexose ring of GlcNAc and parts of the adjoining Gal moiety. These interactions are not possible with type 1 Lewis structures, as the N-acetyl groups of the N-acetyl-glucosamine would be forced away from the specific pocket formed by Tyr 33H. The specificity for type 2 antigens was further enhanced in the hu3S193 complex by solvent-mediated hydrogen bonding of the N-acetyl and CH2OH groups of GlcNAc. Comparison of the Ley antigens of the LLYLec structure with the Ley antibody structure reveal that the Lewis carbohydrate determinants are similar but that the shape of the binding pocket formed by the heavy and light chains and the amino acids involved in binding to Ley of the antibody are very different compared to the CDR loops of the lectin structures (Figure 5B). Fucose 1 of Ley in the LLYlec complex structure is the anchoring glycan (the moiety that is deepest in the binding pocket), whereas, in the antibody complex, fucose 2 is embedded deepest in the pocket.
LLYlec Has an Integrin-Binding RGD Motif
LLYlec has an integrin-binding Arg-Gly-Asp (RGD) motif located on the loop between β strands 5 and 6 (Figure 1), and Arg 112 of the motif has been shown to be critical for Lewis antigen binding (Farrand et al., 2008). The same motif, located in the same position, is found in AAA (Bianchet et al., 2002). We were interested in whether the motif could be functional as an integrin-binding site. First, we superimposed the RGD loop of LLYlec on to the structure of an RGD pentapeptide bound to the integrin αVβ3 (PDB id: 1L5G; Xiong et al., 2002). The superposition resulted in clashes between LLYlec and the integrin, suggesting that, if they do bind, then conformational changes are required of one or both binding partners. There is evidence in the literature that the integrin can undergo large quaternary comformational changes on binding RGD-containing proteins (Lee et al., 1995; Xiong et al., 2002; Takagi et al., 2003).
Small-Angle X-Ray Scattering of Intact LLY
To experimentally determine the position of the LLYlec domain with respect to the LLYCDC, we performed SAXS analysis of the full-length toxin. (More details are provided in the Supplemental Information, including Figure S2.) We calculated an ab initio shape envelope of LLY from the P(r) distribution using dummy atom modeling (Franke and Svergun, 2009). The envelope was elongated and relatively flat but with a slight bend. Computational docking of available CDC crystallographic models, representing LLYCDC, into the shape envelope suggested that the bend was best described by the structure of ALO (Bourdeau et al., 2009). LLYCDC and ALO share 29.6% pairwise sequence identity. These studies also revealed the presence of contiguous unfitted electron density in the plane of the LLYCDC adjacent to its N terminus (Figure 6A). Therefore, a homology model of the LLYCDC was generated with the ALO coordinates, and the SAXS data were used to establish the optimal relative positioning of the LLYlec domain. Simulated annealing and random search methods identified similar LLYlec positions that were consistent with the ab initio shape envelope (Figures 6A and 6B).
Figure 6. Comparison of the Ab Initio Shape Envelope with Computationally Docked and SAXS-Derived LLY Models.
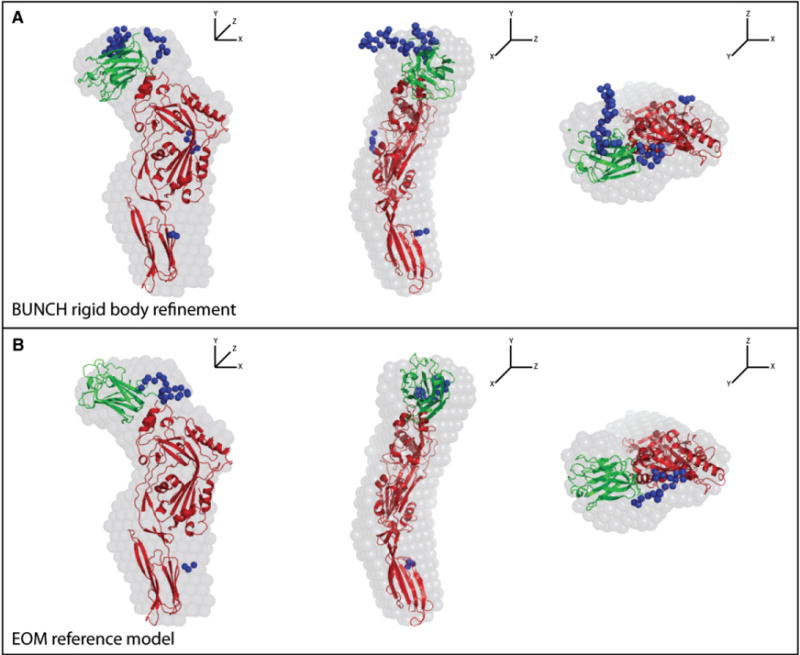
Each panel shows orthogonal views of the fits. The blue spheres represent missing C-terminal residues and loops between secondary structural elements, including the linker peptide between the lectin domain and the rest of the toxin molecule, which were missing in the ALO structure that was used in the modeling. See also Figure S2 for the SAXS data.
(A) Rigid-body refinement of the position of the LLYlec domain with BUNCH (Petoukhov et al., 2005) generated an ensemble of models that were in good agreement with the experimental scattering profile. In all these models, the position of the LLYlec domain corresponded well to the unoccupied density proximal to the N terminus of the LLYCDC domains in the shape envelope. The BUNCH model closest to the average position is shown.
(B) As an alternate method of identifying likely LLYlec domain positions, a large pool of models with randomized LLYlec and LLYCDC orientations, constrained by the intervening linker sequence, were generated and interrogated with the EOM (Bernadó et al., 2007). The individual EOM models in the pool with theoretical scattering profiles that best matched the experimental curve were identified. The EOM model closest to the average position is shown.
Overall, the SAXS analysis indicates that (1) the LLYCDC region is flat and elongated in solution, as seen in the crystal structures of other CDCs; (2) domain 4 is angled with respect to domains 1–3, and the angle is similar to that in ALO; and (3) the LLYlec domain packs its N and C termini against the N terminus of the LLYCDC, thus exposing the Lewis antigen-binding site.
DISCUSSION
To date, the only CDC described to have an N-terminal lectin domain has been LLY. The structures presented here reveal that LLYlec has a F-type lectin fold, first described in the crystal structure of AAA in 2002 (Bianchet et al., 2002). Comparison of the two structures reveals that the core structures are very similar but that there are differences in the CDR loops. These loops have been shown to exhibit a high degree of sequence and conformational variability among the different AAAs so they can recognize different glycans. One of the loops in the AAA domain is longer than in the LLYlec domain and was very rigid. Such features are presumably the reason why AAA lacks affinity for type 2 antigens and prefers to form complexes with type 1 antigens. Structural comparison of LLYlec with SpX-1, the fucose-binding module of the putative Streptococcus pneumoniae family 98 glycoside hydrolase, revealed smaller differences in the loops, and, in contrast to AAA, the SpX-1 domain shows affinity for a number of oligosaccharides. The complex structure of Ley bound to SpX-1 shows that the Lewis antigen is located in the same pocket as in LLYlec, but a shift was observed because of differences in residues of the binding pocket compared to LLYlec and the presence of crystal contacts in SpX-1.
The structures of the complexes with fucose and the blood group antigens Ley and Leb demonstrate that the main binding determinant is one of the fucose rings—fucose 1, which interacts with the three residues, His 85, Arg 112, and Arg 120—with the other fucose ring (fucose 2) interacting with the protein through mainly water-mediated interactions. The equivalent residues are highly conserved in other lectin domains. Arg 112 is part of an RGD motif, which is found in cell adhesion proteins (Pierschbacher et al., 1985) and is a typical integrin-binding motif (Xiong et al., 2002). The presence of the motif in LLYlec suggests that it may have integrin-binding properties. However, integrin binding of the LLYlec domain has not been reported, and our analysis suggests that the LLYlec domain is unlikely to form a complex with integrins, unless large conformational changes take place in both the integrin and the lectin. The other sugar rings of the Ley and Leb antigens make relatively few direct contacts with protein residues.
The SAXS analysis of the full-length toxin suggests that LLYCDC domains 1–4 are in an extended planar arrangement in solution, as seen in crystal structures for other CDCs, with a slight bend between domains 3 and 4, similar in amplitude to that in ALO (Bourdeau et al., 2009). The SAXS modeling predicts that the C terminus of the LLYlec domain is close to the N terminus of domain 1 of the LLYCDC region. The LLYlec domain lies in the plane of the LLYCDC, with the Lewis antigen- binding site distal from the CDC. In this arrangement, the Lewis antigen-binding site would be exposed on the outer surface of the pore based on models of the pneumolysin pore generated by electron microscopy (Gilbert et al., 1999; Tilley et al., 2005).
Farrand et al. (2008) showed that the binding site for saccharides is occluded in the full-length toxin but is revealed on binding of the toxin to the cell surface. However, the SAXS model shows that the Lewis antigen-binding site is exposed in full-length toxin and distal from the rest of the toxin with a distance between the C terminus of LLYlec and the N terminus of the LLYCDC of about 34 Å, a distance that could be straddled by the missing 12-residue linker that joins the two. An alternative model in which the lectin-binding site is buried would increase the distance to about 44 Å but would require a much longer linker to join the two ends and thus is not possible. A plausible explanation for the observation of Farrand et al. is that it was their N-terminal His tag (in particular, a highly charged acidic sequence DDDDK associated with an enterokinase cleavage site), rather than the rest of the toxin, that occluded the Lewis antigen-binding site in the holotoxin.
In healthy humans, Ley is expressed on cells in the upper gastrointestinal tract (such as in the mouth and esophagus). Because S. mitis normally colonizes the mouth, it is possible that Ley expression is required for its establishment and/or maintenance. Because Leb is more widespread, it suggests that Ley is the primary ligand for initial infection and subsequent cell lysis by the bacteria. Recently, LLY was shown to belong to the CD59-binding family of CDCs, together with ILY and vaginolysin (Wickham et al., 2011). These CDCs use CD59, a GPI-anchored protein, as a receptor on the surface of cells rather than membrane cholesterol like the archetypical CDCs. It is intriguing that CD59 has fucose linkages in the outer arms of its N-linked glycan branches; hence, it is possible that the lectin domain of LLY might bind to the N-linked glycan of CD59 as well as through direct protein-protein interactions with domain 4 of the toxin (Wickham et al., 2011). In any case, the presence of the lectin domain in LLY suggests that its role is to enhance host cell recognition by guiding the toxin to fucose-rich sites on the surface of cells. In doing so, the toxin would concentrate at such sites, thus promoting oligomerization and conversion into the prepore state. It is possible that the specific location of the fucose-binding site at one tip of the LLY molecule may act to orient the toxin molecules with respect to each other and the cell surface, thus enhancing head-to-head formation of productive oligomers.
Ley antigens are expressed at high levels in many cancers of epithelial cell origin (e.g., colon, breast, lung, prostate, and ovary) and, therefore, are a promising target for antibody-based immunotherapy (Yuriev et al., 2005). Thus, antibodies have been sought that show a high degree of specificity for type 2 target antigen while avoiding cross-reaction with type 1 antigens. The conjugation of tumor-specific antibodies with cytotoxic molecules has been shown to dramatically improve their clinical utility. This has prompted one group recently to examine the usefulness of conjugating a CDC, listeriolysin O, to a monoclonal antibody that recognizes Lewis Y (Bergelt et al., 2009). Thus, it is intriguing that nature has already engineered one such molecule, LLY, to perform a similar job! The challenges ahead will be: to understand the molecular basis by which the lectin domain regulates LLY cytolytic activity and to explain why some S. mitis strains produce this CDC and whether it contributes to disease and/or the commensal state. Finally, we can engineer the LLY lectin domain so that it specifically recognizes the Ley antigen, which may be useful for the detection and/or targeting of Ley-expressing tumor cells.
EXPERIMENTAL PROCEDURES
Generation and Purification of LLYlec
A mutant of LLYlec (Q190C) with an amino-terminal 6×His tag and a TEV protease cleavage site for His tag removal was used in this study. The fusion protein was expressed in E. coli BL21 (DE3) and purified as detailed in the Supplemental Information. Purified LLYlec was concentrated to 10 mg/ml and stored at −80°C.
Crystallization, Data Collection, and Structure Determination
We performed all crystallizations using the hanging drop vapor diffusion method at 21°C. Protein (2 μl) was mixed with equal volumes of protein and hung over 1 ml of well solution. For data collection, all crystals were dipped quickly and consecutively into 5% (vol/vol), 10% (vol/vol), and 15% (vol/vol) glycerol in crystallization buffer. We collected data in house using an R-axis IV imaging plate detector (Rigaku, Japan) with X-rays from a Micromax 007 X-ray generator (Rigaku, Japan), except for data from the apo LLYlec crystals, which were collected at the MX1 beamline at the Australian Synchrotron (Clayton, Victoria, Australia). Data collection was controlled with Blue-Ice software (McPhillips et al., 2002). All data were processed with the HKL2000 suite (Otwinowski and Minor, 1997), and model building, including selection of water molecules, was performed with COOT (Emsley and Cowtan, 2004). Data were refined with REFMAC 5 (Murshudov et al., 1997) from the CCP4 program suite [CCP4 (Collaborative Computational Project) Program Suite, 1994]. During refinement, we identified a putative Ni2+ ion that was covalently bound to residue His 80 (Ni-N distance of 2.2 Å) and coordinated to five water molecules forming an octahedral geometry about the metal ion. We cannot rule out that the ion may be Mg2+ because of the very high concentrations of this ion in the crystallization buffer, even though this metal rarely forms covalent bonds with nitrogen ligands. If Ni2+, the likely source of this ion was the Ni resin used in the purification of LLYlec. Data statistics for all structures are listed in Table 1.
We set up crystallization trials of apo LLYlec using the protein crystallization screen (Zeelen et al., 1994), with the protein at a concentration of 5 mg/ml. Bipyramidial crystals appeared after 5 days in 2 M MgSO4 and 100 mM Tris buffer, pH 8.5. After screening around this condition, the best crystals were obtained in 2 M MgSO4 and 100 mM Tris buffer, pH 8.2 to 8.6, with maximal dimensions of 0.15 mm × 0.12 mm × 0.12 mm. The crystals diffracted to resolution of 1.9 Å in the tetragonal space group P43212. The structure was solved with the program Phaser (Storoni et al., 2004; McCoy et al., 2007), with one monomer of the fucose-binding module of the putative Streptococcus pneumoniae family 98 glycoside hydrolase (PDB code: 2J1R) used as a search model. The sequence identity between the two domains is 47% (similarity of 67%). The results from Phaser gave a Z score of 27.3. One monomer was found in the asymmetric unit and gave a Matthews coefficient (Matthews, 1968) of 3.30 Da/Å3 and a solvent content of 63%. After omitting a bound glycerol molecule, we used the refined model as a starting model for determining the initial phases for all the complexed carbohydrate structures.
Crystallization of the LLYlec-fucose complex was performed by incubating the protein with 5 mM α-L-fucose (Sigma, Castle Hill, Australia) for 5 hr before setting up the protein crystallization screen (Zeelen et al., 1994). Small crystals were obtained under the same condition as the apo LLYlec crystals but took 14 days to grow instead of 5 days. These crystals exhibited the same shape but were slightly smaller. The difference electron density maps for this complex suggested that fucose was only partially occupied. An estimate of its occupancy was made based on the disappearance of peaks/troughs in difference Fourier maps and was supported by refined ligand B factors being very similar to those of the surrounding side chains. To obtain crystals of the LLYlec-Ley antigen complex, we soaked LLYlec crystals for 5 hr with Ley antigen (Sigma) at a concentration of 5 mM. Similarly, the Leb antigen complex was prepared by soaking apo LLYlec crystals in 5 mM Leb (Sigma) overnight.
The Protein Interfaces, Surfaces and Assemblies server (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) was used for all protein-ligand surface interaction calculations.
SAXS
Hexahis _TEV_LLYT168C, a mutant of LLY, was expressed and purified for SAXS analysis of full-length LLY (see Supplemental Information). We recorded SAXS data on the SAXS/WAXS beamline at the Australian Synchrotron using in-line gel filtration (Gunn et al., 2011). We performed ab initio shape reconstructions using DAMMIF (Franke and Svergun, 2009). Averaged filtered shape envelopes were generated from ensembles of DAMMIF envelopes with DAMAVER (Volkov and Svergun, 2003). We generated theoretical scattering profiles from model coordinates and compared them to experimental data using CRYSOL (Svergun et al., 1995), and the statistical analysis of goodness of fit—Pχ(χ2,ν)—and the relative improvement between fits—PF(F,ν1,ν2)—were performed as described by Mills et al. (2009). Rigid body refinement was carried out with MASSHA (Konarev et al., 2001) and BUNCH (Petoukhov and Svergun, 2005) to identify the likely position of the LLYlec domain. We performed a more extensive search for alternative full-length models consistent with the SAXS data using the ensemble optimized method (EOM) (Bernadó et al., 2007).
SUPPLEMENTAL INFORMATION
Supplemental Information includes two figures and Supplemental Experimental Procedures and can be found with this article online at doi:10.1016/j.str.2011.11.017.
Supplementary Material
Acknowledgments
This research was partly undertaken on the MX1 and SAXS/WAXS beamlines at the Australian Synchrotron, Victoria, Australia. We thank Dr. Julian Adams and the other beamline staff for their assistance. We also thank David Ascher for help with mass spectrometry and Dr. Paul Ramsland for expert comments on our manuscript. We thank Dr. Mike Kuiper for his help in the preparation of the graphical abstract. This work was partly carried out in the Australian Cancer Research Foundation Rational Drug Discovery Facility. This work was also supported by a grant from the National Health and Medical Research Council of Australia (NHMRC) to M.W.P. and funding from the Victorian Government Operational Infrastructure Support Scheme to St Vincent’s Institute. R.K.T. received support from Grant AI037657 from the National Institutes of Health. S.C.F. was supported by a NHMRC Industry Fellowship. M.W.P. is an Australian Research Council Federation Fellow and NHMRC Honorary Fellow.
Footnotes
ACCESSION NUMBERS
The crystal structures have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb/) under the filenames 3LE0 (LLYlec), 3LEI (fucose complex), 3LEG (Ley complex), and 3LEK (Leb complex).
References
- Bergelt S, Frost S, Lilie H. Listeriolysin O as cytotoxic component of an immunotoxin. Protein Sci. 2009;18:1210–1220. doi: 10.1002/pro.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- Bianchet MA, Odom EW, Vasta GR, Amzel LM. A novel fucose recognition fold involved in innate immunity. Nat Struct Biol. 2002;9:628–634. doi: 10.1038/nsb817. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Wang D, Burke RD. Blood group antigen recognition by a Streptococcus pneumoniae virulence factor. J Biol Chem. 2006;281:35263–35271. doi: 10.1074/jbc.M607620200. [DOI] [PubMed] [Google Scholar]
- Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–781. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau RW, Malito E, Chenal A, Bishop BL, Musch MW, Villereal ML, Chang EB, Mosser EM, Rest RF, Tang WJ. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J Biol Chem. 2009;284:14645–14656. doi: 10.1074/jbc.M807631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project Program Suite) The CCP4 Suite: programs for protein crystallography. Acta Crystallogr. D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Delano WL. The PyMol Molecular Graphics System. Palo Alto, CA: Delano Scientific; 2002. [Google Scholar]
- Delbaere LT, Vandonselaar M, Prasad L, Qual JW, Wilson KS, Dauter Z. Structures of the lectin IV of Griffonia simplicifolia and its complex with the Lewis b human blood group determinant at 2.0 A resolution. J Mol Biol. 1993;230:950–965. doi: 10.1006/jmbi.1993.1212. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Farrand S, Hotze E, Friese P, Hollingshead SK, Smith DF, Cummings RD, Dale GL, Tweten RK. Characterization of a streptococcal cholesterol-dependent cytolysin with a Lewis y and b specific lectin domain. Biochemistry. 2008;47:7097–7107. doi: 10.1021/bi8005835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia W, Scott AM, Ramsland PA. A possible role for metallic ions in the carbohydrate cluster recognition displayed by a Lewis Y specific antibody. PLoS ONE. 2009;4:e7777. doi: 10.1371/journal.pone.0007777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J Appl Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert RJ, Jiménez JL, Chen S, Tickle IJ, Rossjohn J, Parker M, Andrew PW, Saibil HR. Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae. Cell. 1999;97:647–655. doi: 10.1016/s0092-8674(00)80775-8. [DOI] [PubMed] [Google Scholar]
- Gowda RM, Ansari AW, Khan IA. Complete endocardial cushion defect (complete atrioventricular canal) manifested in adult life by Streptococcus mitis endocarditis of the common atrioventricular valve. Int J Cardiol. 2003;89:109–110. doi: 10.1016/s0167-5273(02)00459-x. [DOI] [PubMed] [Google Scholar]
- Gunn NJ, Gorman MA, Dobson RCJ, Parker MW, Mulhern TD. Purification, crystallization, small- angle scattering and preliminary X-ray diffraction analysis of the SH2 domain of Csk-homologous kinase. Acta Crystallogr F. 2011;67:336–339. doi: 10.1107/S1744309110053728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall GE, Baddour LM. Apparent failure of endocarditis prophylaxis caused by penicillin-resistant Streptococcus mitis. Am J Med Sci. 2002;324:51–53. doi: 10.1097/00000441-200207000-00008. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Huang IF, Chiou CC, Liu YC, Hsieh KS. Endocarditis caused by penicillin-resistant Streptococcus mitis in a 12-year-old boy. J Microbiol Immunol Infect. 2002;35:129–132. [PubMed] [Google Scholar]
- Imberty A, Varrot A. Microbial recognition of human cell surface glycoconjugates. Curr Opin Struct Biol. 2008;18:567–576. doi: 10.1016/j.sbi.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Jackson MA, Kearns GL. Delayed diagnosis of penicillin-resistant Streptococcus mitis endocarditis following single-dose amoxicillin prophylaxis in a child. Clin Pediatr (Phila) 2004;43:773–776. doi: 10.1177/000992280404300814. [DOI] [PubMed] [Google Scholar]
- Konarev PV, Petoukhov MV, Svergun DI. MASSHA–agraphic system for rigid body modelling of macromolecular complexes against solution scattering data. J Appl Cryst. 2001;34:527–532. [Google Scholar]
- Lee JO, Bankston LA, Arnaout MA, Liddington RC. Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure. 1995;3:1333–1340. doi: 10.1016/s0969-2126(01)00271-4. [DOI] [PubMed] [Google Scholar]
- Lu HZ, Weng XH, Zhu B, Li H, Yin YK, Zhang YX, Haas DW, Tang YW. Major outbreak of toxic shock-like syndrome caused by Streptococcus mitis. J Clin Microbiol. 2003;41:3051–3055. doi: 10.1128/JCM.41.7.3051-3055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, et al. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- Mills RD, Trewhella J, Qiu TW, Welte T, Ryan TM, Hanley T, Knott RB, Lithgow T, Mulhern TD. Domain organization of the monomeric form of the Tom70 mitochondrial import receptor. J Mol Biol. 2009;388:1043–1058. doi: 10.1016/j.jmb.2009.03.070. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macro-molecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nagamune H, Whiley RA, Goto T, Inai Y, Maeda T, Hardie JM, Kourai H. Distribution of the intermedilysin gene among the anginosus group streptococci and correlation between intermedilysin production and deep-seated infection with Streptococcus intermedius. J Clin Microbiol. 2000;38:220–226. doi: 10.1128/jcm.38.1.220-226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkuni H, Todome Y, Okibayashi F, Watanabe Y, Ohtani N, Ishikawa T, Asano G, Kotani S. Purification and partial characterization of a novel human platelet aggregation factor in the extracellular products of Streptococcus mitis, strain Nm-65. FEMS Immunol Med Microbiol. 1997;17:121–129. doi: 10.1111/j.1574-695X.1997.tb01004.x. [DOI] [PubMed] [Google Scholar]
- Ohkuni H, Todome Y, Takahashi H, Nagamune H, Abe J, Ohtsuka H, Hatakeyama H. Antibody titers to Streptococus mitis -derived human platelet aggregation factor (Sm-hPAF) in the sera of patients with Kawasaki disease. In: Sriprakash KS, editor. Proceedings of the 16th Lancefield International Symposium on Streptococci and Streptococcal Diseases. Palm Cove, Australia: Elsevier B.V.; 2006. pp. 71–74. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in the oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Petoukhov MV, Svergun DI. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 2005;89:1237–1250. doi: 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierschbacher MD, Hayman EG, Ruoslahti E. The cell attachment determinant in fibronectin. J Cell Biochem. 1985;28:115–126. doi: 10.1002/jcb.240280205. [DOI] [PubMed] [Google Scholar]
- Polekhina G, Giddings KS, Tweten RK, Parker MW. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc Natl Acad Sci USA. 2005;102:600–605. doi: 10.1073/pnas.0403229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsland PA, Farrugia W, Bradford TM, Mark Hogarth P, Scott AM. Structural convergence of antibody binding of carbohydrate determinants in Lewis Y tumor antigens. J Mol Biol. 2004;340:809–818. doi: 10.1016/j.jmb.2004.05.037. [DOI] [PubMed] [Google Scholar]
- Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell. 1997;89:685–692. doi: 10.1016/s0092-8674(00)80251-2. [DOI] [PubMed] [Google Scholar]
- Rossjohn J, Polekhina G, Feil SC, Morton CJ, Tweten RK, Parker MW. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J Mol Biol. 2007;367:1227–1236. doi: 10.1016/j.jmb.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hatada M, Iwanaga S, Kawabata S. A newly identified horseshoe crab lectin with binding specificity to O-antigen of bacterial lipopolysaccharides. J Biol Chem. 1997;272:30703–30708. doi: 10.1074/jbc.272.49.30703. [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Svergun DI, Barberato C, Koch MHJ. CRYSOL – a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- Takagi J, Strokovich K, Springer TA, Walz T. Structure of integrin α5β1 in complex with fibronectin. EMBO J. 2003;22:4607–4615. doi: 10.1093/emboj/cdg445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley SJ, Orlova EV, Gilbert RJ, Andrew PW, Saibil HR. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell. 2005;121:247–256. doi: 10.1016/j.cell.2005.02.033. [DOI] [PubMed] [Google Scholar]
- Tweten RK. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham SE, Hotze EM, Farrand AJ, Polekhina G, Nero TL, Tomlinson S, Parker MW, Tweten RK. Mapping the intermedilysin-human CD59 receptor interface reveals a deep correspondence with the binding site on CD59 for complement binding proteins C8α and C9. J Biol Chem. 2011;286:20648–20657. doi: 10.1074/jbc.M111.237446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin α Vbeta3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- Xu L, Huang B, Du H, Zhang XC, Xu J, Li X, Rao Z. Crystal structure of cytotoxin protein suilysin from Streptococcus suis. Protein Cell. 2010;1:96–105. doi: 10.1007/s13238-010-0012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuriev E, Farrugia W, Scott AM, Ramsland PA. Three-dimensional structures of carbohydrate determinants of Lewis system antigens: implications for effective antibody targeting of cancer. Immunol Cell Biol. 2005;83:709–717. doi: 10.1111/j.1440-1711.2005.01374.x. [DOI] [PubMed] [Google Scholar]
- Zeelen JP, Hiltunen JK, Ceska TA, Wierenga RK. Crystallization experiments with 2-enoyl-CoA hydratase, using an automated ‘fast-screening’ crystallization protocol. Acta Crystallogr D Biol Crystallogr. 1994;50:443–447. doi: 10.1107/S0907444994001277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.