

Abstract
The production of human induced pluripotent stem cells (hiPSCs) has greatly expanded the realm of possible stem cell-based regenerative medicine therapies and has particularly exciting potential for autologous therapies. However, future therapies based on hiPSCs will first have to address not only similar regulatory issues as those facing human embryonic stem cells with the US FDA and international regulatory agencies, but also hiPSCs have raised unique concerns as well. While the first possible clinical use of hiPSCs remains down the road, as a field it would be wise for us to anticipate potential roadblocks and begin formulating solutions. In this article, I discuss the potential regulatory issues facing hiPSCs and propose some potential changes in the direction of the field in response.
Keywords: FDA, hiPSC, induced pluripotent stem cell, iPS cell, stem cell, translational medicine
Background
The field of cellular reprogramming and human induced pluripotent stem cells (hiPSCs) continues to mature half a dozen years after mouse reprogramming was first reported [1]. The explosion of excitement surrounding hiPSCs stemmed from their potential to hurdle two crucial issues facing human embryonic stem cells (hESCs): the use of human blastocysts and immune rejection [2,3]. It is also notable that hESCs can now be produced without blastocyst destruction using, for example, the blastomere technology of Advanced Cell Technology (ACT) used to produce hESC lines and then subsequently retinal pigmented epithelial (RPE) cells. Since their first production, a number of important issues have arisen regarding the possible future clinical use of hiPSCs [4,5], including potentially suboptimal genomic and epigenomic states in hiPSCs, as well as some evidence of immunogenicity of mouse induced pluripotent stem cells (iPSCs) in syngeneic recipients. In this article, I discuss the main regulatory issues that hiPSC-based therapies are predicted to face with the US FDA and propose some potential changes in the direction of the hiPSC field to anticipate these possible future regulatory hurdles (Figure 1).
Figure 1. Scientific and regulatory steps required for human embryonic stem cell- and human induced pluripotent stem cell-based therapies in the early development of cell therapies from bench to bedside.
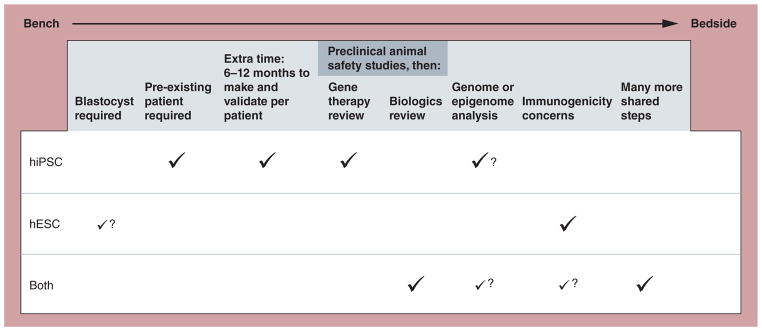
A check mark indicates a required step. A question mark indicates a potentially required future step. For ‘Blastocyst required’ for hESCs, a small check mark with a question mark indicates that while most hESC lines are still derived from blastocysts, new technology using alternative sources, such as most notably Advanced Cell Technology’s blastomere technology, are emerging. For ‘Genome and epigenome analysis’, the small check mark for ‘Both’ reflects the fact that such validation could be required for hESCs as well, but that the current science suggests a more pressing need regarding this step for hiPSCs. The small check mark for ‘Both’ for ‘Immunogenicity concerns’ reflects two factors: the uncertainty at this time whether hiPSCs will always be recognized as ‘self’ in otherwise syngeneic recipients; and the potential allogenic use of hiPSC-derived therapies.
hESC: Human embryonic stem cell; hiPSC: Human induced pluripotent stem cell.
The safety of hiPSC-based therapies
Based on historical precedents preceding even the first production of hESCs, it seems certain that one of the primary concerns of the FDA regarding clinical use of hiPSC-based biologics is going to be safety, particularly as it relates to how the methods used to produce hiPSCs impact safety. In this regard, it is important to distinguish between two main types of hiPSC-based therapeutic products: the use of hiPSCs themselves for transplantation versus employing products made from hiPSCs, such as differentiated cells or proteins produced by the hiPSCs. The former approach is widely viewed as greatly more challenging given the teratoma-forming activity of hiPSCs. To date, the latter approach has been the one primarily pursued in the form of differentiated derivatives based on hESCs. For example, ACT has two ongoing combined Phase I/II clinical trials for treating macular dystrophy using differentiated RPE cells, and Geron (studies now halted) used hESC-derived oligodendrocyte precursors to treat spinal cord injuries. The experiences of ACT and Geron are instructive for clinical applications of hiPSCs in terms of the requirements for preclinical data prior to approving Investigational New Drugs (INDs) and also for early-phase clinical trials based on hESCs. One can imagine in the future similar kinds of studies based on hiPSC-derivative differentiated cells as well. Another important distinction related to hiPSC therapies in development is their autologous and allogeneic use. While in principle hiPSCs are from one perspective inherently most ideal for autologous therapies, in practice hiPSCs could be used for both autologous and allogeneic therapies. Indeed, it is even possible that the first clinical use of hiPSC-derived therapies could be allogeneic in nature.
For both ACT and Geron, the FDA has been appropriately fastidious in terms of the requirements for preclinical data prior to approving INDs and also for early-phase clinical trials based on hESCs. An informative example of this is the nearly year-long delay for Geron because of what turned out to be cysts at the sites of injection. While the cysts appeared benign and the trial was ultimately allowed to proceed, I believe the caution was in order given the potential risks of uncontrolled cellular behavior in transplant recipients.
Novocell (now Viacyte) is a biotechnology company currently conducting preclinical studies for an encapsulated hESC-based pancreatic β-cell progenitor (Pro-Islet™) therapy for Type 1 diabetes. In preclinical studies, the therapy effectively induced insulin-sensitivity and stable blood sugar in mice [6]; however, Viacyte’s diabetes therapy still remains in the preclinical phases of development. By contrast, ACT is now well into its clinical trials and recently reported no adverse outcomes in the first few patients in its trials, providing encouraging early results in terms of safety [7]. Why is ACT moving more rapidly than Viacyte? At least one difference is intrinsic to the target pathology, with Type 1 diabetes being an autoimmune disease, providing Viacyte with some unique challenges such as recipient rejection of a cellular therapy. While hESC-based, the case of Viacyte is instructive for hiPSCs as well, since the use of hiPSC therapies for diabetes would also face similar challenges. While hESC-based therapies have faced and continue to face significant regulatory hurdles, as illustrated by the above examples, it is probable that the FDA will need to be even stricter with hiPSC-based therapies given the extra steps required to produce hiPSCs relative to the derivation of hESCs.
I predict that the FDA will view hiPSCs in the context of their experiences with hESC-based products, with many of the same issues to be addressed for both. The most prominent concern is safety in the form of either tumorigenesis (e.g., teratoma) or production of undesired tissues, given the pluripotency of hiPSCs. However, hiPSCs face unique challenges as well, including the methods used to produce them and the predicted instability. In this regard, it is well documented that hESCs tend to accumulate mutations over time in culture, including many related to oncogenes [8–15], and the same was predicted to be true for hiPSCs. One type of genomic change – copy number variations (CNVs) – occurs when individual cells have gain or loss of specific genes. For example, cancer cells often have CNVs reflecting gains of oncogenes and losses of tumor suppressor genes. Interestingly, the number of CNVs in hiPSCs, which was higher than in hESCs, was observed to fall over time in culture [16], suggesting many CNVs were somehow selected against. Importantly, the levels of CNVs in hiPSCs were generally equal to or higher than in hESCs, indicating that in this regard hiPSCs do not have an advantage over hESCs. Furthermore, the drift of CNV numbers in hiPSCs at the same time also demonstrates their inherent changeability in culture, an undesirable property from a therapeutic standpoint. Of particular concern is the fact that CNVs in hiPSCs and hESCs both frequently involved gains in copy numbers of oncogenes. Methods to enhance iPSC stability during production and culture have been suggested, including addition of factors during reprogramming that promote genome stability [4].
The epigenetic state or ‘epigenome’ of hiPSCs is remarkably similar to hESCs, but also contains imperfections and ‘memories’ [17–19] of unknown biological function. During cellular reprogramming, the somatic cell epigenome is reprogrammed as well. However, a variety of differences in the epigenome are apparent in hiPSCs as compared with hESCs. This incomplete epigenomic reprogramming includes regions of persistent DNA methylation and histone modifications that are distinct from those observed in hESCs. The inherently more dynamic state of the epigenome versus the genome makes it reasonable to predict that the hiPSC epigenome will prove changeable during large-scale culturing as well. While it is unclear whether in the future the FDA will require epigenomic data as part of the approval process of hiPSC-based therapies, such data could prove highly informative in the evaluation of biologics and potentially in the regulatory decision-making processes. Given these factors, how might the epigenomic state of hiPSCs or their derivatives be most effectively assessed? Current state-of-the-art technology for globally assessing the epigenome is chromatin immunoprecipitation sequencing (ChIP-Seq) using antibodies for specific histones or methylated DNA. As sequencing technology advances, it is reasonable to expect that ChIP-Seq will become faster and cheaper, making it potentially more applicable to preclinical screening of stem cell lines (reviewed in [20]). Given that epigenomic reprogramming is intrinsic to the production of hiPSCs and that hiPSCs are not identical to hESCs, I recommend some level of epigenetic assessment be incorporated into the clinical hiPSC validation process. Other potential levels of validation that are predicted to be informative include proteomics and metabolomics profiling [21,22], as the proteomes and metabolomes of iPSCs are not identical to hESCs.
An additional challenge regarding hiPSC safety is the relative dearth of safety-related studies that have strong clinical relevance [4]. With few exceptions, the tumorigenicity of hiPSC lines has been evaluated using classic so-called ‘teratoma assays’. In these assays, hiPSCs are most often injected subcutaneously into immunodeficient mouse models. While such assays are effective tools for evaluating the pluripotency of stem cell lines, teratoma assays have a number of weaknesses. The lack of standardization in how they are performed [23] and their dissimilarity to the context of how hiPSCs would actually be transplanted in a true human clinical setting are problematic. For example, most human patients would be immunocompetent and injection of cells subcutaneously may have little relevance to any potential human tissue into which hiPSCs may be transplanted in the clinical setting. What are potential alternatives to teratoma assays? One intriguing approach, PluriTest™ (Scripps Research Institute), relies on gene expression profiling that accurately assesses pluripotency without the use of teratoma assays [24]. It may be possible to glean more precise and useful preclinical information on pluripotency signatures through genomics approaches as well, as discussed below.
There currently is no regulatory mandate that hiPSCs should be produced using non-genetic methods. If genetic methods are used to make hiPSCs intended for clinical use, as a consequence, the potential hiPSC-based therapy in question would be both a cellular and gene therapy product. What this means is that any such genetically based hiPSC therapy would be subject to significantly more challenging regulatory hurdles than, for example, hESCs or hiPSCs made without genetic modification. Thus, while the FDA has not publicly banned or even advised against the future clinical use of genetically produced hiPSCs as therapies outright, scientists wanting to translate such therapies to the bedside may be somewhat daunted by the extra regulatory hurdles. However, the use of genetically modified cells has precedent and may prove valuable. For example, ReNeuron’s fetal brain-derived cellular product RN001, approved by UK regulators for clinical trials in stroke patients, was genetically modified, but it is notable that the company is performing the trial outside the USA after a prolonged period of FDA review.
The use of genetic methods, such as integrating viruses to make hiPSCs, present potentially more intrinsic problems in the form of cells that may be predisposed to be less safe due to the effects of transgenes [25]. For example, the use of Myc genes has long been a concern in a murine context due to its demonstrated ability to cause tumors in hiPSC-derived mice [26], but whether endogenous or exogenous, Myc proteins fulfill essential roles in both embryonic stem cells and hiPSCs [27,28] and pose potential risks. Nonetheless, the search for completely nongenetic, reasonably efficient hiPSC production technologies may not be as entirely essential from a regulatory perspective as is widely assumed in the field. In addition, it may be possible to replace c-Myc or N-Myc with the potentially safer L-Myc or with mutant forms of Myc that are reportedly still functional for reprogramming but far less oncogenic [29]. Alternatively, Myc may be replaced with other genes that appear safer, such as UTF1, which has already been shown to effectively substitute for Myc [30], or others, such as Lin28, Tbx3 or Sal4. It is notable that the field has greatly advanced in the last few years, as reflected in the declining number of reprogramming factors required to produce hiPSCs, pointing to Oct4 being the most essential. The use of small-molecule drugs during reprogramming has greatly added to the efficiency of such methods using fewer factors. More broadly, the field is shifting towards the use of nongenetic approaches to hiPSC production, such as episomal vectors and viruses such as adenovirus, which may ultimately facilitate most future hiPSCs being made using non-genetic methods [31–33]. However, at this time, genetic approaches remain by far the most efficient and most widely used. Importantly, the issue of genetic versus nongenetic hiPSC production becomes largely moot in the case of hiPSC-produced drugs used for therapies.
The economics of hiPSCs
One of the critical concerns regarding hiPSCs is their potential cost. For example, what would it cost to analyze the genomes of many hiPSC lines? Sequencing the entire genomes or the exomes of a cohort of hiPSCs in the pipeline for potential use as therapies is potentially reasonable in 2012 in terms of cost. In addition, the cost of whole-genome sequencing is plummeting [34], with the cost of sequencing of a whole genome approaching US$1000, whereas in contrast the first sequencing of a human genome was a multibillion dollar endeavor. Pushing the limits further, the genomics company Oxford Nanopore reports a disposable flash drive-like USB-based machine that can purportedly sequence genomes in minutes.
The expenses and times related to such genomic validation in fact pale in comparison to the costs and efforts of rodent-based preclinical safety studies, which can involve thousands of animals and millions of dollars. Such studies are crucial for developing new hiPSC-based biologics. An important question is how the economics of hiPSCs compare with hESCs. The best prediction at this time is that hiPSCs will be significantly more expensive per patient than hESCs. However, a crucial factor in estimating cost is addressing the predicted extent to which hiPSCs can in effect ‘ride the coat tails’ of hESCs. An interesting hypothetical scenario in this regard would be an autologous hiPSC therapy using the same final product (e.g., RPE cells) that a separate team had produced from hESCs. Could the hiPSC-derived RPE cells benefit from the already existent FDA review of the hESC-derived RPE cells? I predict that hiPSC products will benefit from previous hESC review, but this will not lead to an automatic approval.
Will the FDA go genomic on stem cells?
There is a growing consensus in the stem cell field that the current status quo of karyotyping is simply not sensitive enough to evaluate the genome integrity of stem cells. Subkaryotypic genomic changes occur in stem cells. As next-generation sequencing technology has rapidly advanced, whole-genome sequencing has become more practical and would seem a more powerful alternative to karyotyping. While the main focus of next-generation sequencing has been for the identification of mutations, for example in cancer, as well as whole-genome sequencing of various organisms, single nucleotide polymorphism analysis and evolutionary studies [35–38], it has great importance for the stem cell field as well. The potential importance of such sequencing is illustrated by a group of papers from 2010 to 2011 indicating that hiPSCs contain varying levels and types of mutations [16,39–42]. Some mutations were already present in the parental cells from which the hiPSCs were derived, but other mutations occurred specifically during the hiPSC production process. While many of these mutations are linked in the genome to cancer-related genes, their functional significance remains unknown.
The predicted new genomic regulatory hurdles raise some interesting questions. What information will the FDA specifically require with regard to the hiPSC genome? What reference will be used as the basis for determining whether a given stem cell line has undergone genomic changes of concern? How do we decide what type of genomic changes are acceptable? Will they require whole-genome sequencing or, as I think is more likely, only exome sequencing? Will the FDA require sequencing before and after large-scale production of hiPSC-based therapies to account for potential ‘drift’ during the many cell divisions needed to produce billions of hiPSCs? While the FDA has not publicly issued guidance on genomic sequencing, given its potential value as a screening tool, it can be reasonably expected that in the coming years genomic sequenced data will be encouraged, if not outright mandated, by the FDA. Beyond sequencing, other assays, such as array comparative genomic hybridization and single nucleotide polymorphism arrays for high-resolution virtual karyotyping, could be far superior or complimentary to traditional karyotyping. However, in the end, I would argue that whole-exome sequencing, given its power and increasing practicality, is most likely to become the standard in the field.
Differences between regulatory issues for hiPSCs versus hESCs: a new therapy to validate for every patient?
One of the key differences, from a clinical perspective, which theoretically makes hiPSCs potentially far better than hESCs as the basis for treating human disease, is that hiPSCs can be used for autologous therapies. For this reason, there is the anticipation that hiPSC-based therapies could be given to specific patients without immunosuppression. However, the potential autologous nature of hiPSCs is a double-edged sword, as it also presents unique and potentially challenging issues from a regulatory standpoint. If a hypothetical biotechnology company has a candidate hiPSC-based therapy for a specific disease, how would the FDA handle evaluating such a therapy since the reality is that the therapy would be unique for every patient? In this regard, the inherent heterogeneity between each patient’s therapy is a potential major challenge for translating hiPSCs to the clinic, likely leading the FDA to require such therapies to meet a higher standard of safety and stability, with at least some component of validation repeatedly required for every hiPSC line derived from each new patient. By contrast, batch-prepared hESCs can be given to multiple patients with a single round of validation.
Differences between regulatory issues for hiPSCs versus hESCs: timing & efficacy
Another unique challenge that goes along with the benefit of potential hiPSC-based, autologous therapies is timing. To realize the power of hiPSCs, we have to expect patients to wait a period of months for their treatments, which have to be made de novo for every individual from that individual’s own cells. hESC-based therapies can be batch prepared in advance and taken through quality control/assurance before the target patient is even known. By contrast, the production and validation of hiPSC-based treatments would have to be initiated anew with every new patient. What this means from a clinically realistic perspective is that the time from the initial identification of the patient to their potential treatment with a hiPSC-based therapy would almost certainly be at a minimum of 6–12 months. As such, the autologous use of hiPSCs for the treatment of serious, acute injuries such as heart attacks, stroke, spinal cord injury and acute brain injury, among others, is not likely to be realized, barring some fundamental game changer in terms of how hiPSCs are produced and validated. As a result, allogeneic adult stem cell-based therapies are a more promising basis for treatment of acute injuries, with potential regenerative functions related to both cell replacement and their immunomodulatory properties. Furthermore, at least compared with hiPSCs, batch-prepared hESCs or their derivative differentiated progenitor cells also have an inherent potential advantage, specifically in terms of timing, as they can be cryo-preserved in large quantities in advance for use on an as-needed basis.
Another important question relates to the differentiation potential of hiPSCs. While bona fide hiPSCs by definition are pluripotent, based on the current standards in the field, there are varying degrees of pluripotency, and some cell lines may have predispositions towards or against specific lineages. Are hiPSCs pluripotent in the same sense as hESCs? For example, would RPE cells produced from hiPSCs essentially be equivalent to RPE cells made from hESCs? What about cardiac cells derived from hiPSCs? Would they behave similarly to those derived from hESCs? Even prior to the existence of hiPSCs, concerns were raised over the apparent immature functional nature of hESC-derived differentiated cells, such as cardiac cells [43]. Because at this time it remains mostly unknown as to how specific cells differentiated from hiPSCs stack up in terms of their functional maturity, this remains an important open area to be addressed in the field.
What will it take to get hiPSCs to an IND & then the bedside? Lessons from the path of hESCs
As part of the IND process, preclinical data on hiPSC-based therapies would be required, which is very much similar to what the FDA required of ACT and Geron for their hESC-based therapies. The main issues to be addressed for a hiPSC IND would include the following: the administration methodology and potential tissue injury at the site of administration; potential systemic effects and off-target tissue localization; ectopic tissue growth; teratoma formation; and malignant cancer formation. Each of these issues applies to both hESCs and hiPSCs, and will have to be addressed for hiPSCs in much the same manner as has been the case for hESCs. Both short- and long-term (12–24 months post-transplant) safety studies in rodents will need to be presented to the FDA. Such studies are likely to bear little if any resemblance to classical subcutaneous teratoma assays in immunodeficient mice. The administration of the hiPSC-based therapy in the preclinical studies, which will have to match the modality to be used in human patients as closely as possible, will need to be carefully tested for efficacy and safety. Furthermore, the animal model system must also model the human disease state as closely as possible. For example, Geron used a rat model of spinal cord injury to test their hESC-based therapy for safety and efficacy [44–46]. A further important consideration is that for certain diseases, rodents are poor models. For instance, for coronary artery disease and myocardial infarction, larger and more expensive animals such as pigs are far superior models. If the human patients who will receive the hypothetical hiPSC-based treatment will not be immunocompromised, then preclinical studies should utilize at least partially immunocompetent animals given transient immunosuppression, as has been used for human patients [7].
The key differences in the preclinical pathway for hiPSCs are predicted to be that if the hiPSCs were produced using any aspect of gene introduction, said product would also have to undergo gene therapy review by the NIH Recombinant DNA Advisory Committee, and that hiPSCs may require scrutiny due to their potentially more dynamic genomic and epigenomic states. Certainly, within the context of preclinical work leading up to an IND in which the product (e.g., cardiac cells) is not in the same lineage as the parental cells from which the hiPSCs were derived (e.g., skin), potential epigenetic instability will be important to monitor.
Another major issue is the large-scale production of the hiPSC cultures and derivative cultures required for treatment of many diseases. Scaling of therapies based on both hiPSCs and hESCs faces the challenges of both rapidly producing large numbers of cells (billions) and also retaining the desired functional properties. In this regard, significant advances have occurred [47–49], such as growth of suspension cultures in spinner flasks in defined serum-free media.
Future perspective: some suggested course corrections
How does the stem cell field most wisely get from point A, where we are now, to point B, where patients receive safe and effective hiPSC-based therapies? The current direction of the hiPSC subfield is a strong, albeit less than ideal one from a clinical perspective. An enormous amount of resources have gone into research and publications on a host of different methods to produce hiPSCs. However, at the same time, some fundamental questions regarding hiPSCs have only at best been addressed in a limited fashion, such as: hiPSC tumorigenicity in a clinically relevant transplantation setting; hiPSC stability in batch-scale production (billions of cells from many rounds of cell division); and hiPSC immunogenicity, which is not as simple (i.e., nonexistent) as once thought [50]. In addition, we do not currently have a benchmark for how much in the way of genomic change (and what kinds of genomic changes) in cells is clinically acceptable. A continuing failure to thoroughly address these questions may constrain the clinical relevance of the tremendous amount of hiPSC research that is ongoing. Instead, we need to anticipate these hurdles (Figure 1) and devise solutions now as we continue hiPSC research. On the tumorigenicity front, more clinically relevant studies need to be carried out using a variety of hiPSC lines so that we are aware of the malignant potential, if any, of hiPSCs. In terms of batch preparation, hiPSCs need to be tested for biological properties and gene expression, as well as genomic and ideally epigenomic signatures, before and after expansion. In relation to immunogenicity, far more studies including not in the context of teratoma (a limitation of the Zhao study [50]) need to be conducted to determine whether hiPSCs are immunogenic in autologous settings.
As much as the hiPSC reprogramming feat itself is inherently a landmark and has opened doors to new, creative ways of thinking about cell biology, what took our collective breath away a half dozen years ago was the clinical potential of hiPSCs, with the goal of making this potential a reality. That goal remains attainable, but in a time of limited research funding resources, we need to appropriately prioritize how we are conducting research on hiPSCs. Trials for hiPSCs may not be so far off as many have previously anticipated, with researchers in Japan possibly as little as 1 year away from applying for Phase I trials of a hiPSC-derived therapy. It is perhaps no coincidence that one of the first, if not the first, hiPSC trials will be for age-related macular degeneration, building on the path forged by ACT. Given the rapid approach of hiPSC trials, even if not in the USA, there is a pressing need to address the issues discussed in this review.
Executive summary.
Preclinical safety studies of human induced pluripotent stem cell (hiPSC)-based therapies are essential, but there are few reports of such studies.
Realistic assessments of the costs of potential hiPSC-based treatments are of great importance.
The emerging area of genomics may prove critical for validation of stem cell therapies.
hiPSCs face many of the same regulatory issues as human embryonic stem cells, but some possible unique challenges as well.
For moving hiPSC-based technology to the clinic, there are lessons to be learned from the regulatory path of human embryonic stem cells.
More focus on a clinical pathway is needed for hiPSC research.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
This work was supported by grant RN2-00922 from the California Institute for Regenerative Medicine. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1▪▪.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. First induced pluripotent stem cell (iPSC) paper of any kind. [DOI] [PubMed] [Google Scholar]
- 2▪.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. Describes the production of human iPSCs (hiPSCs) [DOI] [PubMed] [Google Scholar]
- 3▪.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. Describes the production of hiPSCs using a system that is distinct from Yamanaka et al. [DOI] [PubMed] [Google Scholar]
- 4.Barrilleaux B, Knoepfler PS. Inducing iPSCs to escape the dish. Cell Stem Cell. 2011;9(2):103–111. doi: 10.1016/j.stem.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okita K, Yamanaka S. Induced pluripotent stem cells: opportunities and challenges. Philos Trans R Soc Lond B Biol Sci. 2011;366(1575):2198–2207. doi: 10.1098/rstb.2011.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroon E, Martinson LA, Kadoya K, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26(4):443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz SD, Hubschman JP, Heilwell G, et al. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet. 2012;379(9817):713–720. doi: 10.1016/S0140-6736(12)60028-2. [DOI] [PubMed] [Google Scholar]
- 8.Spits C, Mateizel I, Geens M, et al. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat Biotechnol. 2008;26(12):1361–1363. doi: 10.1038/nbt.1510. [DOI] [PubMed] [Google Scholar]
- 9.Lefort N, Feyeux M, Bas C, et al. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat Biotechnol. 2008;26(12):1364–1366. doi: 10.1038/nbt.1509. [DOI] [PubMed] [Google Scholar]
- 10.Abeyta MJ, Clark AT, Rodriguez RT, Bodnar MS, Pera RA, Firpo MT. Unique gene expression signatures of independently-derived human embryonic stem cell lines. Hum Mol Genet. 2004;13(6):601–608. doi: 10.1093/hmg/ddh068. [DOI] [PubMed] [Google Scholar]
- 11.Maitra A, Arking DE, Shivapurkar N, et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet. 2005;37(10):1099–1103. doi: 10.1038/ng1631. [DOI] [PubMed] [Google Scholar]
- 12.Narva E, Autio R, Rahkonen N, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010;28(4):371–377. doi: 10.1038/nbt.1615. [DOI] [PubMed] [Google Scholar]
- 13.Martins-Taylor K, Xu RH. Concise review: genomic stability of human induced pluripotent stem cells. Stem Cells. 2012;30(1):22–27. doi: 10.1002/stem.705. [DOI] [PubMed] [Google Scholar]
- 14.Ross AL, Leder DE, Weiss J, Izakovic J, Grichnik JM. Genomic instability in cultured stem cells: associated risks and underlying mechanisms. Regen Med. 2011;6(5):653–662. doi: 10.2217/rme.11.44. [DOI] [PubMed] [Google Scholar]
- 15.Elliott AM, Elliott KA, Kammesheidt A. Array-comparative genomic hybridization characterization of human pluripotent stem cells. Methods Mol Biol. 2012;873:261–267. doi: 10.1007/978-1-61779-794-1_17. [DOI] [PubMed] [Google Scholar]
- 16.Hussein SM, Batada NN, Vuoristo S, et al. Copy number variation and selection during reprogramming to pluripotency. Nature. 2011;471:58–62. doi: 10.1038/nature09871. [DOI] [PubMed] [Google Scholar]
- 17.Lister R, Pelizzola M, Kida YS, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471(7336):68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18▪▪.Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467(7313):285–290. doi: 10.1038/nature09342. Demonstrated that iPSCs possess epigenetic memory of their cells of origin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bar-Nur O, Russ HA, Efrat S, Benvenisty N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 2011;9(1):17–23. doi: 10.1016/j.stem.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Rada-Iglesias A, Wysocka J. Epigenomics of human embryonic stem cells and induced pluripotent stem cells: insights into pluripotency and implications for disease. Genome Med. 2011;3(6):36. doi: 10.1186/gm252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panopoulos AD, Yanes O, Ruiz S, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22(1):168–177. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williamson AJ, Whetton AD. The requirement for proteomics to unravel stem cell regulatory mechanisms. J Cell Physiol. 2011;226(10):2478–2483. doi: 10.1002/jcp.22610. [DOI] [PubMed] [Google Scholar]
- 23.Muller FJ, Goldmann J, Loser P, Loring JF. A call to standardize teratoma assays used to define human pluripotent cell lines. Cell Stem Cell. 2011;6(5):412–414. doi: 10.1016/j.stem.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Muller FJ, Schuldt BM, Williams R, et al. A bioinformatic assay for pluripotency in human cells. Nat Methods. 2011;8(4):315–317. doi: 10.1038/nmeth.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung Y, Bauer G, Nolta JA. Concise review: induced pluripotent stem cell-derived mesenchymal stem cells: progress toward safe clinical products. Stem Cells. 2012;30(1):42–47. doi: 10.1002/stem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 27▪.Varlakhanova NV, Cotterman RF, Devries WN, et al. myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation. 2010;80(1):9–19. doi: 10.1016/j.diff.2010.05.001. First paper to report the function of endogenous myc in pluripotent stem cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knoepfler PS. Why myc? An unexpected ingredient in the stem cell cocktail. Cell Stem Cell. 2008;2(1):18–21. doi: 10.1016/j.stem.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa M, Takizawa N, Narita M, Ichisaka T, Yamanaka S. Promotion of direct reprogramming by transformation-deficient Myc. Proc Natl Acad Sci USA. 2010;107(32):14152–14157. doi: 10.1073/pnas.1009374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Yin X, Qin H, et al. Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell. 2008;3(5):475–479. doi: 10.1016/j.stem.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Cheng L, Hansen NF, Zhao L, et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell. 2012;10(3):337–344. doi: 10.1016/j.stem.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okita K, Hong H, Takahashi K, Yamanaka S. Generation of mouse-induced pluripotent stem cells with plasmid vectors. Nat Protoc. 2010;5(3):418–428. doi: 10.1038/nprot.2009.231. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324(5928):797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonetta L. Whole-genome sequencing breaks the cost barrier. Cell. 2010;141(6):917–919. doi: 10.1016/j.cell.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 35.Ross JS, Cronin M. Whole cancer genome sequencing by next-generation methods. Am J Clin Pathol. 2011;136(4):527–539. doi: 10.1309/AJCPR1SVT1VHUGXW. [DOI] [PubMed] [Google Scholar]
- 36.Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12(7):499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- 37.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12(6):443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brockhurst MA, Colegrave N, Rozen DE. Next-generation sequencing as a tool to study microbial evolution. Mol Ecol. 2011;20(5):972–980. doi: 10.1111/j.1365-294X.2010.04835.x. [DOI] [PubMed] [Google Scholar]
- 39▪▪.Mayshar Y, Ben-David U, Lavon N, et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. Reported chromosomal changes in hiPSCs. [DOI] [PubMed] [Google Scholar]
- 40.Laurent LC, Ulitsky I, Slavin I, et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell. 2011;8:106–118. doi: 10.1016/j.stem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gore A, Li Z, Fung H-L, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ben-David U, Mayshar Y, Benvenisty N. Large-scale analysis reveals acquisition of lineage-specific chromosomal aberrations in human adult stem cells. Cell Stem Cell. 2011;9(2):97–102. doi: 10.1016/j.stem.2011.06.013. [DOI] [PubMed] [Google Scholar]
- 43.Rajala K, Pekkanen-Mattila M, Aalto-Setälä K. Cardiac differentiation of pluripotent stem cells. Stem Cells Int. 2011;2011:383709. doi: 10.4061/2011/383709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keirstead HS, Nistor G, Bernal G, et al. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25(19):4694–4705. doi: 10.1523/JNEUROSCI.0311-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harper JM, Krishnan C, Darman JS, et al. Axonal growth of embryonic stem cell-derived motoneurons in vitro and in motoneuron-injured adult rats. Proc Natl Acad Sci USA. 2004;101(18):7123–7128. doi: 10.1073/pnas.0401103101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharp J, Frame J, Siegenthaler M, Nistor G, Keirstead HS. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants improve recovery after cervical spinal cord injury. Stem Cells. 2010;28(1):152–163. doi: 10.1002/stem.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krawetz R, Rancourt DE. Suspension bioreactor expansion of undifferentiated human embryonic stem cells. Methods Mol Biol. 2012;873:227–235. doi: 10.1007/978-1-61779-794-1_14. [DOI] [PubMed] [Google Scholar]
- 48.Baharvand H, Larijani MR, Yousefi M. Protocol for expansion of undifferentiated human embryonic and pluripotent stem cells in suspension. Methods Mol Biol. 2012;873:217–226. doi: 10.1007/978-1-61779-794-1_13. [DOI] [PubMed] [Google Scholar]
- 49.Chen VC, Couture SM, Ye J, et al. Scalable GMP compliant suspension culture system for human ES cells. Stem Cell Res. 2012;8(3):388–402. doi: 10.1016/j.scr.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 50▪.Zhao T, Zhang Z-N, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474(7350):212–215. doi: 10.1038/nature10135. First report to question the absent immunogenicity of iPSCs. [DOI] [PubMed] [Google Scholar]