

Abstract
A combination of weakly coordinating auxiliaries and ligand acceleration allows for the development of both ortho- and meta-selective C–H olefination of phenol derivatives. These reactions demonstrate the feasibility of directing C–H functionalizations when functional groups are distal to target C–H bonds. The meta-C–H functionalization of electron-rich phenol derivatives is unprecedented and orthogonal to previous electrophilic substitution of phenols in terms of regioselectivity. These methods are also applied to functionalize α-phenoxyacetic acids, a fibrate class of drug scaffolds.
1. Introduction
Directed C–H activation via 5-, 6-membered cyclopalladation has recently emerged as a versatile tool for developing synthetic transformations.1 However, activation of C–H bonds that are more than five bonds away from the chelating atom are still rare due to the difficulty of forming larger membered palladacycles.2 Alternative strategies relying on the proximity between remote C–H bonds and highly reactive species such as radical or meta-oxo species have been reported.3 In contrast to the directed metalation processes, these strategies do not produce systematic and predictable structural patterns at this stage of development. On the other hand, directed metalation of C–H bonds at certain geometric positions that are resistant to the assembly of cyclic transition states due to ring strain has limited success, as exemplified by the lack of examples of directed meta-C–H activation of arenes until recently.2b Development of meta-C–H activation of broad range of synthetically useful substrates will significantly enhance the versatility of directed C–H activation reactions in synthetic applications.
Phenol and their derivatives have drawn intensive efforts from the community of C–H functionalizations due to their broad synthetic utility. Early studies by Rawal and Miura used the phenol to direct arylation of biphenyls with ArI and Pd(0)/PPh3 catalysts.4 Bedford and others succeeded in using catalytic amount of phosphites as directing groups to promote Rh(I)-catalyzed ortho-arylation of phenols (Fig 1).5 Hartwig reported Ir(I)-catalyzed borylation of hydrosilyl ethers of phenols.6 Recent emergence of C–H activation reactions with Pd(II),7 Rh(III),8 Ru(II)9 catalysts using weakly coordinating carbonyls have led to a range of ortho-C–H functionalization reactions of phenol derivatives (Fig 1). Due to the strong electron-donating ability of the phenoxyl group, meta-selective C–H activation is especially challenging and yet synthetically enabling.
Figure 1.
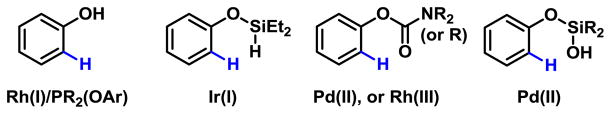
Previous ortho-C–H Functionalizations of Phenol
Herein we report two approaches for achieving ortho- and meta-C–H olefination of phenol derivatives respectively through remote C–H activation promoted by weak coordination and ligand acceleration (Fig 2). Removal of the acetic acid auxiliaries affords synthetically valuable ortho- or meta-olefinated phenols. This protocol was successfully applied to the parent α-phenoxycarboxylic acids of drug molecules fenofibrate, clofibrate and etofibrate (Fig 3).
Figure 2.
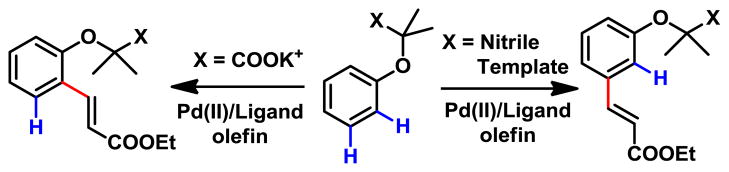
Remote ortho- and meta-C–H Functionalizations
Figure 3.
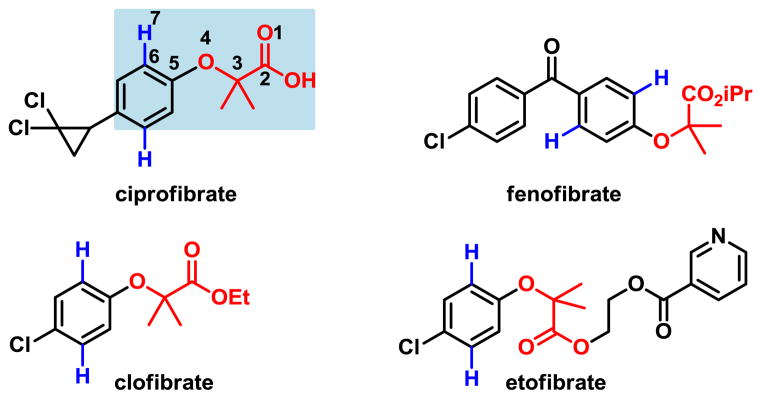
Drug Molecules Based on α-Phenoxyacetic Acids
2. Results and Discussion
We initially aimed to develop ortho-C–H functionalization methods for late-stage diversification of α-phenoxyacetic acids, important pharmacophores found in the fibrate class of lipid-lowering agents (Figure 3).10 Despite a number of elegant directing groups previously developed for the ortho-C–H functionalizations of phenols,5–9 the distance between the C–H bonds and the functional groups in α-phenoxyacetic acids presents a challenge. Although we have developed various ortho-C–H funcationalizations of phenylacetic acids via a relatively distal directing group, the ortho- C–H bonds and the chelating atom of the carboxyl group in α-phenoxyacetic acids are further apart (six bonds away) as shown in ciprofibrate (Figure 3). The presence of the α-oxygen atom in the side chain could also be problematic as Pd(II) could form a bis-chelation with this oxygen atom and the directing group, which could prevent Pd center from aligning with C–H bonds with a relatively small dihedral angle, a requisite for facile C–H activation.
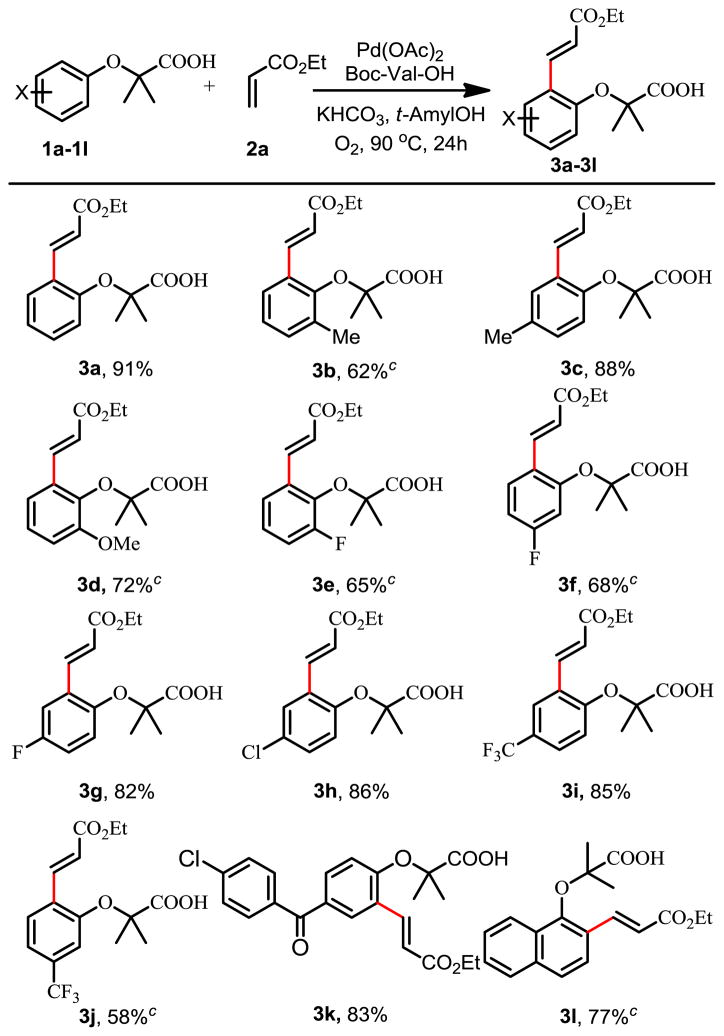
Encouraged by our recently observed ligand acceleration,11 we began to identify ligands that can cooperate with the weak coordination of the distal carbonyl in COOK moiety to promote the C–H activation reactivity. Guided by our previous C–H olefination of phenyl acetic acids11 and early olefination reactions,12 we established conditions for the ortho-olefination of α-phenoxyacetic acids. Thus, substrate 1a was reacted with 2 equiv of ethyl acrylate under 1 atm of O2 in the presence of 5 mol% of Pd(OAc)2, 10 mol% Boc-Val-OH, 2 equiv of KHCO3 in t-amyl alcohol at 90 °C for 24 hours to give 90% of the mono-olefinated product 3a (Table 1). The high mono-selectivity can be attributed to the steric buttress imparted by the α,α-dimethyl groups. Interestingly, the absence of the α,α-dimethyl groups resulted in a significant loss of reactivity leading to poor yields, presumably due to the loss of favorable Thrope-Ingle effect (less than 10%, see SI). The replacement of O2 with air dropped the yield to 34%.
Table 1.
|
Reaction conditions: 1a–1l (0.1 mmol), 2a (0.2 mmol), Pd(OAc)2 (5 mol %), Boc-Val-OH (10 mol %), KHCO3 (0.2 mmol), O2 (1 atm), t-AmylOH (1 mL), 90 °C, 24h.
Isolated yield.
Pd(OAc)2 (10 mol %), Boc-Val-OH (20 mol %).
This reaction tolerates electron-donating substituents such as methyl and methoxyl (3b–d). The naphthalene ring in example 3l is selectively olefinated at the 2-position. Olefination also proceeds smoothly with a wide range of electron deficient arenes, including those with fluoro (3e–g), chloro (3h), trifluoromethyl (3i, 3j) and keto functionality (3k). This method can be used to directly functionalize medicinally useful α-phenoxyacetic acids or prepare ortho-substituted phenols after removal of the acetic acid directing groups (Scheme 1). Notably, substrates 1h and 1k are derived from drug molecules clofibrate and fenofibrate respectively.
Scheme 1.
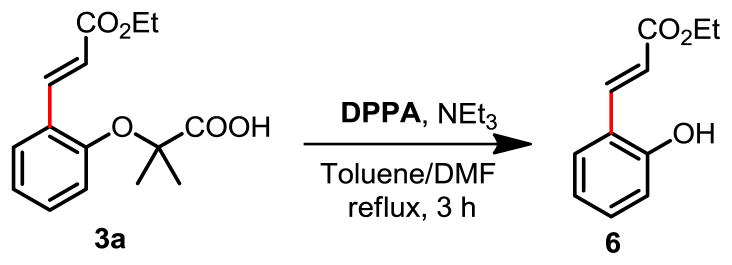
The removal of Directing Groupa
a Reaction conditions: 3a (0.4 mmol), DPPA (0.4 mmol), Et3N (0.4 mmol), toluene (5 mL), DMF (0.5 mL), refluxed 3h. DPPA: diphenylphosphoryl azide. Yield: 71%.
Prompted by our recent development of meta-selective C–H olefination reactions of hydrocinnamic acids,2b we wondered if the end-on coordinating nitrile template can be modified to direct meta-C–H olefination of α-phenoxyacetic acids. We were pleased to find that olefination of 4a proceeded with high meta-selectivity under our previous conditions to give the mono- and di-olefinated products in 60% and 29% yields respectively (Table 2). Meta-substitutions of phenol derivatives complement the ortho- and para- electrophilic substitutions, thus providing a new strategy to construct densely substituted arenes. This reaction is compatible with electron-donating groups (5b–f), as well as electron-withdrawing groups (5g–m). High mono-selectivities were obtained with ortho-substituted substrates (5b, 5e, 5g, 5j, 5l). Importantly the template overrides the electronic influence of substituents on the arenes. The regioselectivity observed with 5l is most striking as olefination occurs at the position that is meta to alkoxy and para to trifluoromethyl group. The use of a directing group to govern meta-selectivity is fundamentally a different approach compared to previously developed meta-C–H functionalizations where the electronic or steric bias of arene substrates plays a decisive role.13–17
Table 2.
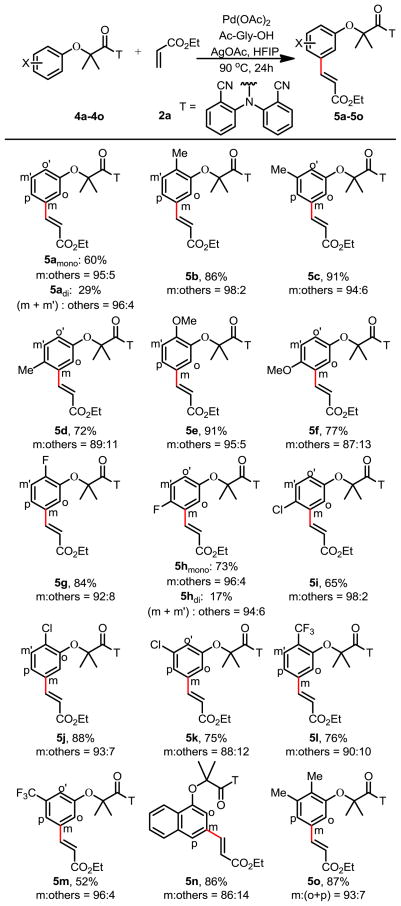
|
Reaction conditions: 4a–4o (0.1 mmol), 2a (0.2 mmol), Pd(OAc)2 (10 mol %), Ac-Gly-OH (20 mol %), AgOAc (0.3 mmol), HFIP (1 mL), 90 °C, 24h.
Isolated yield.
Regioselectivity were determined by 1H NMR analysis of the crude product and confirmed by one-dimensional selective NOESY experiments; the variance is estimated to be within 5%.
To further demonstrate the synthetic utility of this method, we performed meta-olefination of an ortho-brominated substrate 5p (Table 3). We were pleased to find that a wide range of olefins including disubstituted olefins are reactive. Coupling of 4p with trans-2-butenoate gave the desired product 5s stereospecifically. It is worth noting that di-substituted olefins are generally not compatible with directed C–H olefination reactions; only rare examples have been shown to date.2b Additionally, the bromine functionality is a highly versatile handle that allows for subsequent transformations. Finally, the nitrile template can be removed by hydrolysis2b to give the medicinally useful meta-substituted α-phenoxyacetic acids. The acetic acid moiety attached to the phenoxy can then be removed by treatment with DPPA to afford the meta-substituted phenol (Scheme 1).
Table 3.
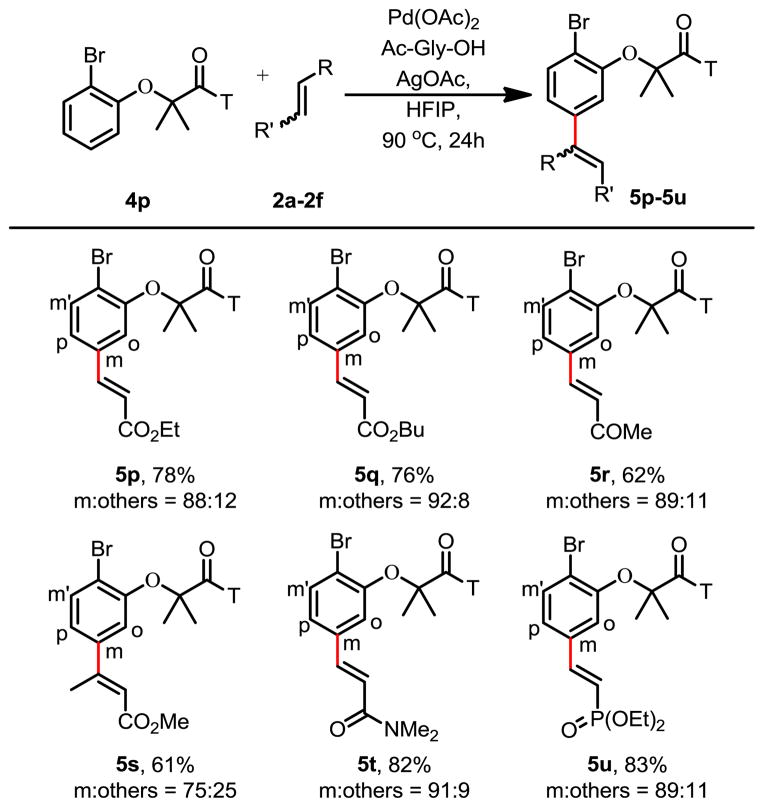
|
Reaction conditions: 4p (0.1 mmol), 2a–2f (0.2 mmol), Pd(OAc)2 (10 mol %), Ac-Gly-OH (20 mol %), AgOAc (0.3 mmol), HFIP (1 mL), 90 °C, 24h.
Isolated yield.
Regioselectivity were determined by 1H NMR analysis of the crude product and confirmed by one-dimensional selective NOESY experiments; the variance is estimated to be within 5%.
Extensive studies on C–H olefination have shown that electron-deficient olefins are in general more effective coupling partners while electron-rich olefins tend to react with Pd(II) catalyst via the Wacker oxidation pathway with rare exceptional examples.18 To examine the scope of this meta-selective olefination reaction with respect to the olefins, we tested alkenes and styrenes as the coupling partners and found only those styrenes with an electron-withdrawing group attached to the arenes reacted to some extent to give the desired product in moderate yields (Table 4). Further improvement of the C–H olefination with electron-rich olefins such as simple styrene and hexane will require the modification of the ligand.
Table 4.
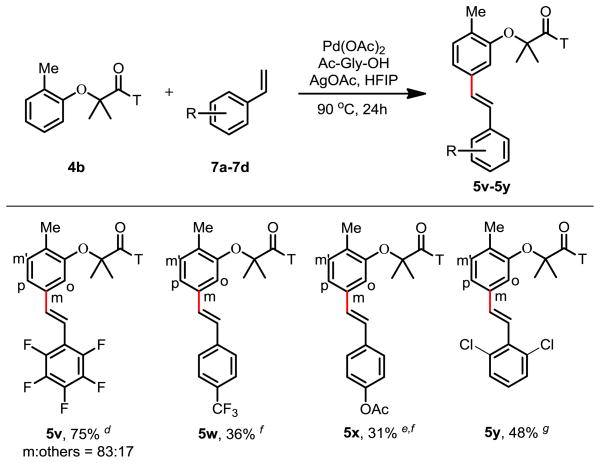
|
Reaction conditions: 4b (0.1 mmol), 7a–7c (0.2 mmol), Pd(OAc)2 (10 mol %), Ac-Gly-OH (30 mol %), AgOAc (0.3 mmol), HFIP (1 mL), 90 °C, 24h.
Isolated yield.
Regioselectivity were determined by 1H NMR analysis of the crude product and confirmed by one-dimensional selective NOESY experiments; the variance is estimated to be within 5%.
Ac-Gly-OH (20 mol%).
30 h.
Only one major regioisomer was observed.
NMR yield.
While the detailed mechanism and origin of the meta-selectivity remains to be elucidated by computational, kinetic as well as structural studies, We have also observed a significant isotope effect (kH/kD = 3.8) of the nitrile-directed meta-selective C–H olefination with analogous substrates (see supporting information), suggesting that the C–H cleavage may be involved in the rate-determining step. A tentative catalytic cycle can also be proposed based on our previous studies (Fig 4).
Figure 4.

Catalytic Cycle of meta-C–H Olefination
3. Conclusion
In summary, we have developed two new approaches for ortho- and meta-C–H functionalization of phenol derivatives. These C–H activation reactions feature rare cyclometalation processes of C–H bonds directed by distal chelating atoms (six bonds away). The selective functionalizations of phenol derivatives at the meta-positions are unprecedented and especially useful as the regioselectivity is orthogonal to the electrophilic substitution. The methods are also applied to functionalize α-phenoxyacetic acids, a fibrate class of drug scaffolds.
4. Experimental Section
4.1. General Information
All commercial reagents were purchased from Sigma-Aldrich, Fluka, Alfa Aesar, TCI, Oakwood, and Acros of the highest purity grade. They were used without further purification unless specified. Palladium acetate, silver acetate was purchased from Sigma-Aldrich. The amino acid ligands were bought from Novabiochem, Bachem and Sigma-Aldrich. 2,2′-azanediyldibenzonitrile was prepared by literature methods.19 1H and 13C NMR spectra were recorded on Bruker AV 400, Varian Inova 400 (400 MHz and 100 MHz, respectively), Bruker DRX 500 (500 MHz and 125 MHz, respectively) and Bruker DRX 600 (600 MHz and 150 MHz, respectively) instruments. The peaks were internally referenced to TMS (0.00 ppm) or residual undeuterated solvent signal. The following abbreviations were used to explain multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, and br = broad. High resolution mass spectra were recorded at the Center for Mass Spectrometry, The Scripps Research Institute.
4.2 General procedure for Pd(II)-catalyzed ortho-C–H olefination of α-phenoxyacetic acids (1a–1l)
A 50 mL Schlenk-type tube (with a Teflon high pressure valve and side arm) equipped with a magnetic stir bar was charged with acid 1 (0.20 mmol, 1.0 equiv.), Pd(OAc)2 (2.3 mg, 0.010 mmol, 5 mol %), Boc-Val-OH (4.3 mg, 0.020 mmol, 10 mol %), KHCO3 (40 mg, 0.40 mmol, 2.0 equiv.), ethyl acrylate 2a (0.40 mmol, 2.0 equiv.), and t-AmylOH (2.0 mL). The reaction tube was evacuated and backfilled with O2 (5-times, balloon) and heated to 90 °C for 24 hours under vigorous stirring. The reaction vessel was then cooled to 0 °C in an ice bath. A 2.0 N HCl solution (5 mL) was then added, and the mixture was extracted with EtOAc (3 × 20 mL). The organic layers were combined, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The resulting residue was purified by preparative TLC using hexanes/EtOAc as the eluent to afford the product (3a–3l).
4.3 General procedure for Pd(II)-catalyzed meta-C–H olefination of phenol derivative (4a–4p)
A 35 mL sealed tube (with a Teflon cap) equipped with a magnetic stir bar was charged with amide 4 (0.10 mmol, 1.0 equiv.), Pd(OAc)2 (2.3 mg, 0.010 mmol, 10 mol %), Ac-Gly-OH (2.4 mg, 0.020 mmol, 20 mol %) and AgOAc (50 mg, 0.30 mmol, 3.0 equiv.). HFIP (0.60 mL) was added to the mixture, followed by ethyl acrylate 2a (2.0 equiv.) and then another 1.0 mL of HFIP. The tube was then capped and submerged into a pre-heated 90 °C oil bath. The reaction was stirred for 24 h and cooled down to room temperature. The crude reaction mixture was diluted with EtOAc (2 mL) and filtered through a short pad of Celite. The sealed tube and Celite pad were washed with an additional 15 mL of EtOAc. The filtrate was concentrated in vacuo, and the resulting residue was purified by preparative TLC using hexanes/EtOAc as the eluent. The positional selectivity was determined by 1H NMR analysis of the unpurified reaction mixture.
4.4 Procedure for removal of template20
To a 50 mL of flask, 3a (111.2 mg, 0.4 mmol) was dissolved in anhydrous toluene (5 mL) and DMF (0.5 mL), then Et3N (46.4 mg, 0.4 mmol) and DPPA (110 mg, 0.4 mmol) were added and the mixture was refluxed for 3h. 30 mL of water was added and continue to reflux for 2h. After the mixture cool to room temperature, the solution was acidified with 2N HCl solution (5 mL) and extracted with EtOAc (3 × 5 mL). The combined organic phases were washed with brine, dried over anhydrous MgSO4 and evaporated. The residue was purified via column chromatography on silica with hexane and EtOAc (10:1) as eluents to afford product A21 in 71% yield (54.3 mg). 1H NMR (400 MHz, CDCl3) δ: 8.05 (d, J = 16.2 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.26–7.21 (m, 1H), 6.93–6.85 (m, 2H), 6.80 (br, 1H), 6.65 (d, J = 16.2 Hz, 1H), 4.30 (q, J = 7.2 Hz, 2H), 1.35 (t, J = 7.2 Hz, 3H); 13C NMR (600 MHz, CDCl3) δ: 169.3, 158.3, 141.5, 132.2, 130.1, 122.6, 121.5, 119.2, 117.3, 61.6, 15.2.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute, the National Institutes of Health (NIGMS 1 R01 GM102265-01), and Eli Lilly for financial support. X.-G.Z. is a visiting scholar from Wenzhou University and was sponsored by the China Scholarship Council and the National Natural Science Foundation of China (21002070). We thank Li Wan for experimental assistance.
Footnotes
Supporting Information Available. Detailed experimental procedures, characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem, Int Ed. 2009;48:5094. doi: 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Engle KM, Mei TS, Wasa M, Yu JQ. Acc Chem Res. 2012;45:788. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Li JJ, Giri R, Yu JQ. Tetrahedron. 2008;64:6979. [Google Scholar]; (b) Leow D, Li G, Mei TS, Yu JQ. Nature. 2012;486:518. doi: 10.1038/nature11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For early discussion and other approaches for remote C–H activation: Breslow R. Acc Chem Res. 1980;13:170.Schwarz H. Acc Chem Res. 1989;22:282.Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312:1941. doi: 10.1126/science.1127899.
- 4.(a) Hennings DD, Iwasa S, Rawal VH. J Org Chem. 1997;62:2. doi: 10.1021/jo961876k. [DOI] [PubMed] [Google Scholar]; (b) Satoh T, Kawamura Y, Miura M, Nomura M. Angew Chem, Int Ed. 1997;36:1740. [Google Scholar]; (c) Miura M, Tsuda T, Satoh T, Nomura M. Chem Lett. 1997:1103. [Google Scholar]
- 5.(a) Bedford RB, Coles SJ, Hursthouse MB, Limmert ME. Angew Chem Int Ed. 2003;42:112. doi: 10.1002/anie.200390037. [DOI] [PubMed] [Google Scholar]; (b) Oi S, Watanabe S, Fukita S, Inoue Y. Tetrahedron Lett. 2003;44:8665. [Google Scholar]; (c) Bedford RB, Limmert ME. J Org Chem. 2003;68:8669. doi: 10.1021/jo030157k. [DOI] [PubMed] [Google Scholar]
- 6.Ir(I)-catalyzed C–H activation of hydrosilyl ether of Phenols: Boebel TA, Hartwig JF. J Am Chem Soc. 2008;130:7534. doi: 10.1021/ja8015878.
- 7.Pd(II)-catalyzed C–H functionalizations of Phenol derivatives: Huang C, Gevorgyan V. J Am Chem Soc. 2009;131:10844. doi: 10.1021/ja904791y.Zhao X, Yeung CS, Dong VM. J Am Chem Soc. 2010;132:5837. doi: 10.1021/ja100783c.Xiao B, Fu Y, Xu J, Gong TJ, Dai JJ, Yi J, Liu L. J Am Chem Soc. 2010;132:468. doi: 10.1021/ja909818n.Huang C, Chattopadhyay B, Gevorgyan V. J Am Chem Soc. 2011;133:12406. doi: 10.1021/ja204924j.Huang C, Ghavtadze N, Chattopadhyay B, Gevorgyan v. J Am Chem Soc. 2011;133:17630. doi: 10.1021/ja208572v.Xiao B, Gong TJ, Liu ZJ, Liu JH, Luo DF, Xu J, Liu L. J Am Chem Soc. 2011;133:9250. doi: 10.1021/ja203335u.John A, Nicholas KM. J Org Chem. 2012;77:5600. doi: 10.1021/jo300713h.
- 8.Rh(III)-catalyzed C–H activation of Phenol derivatives: Gong TJ, Xiao B, Liu ZJ, Wan J, Xu J, Luo DF, Fu Y, Liu L. Org Lett. 2011;13:3235. doi: 10.1021/ol201140q.
- 9.Ru(II)-catalyzed C–H activation of Phenol derivatives: Ackermann L, Diers E, Manvar A. Org Lett. 2012;14:1154. doi: 10.1021/ol3000876.
- 10.(a) Razavi B, Song W, Cooper WJ, Greaves J, Jeong J. J Phys Chem. 2009;113:1287. doi: 10.1021/jp808057c. [DOI] [PubMed] [Google Scholar]; (b) Chuang S, Velkov T, Horne J, Wielens J, Chalmers DK, Porter CJH, Scanlon MJ. J Med Chem. 2009;52:5344. doi: 10.1021/jm801349e. [DOI] [PubMed] [Google Scholar]
- 11.(a) Engle KM, Wang DH, Yu JQ. J Am Chem Soc. 2010;132:14137. doi: 10.1021/ja105044s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2010;49:6169. doi: 10.1002/anie.201002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Moritanl I, Fujiwara Y. Tetrahedron Lett. 1967;8:1119. [Google Scholar]; (b) Fujiwara Y, Noritani I, Danno S, Asano R, Teranishi S. J Am Chem Soc. 1969;91:7166. doi: 10.1021/ja01053a047. [DOI] [PubMed] [Google Scholar]; (c) Jia C, Kitamura T, Fujiwara Y. Acc Chem Res. 2001;34:63. doi: 10.1021/ar000209h. [DOI] [PubMed] [Google Scholar]
- 13.Cho J-Y, Tse MK, Holmes D, Maleczka RE, Jr, Smith MR., III Science. 2002;295:305. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- 14.Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J Am Chem Soc. 2002;124:390. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- 15.Zhang YH, Shi BF, Yu JQ. J Am Chem Soc. 2009;131:5072. doi: 10.1021/ja900327e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phipps RJ, Gaunt MJ. Science. 2009;323:1593. doi: 10.1126/science.1169975. [DOI] [PubMed] [Google Scholar]
- 17.Saidi O, Marae J, Ledger AEW, Liu PM, Mahon MF, Kociok-Köhn G, Whittlesey MK, Frost CG. J Am Chem Soc. 2011;133:19298. doi: 10.1021/ja208286b. [DOI] [PubMed] [Google Scholar]
- 18.Wang DH, Engle KM, Shi BF, Yu JQ. Science. 2010;327:315. doi: 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorvin JH. J Chem Soc, Perkin Trans1. 1988:1331. [Google Scholar]
- 20.Mirk D, Waldvogel SR. Tetrahedron Lett. 2004;45:7911. [Google Scholar]
- 21.Park JO, Youn SW. Org Lett. 2010;12:2258. doi: 10.1021/ol100610v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.