

Abstract
Purpose
The aim of this study was to evaluate the risk of Parkinson disease using clinical and demographic data alone and when combined with information from genes associated with Parkinson disease.
Methods
A total of 1,967 participants in the dbGAP NeuroGenetics Research Consortium data set were included. Single-nucleotide polymorphisms associated with Parkinson disease at a genome-wide significance level in previous genome-wide association studies were included in risk prediction. Risk allele scores were calculated as the weighted count of the minor alleles. Five models were constructed. Discriminatory capability was evaluated using the area under the curve.
Results
Both family history and genetic risk scores increased risk for Parkinson disease. Although the fullest model, which included both family history and genetic risk information, resulted in the highest area under the curve, there were no significant differences between models using family history alone and those using genetic information alone.
Conclusion
Adding genome-wide association study–derived genotypes, family history information, or both to standard demographic risk factors for Parkinson disease resulted in an improvement in discriminatory capacity. In the full model, the contributions of genotype data and family history information to discriminatory capacity were similar, and both were statistically significant. This suggests that there is limited overlap between genetic risk factors identified through genome-wide association study and unmeasured susceptibility variants captured by family history. Our results are similar to those of studies of other complex diseases and indicate that genetic risk prediction for Parkinson disease requires identification of additional genetic risk factors and/or better methods for risk prediction in order to achieve a degree of risk prediction that is clinically useful.
Keywords: genetics, Parkinson disease, risk prediction
INTRODUCTION
Parkinson disease (PD) is the second most common neurodegenerative disorder in the aged population, with a prevalence of 1% at age 65 that rises to 3% by age 75.1 The cardinal motor features of PD are resting tremor, bradykinesia, rigidity, and postural instability. A definitive diagnosis of PD can only be made at autopsy, and the accuracy of clinical diagnosis varies between 76% and 99%.2 A number of nonmotor problems can also arise during the course of the disease, including cognitive impairment, psychosis, sleep disturbances, depression, and autonomic dysfunction.
Several environmental factors have been postulated to contribute to the development of PD, including long-term exposure to heavy metals and pesticides, although these associations are far from conclusive. Clinical indicators that have been repeatedly and reliably associated with developing PD are advanced age and male sex. Smoking cigarettes, drinking coffee, and using nonsteroidal anti-inflammatory drugs are protective factors against developing PD.1,3 However, unlike diabetes or cardiovascular disease, there are no markers in the blood that can be used to prognosticate risk for PD.
PD was once thought to be completely environmental in etiology. However, mutations in at least six genes, LRRK2, PARK2, PARK7, PINK1, VPS35, and SNCA, are now known to cause monogenic forms of the disease.4–7 Furthermore, common variants in several genes including MAPT and SNCA have consistently been demonstrated to associate with typical, late-onset PD.8–16 Genotype data from these genes and others can be combined to create a genetic risk score that may better predict PD risk than relying on clinical and demographic data alone. Thus, the goal of this project was to compare the impact of adding family history, which reflects shared genetic and environmental factors, and specific genetic markers for new and established candidate genes with a model that includes only established clinical and environmental risk factors for PD.
MATERIALS AND METHODS
Study sample
The study population was derived from 2,000 patients with PD and 1,986 controls enrolled through the NeuroGenetics Research Consortium (NGRC), which includes movement disorder clinics in Albany, NY; Atlanta, GA; Portland, OR; and Seattle, WA; and was downloaded from dbGAP (phs000126.v1.p1). All patients met UK PD Society Brain Bank clinical diagnostic criteria for PD as determined by a movement disorder specialist17 and were consecutively recruited except that patients who had an age at onset <20 years or whose race was not solely classified as “white” (by self-report) were excluded from the sample. Data on smoking behavior were collected at all sites using a standardized questionnaire. Controls had no history of parkinsonism and were either spouses of patients with PD or community volunteers.
Genotypes were derived from a genome-wide association study (GWAS) previously performed on the NGRC case–control sample.11 The NGRC GWAS data set included 811,597 single-nucleotide polymorphisms (SNPs) assayed on the Illumina HumanOmni1-Quad_v1-0_B genotyping array (Illumina, San Diego, CA). Ungenotyped SNPs in our regions of interest were imputed using the software program IMPUTE2 version 2 with the methods described by Howie et al.18 To ensure that rare variants were adequately covered, we used two phased reference panels from HapMap3 and 1000 Genomes pilot data with release dates of February 2009 and June 2010, respectively. A genotype probability of 80% or greater was used to call the most likely genotype for each SNP. LRRK2 G2019 S was genotyped separately as previously described.19
To determine which SNPs to include for risk prediction, we first constructed a list of 46 SNPs that were reported to be associated with PD at a genome-wide level of significance in one or more previous GWA studies.8–16 Of these 46 SNPs, 21 were directly genotyped in the original NGRC data set and the remainder (n = 25) were imputed using the HapMap3 and 1000 Genomes reference panels. Seven SNPs with >5% missing data were excluded; all of these were imputed SNPs. We also included the genotype of the LRRK2 G2019S mutation. Multicollinearity was assessed for all pairs of variants. In the case of a pair with strong correlation (r2 ≥ 0.80), the variant with more missing data was excluded. Seven SNPs were excluded for collinearity. A total of 33 variants were eligible for model inclusion.
Statistical analysis
Family history was missing in 62% of participants from the Oregon site, including 91% of controls. Furthermore, cases from this site reported a positive family history of PD (36%) more often than cases from the other three sites (15–26%); thus, all participants from Oregon, totaling 1,402, were excluded from the analysis. Family history information was not different among cases and controls (Table 1) and appeared to be missing at random for the remaining three sites, although there were some differences in total percentage missing across sites. A total of 617 of the remaining participants were missing data on one or more genetic variants of interest and were excluded from the analysis. Smoking behavior was missing in 354 participants and was imputed in these participants using logistic regression including the covariates age and sex. Family history was coded using a group of four dummy variables: one dummy variable indicated whether family history was missing or unknown, and the remaining three variables indicated family history in a first-, second-, or third-degree relative, noting family history in the closest relative. Known, negative family history was the reference group. Age was coded as age at time of blood draw. The 1,967 participants available for analysis were randomly divided into “training” and “test” data sets. The “training” data set consisted of 543 patients with PD and 435 controls. The “test” data set included 594 patients with PD and 395 controls.
Table 1.
Characteristics of participants
| Training set |
Test set |
|||
|---|---|---|---|---|
| Cases (n = 543) | Controls (n = 435) | Cases (n = 594) | Controls (n = 395) | |
| Age in years (mean ± SE) | 67.5 ± 0.47 | 65.9 ± 0.58 | 67.1 ± 0.44 | 65.0 ± 0.59 |
| Male sex (%) | 66.9 | 37.9 | 69.4 | 36.7 |
| Ever smoker (%) | 46.6 | 44.6 | 45.8 | 48.9 |
| Self-reported family history (%) | ||||
| Unknown | 11.4 | 10.3 | 9.9 | 10.1 |
| First-degree relative | 9.6 | 3.5 | 13.3 | 3.8 |
| Second-degree relative | 7.7 | 3.0 | 5.2 | 3.5 |
| Third-degree relative | 2.0 | 1.2 | 2.5 | 1.0 |
| Risk allele score (mean ± SE) | ||||
| Model 3/5 | 0.07 ± 0.01 | −0.04 ± 0.02 | 0.07 ± 0.02 | −0.02 ± 0.02 |
| Model 4 | 0.18 ± 0.03 | −0.19 ± 0.03 | 0.09 ± 0.03 | −0.13 ± 0.03 |
Characteristics for cases and controls were compared using a two-sample t-test with equal variance for age and Pearson χ2 tests for categorical variables in Stata version 12 (Stata, College Station, TX).
Risk-prediction analyses were conducted in R version 2.15.0. Five risk models were constructed using logistic regression. All five models included the following baseline covariates: sex, age, and smoking status (ever vs. never). Model 1 is the baseline model with only the baseline covariates. Model 2 also included whether family history was known or unknown and the degree of family history of PD. Model 3 added to the baseline model a risk allele score constructed from the following SNPs: SNCA rs11931074, SNCA rs356220, MAPT rs1800547, and the LRRK2 G2019S mutation (rs34637584). SNPs in these genes were chosen because SNCA and MAPT are the most consistently replicated PD susceptibility genes.11 LRRK2 G2019S was selected because it accounts for 1–2% of PD in populations of European origin.19 Model 4 included a risk allele score constructed from all 33 SNPs and the baseline variables to evaluate the improvement of risk prediction with additional genetic information. Model 5 included covariates from the baseline model, whether family history was known or unknown and the degree of family history, and the weighted risk allele score constructed from four variants used in model 3. Risk allele scores were calculated as the sum of the minor alleles weighted by the β coefficient of that allele from a multivariate logistic regression of genetic covariates only. Each model’s discriminatory capability was evaluated using the C-statistic, which is the area under the curve (AUC) of receiver operating characteristic analyses; in the receiver operating characteristic, the sensitivity and specificity are both based on the classification of PD cases and controls, given the risk predicted from the logistic model. A C-statistic ranges from 0.5 (no predictive ability) to 1 (perfect predictive ability). We used DeLong’s test for two correlated receiver operating characteristic curves from the pROC R package to test for statistically significant differences in AUC obtained from each model.20
RESULTS
Participant characteristics and association with PD
A total of 1,967 participants were included in analyses. Table 1 shows the characteristics of these participants. As compared with controls, cases were mostly male, had a known family history of PD in first- and second-degree relatives, and were slightly older.
In model 1 multivariate analysis, men were three times more likely to have PD as compared with women (odds ratios (OR): 3.29 95% confidence interval (CI:) (2.52–4.31)). Neither age nor smoking was significantly associated with PD in this model (Table 2).
Table 2.
PD risk prediction regression estimates by model using the training set
| Model 1 |
Model 2 |
Model 3a |
Model 4b |
Model 5a |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Covariate | OR (95% CI) |
P value | OR (95% CI) |
P value | OR (95% CI) |
P value | OR (95% CI) |
P value | OR (95% CI) |
P value |
| Age | 1.01 0.99–1.02 |
0.182 | 1.01 0.98–1.00 |
0.26 | 1.01 0.99–1.02 |
0.13 | 1.01 0.99–1.02 |
0.12 | 1.01 0.99–1.02 |
0.12 |
| Male sex | 3.29 2.52–4.31 |
<0.001 | 3.39 2.58–4.45 |
<0.001 | 3.20 2.44–4.19 |
<0.001 | 3.21 2.43–4.24 |
<0.001 | 3.28 2.49–4.32 |
<0.001 |
| Smoking | 0.91 0.69–1.18 |
0.469 | 0.92 0. 70–1.21 |
0.54 | 0.90 0.69–1.18 |
0.003 | 0.91 0.69–1.20 |
0.50 | 0.92 0.69–1.22 |
0.52 |
| Risk allele score |
—
— |
—
— |
—
— |
—
— |
2.57 1.72–3.83 |
<0.001 | 2.62 2.07–3.30 |
<0.001 | 2.39 1.60–3.59 |
<0.001 |
| Family history: unknown |
—
— |
—
— |
1.35 0.88–2.09 |
0.16 |
—
— |
—
— |
—
— |
—
— |
1.28 0.83–1.97 |
0.27 |
| Family history: first degree |
—
— |
—
— |
3.59 1.94–6.64 |
<0.001 |
—
— |
—
— |
—
— |
—
— |
3.39 1.81–6.28 |
<0.001 |
| Family history: second degree |
—
— |
—
— |
3.25 1.67–6.32 |
0.001 |
—
— |
—
— |
—
— |
—
— |
3.19 1.60–6.25 |
0.001 |
| Family history: third degree |
—
— |
—
— |
2.07 0.68–6.32 |
0.20 |
—
— |
—
— |
—
— |
—
— |
1.88 0.61–5.83 |
0.28 |
CI, confidence interval; OR, odds ratio; PD, Parkinson disease; SNP, single-nucleotide polymorphism.
Included four SNPs: SNCA rs11931074, SNCA rs356220, MAPT rs1800547, and the LRRK2 G2019S mutation (rs34637584).
Included all 33 SNPs with known association with PD.
Model 2 evaluated family history of PD adjusted for the covariates included in model 1. Those who reported a family history of PD in a first- or second-degree relative were nearly four and three times, respectively, more likely to have PD as compared with those without a family history of PD in first-, second-, or third-degree relatives (OR: 3.59, 95% CI: (1.94–6.64) and OR: 3.25, 95% CI: (1.67–6.32), respectively). Family history in a third-degree relative was not associated with PD in this model (Table 2).
Characteristics of genetic variants
The characteristics of the genetic variants are shown in Supplementary Table S1 online. Minor allele frequencies for SNPs in our control sample were similar to those in the HapMap CEU population, and Hardy–Weinberg equilibrium was not significantly violated in the controls (>0.10). Three-quarters of risk alleles were common, with minor allele frequencies >10%. The minor alleles of the SNCA rs11931074, SNCA rs356220, TMEM175 rs6599388, LRRK2 rs1491942, LRRK2 rs34637584, GAK rs11248051, and HLA-DRA rs3129882 variants were associated with a significantly increased risk of PD in univariate analyses (Supplementary Table S1 online). The minor allele of the MAPT rs1800547 variant conferred a decreased risk for PD (Supplementary Table S1 online).
A multivariate analysis was conducted on a subset of four variants from three established PD genes to create the weighted risk allele score used in models 3 and 5. In this analysis, SNCA rs356220, LRRK2 rs34637584, and MAPT rs1800547 remained associated with PD, but SNCA rs11931074 did not. (Supplementary Table S2 online).
A fuller multivariate analysis was conducted using all 33 SNPs from 25 genes to create the weighted risk allele score used in model 4. In this analysis, HLA-DRA rs3129882 was associated with increased risk, whereas FAM47E rs6812193, BST1 rs11724635, and MAPT rs1800547 were associated with decreased risk of PD (Supplementary Table S3 online).
Risk allele score and risk for PD
Figure 1 shows the distribution of the weighted risk allele score by case–control status for models 3 and 5 (Figure 1a) and model 4 (Figure 1b). Histograms for both models in controls and cases are normally distributed and overlap each other extensively, although the distribution for cases is shifted slightly to the right.
Figure 1.
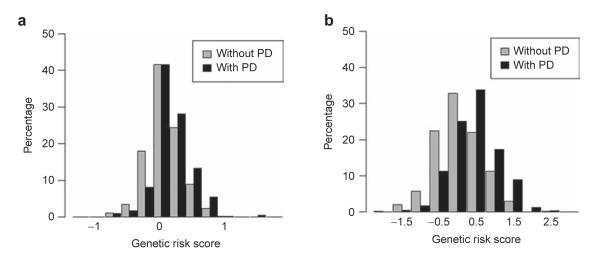
Distribution of weighted risk allele score by case–control status for (a) model 3/5 and (b) model 4. PD, Parkinson disease.
In model 3, the risk allele score constructed from four variants was included in risk prediction along with age, sex, and smoking. The risk allele score was associated with an approximately three-fold increase in risk for PD for every one unit increase in risk allele score (OR: 2.57 95%, CI: (1.72–3.83)). Model 4 included a weighted risk allele score constructed from 33 variants in addition to age, sex, and smoking. The weighted risk allele score was also associated with a nearly threefold increase in risk of PD for every one unit increase in risk allele score (OR: 2.62, 95% CI: (2.07–3.30)) (Table 2). Including either risk allele score did not attenuate the association of sex with PD observed in model 1.
Model 5 was the largest model and added the weighted risk allele score created from four variants to model 2 covariates. The weighed risk allele score remained significantly associated with PD after adjusting for family history (Table 2). Similarly, the OR for family history was not attenuated by adding the risk allele score.
Discriminatory ability
The receiver operating characteristic curves for all models are shown in Figure 2. Our first model, which included only age, sex, and smoking, had a discriminatory capacity of 0.6534 (95% CI: (0.6183–0.6885)) in the training set and was replicated in the test set (AUC: 0.6831, 95% CI: (0.6484–0.7177), pcompared to model 1 training = 0.2382). Adding family history, but no genetic markers (model 2), significantly increased discriminatory capacity to 0.6847 (95% CI: (0.6513–0.7181), pcompared to model 1 training set <0.001) in the training set and was replicated successfully with an AUC of 0.7117 (95% CI: (0.6789–0.7446), pcompared to model 1 test set = 0.002, pcompared to model 2 training set = 0.2581) in the test set. Alternatively, we added a risk allele score constructed from variants within the SNCA, LRRK2, and MAPT genes in model 3; this model was replicated in the test set and significantly increased the discriminatory capacity as compared with model 1 in the training and test sets (pcompared to model 3 training set = 0.5048, AUC = 0.6886, 95% CI: (0.6552–0.722), pcompared to model 1 training set = 0.001; AUC = 0.7047, 95% CI: (0.6712–0.7382), pcompared to model 1 test set = 0.044). Adding the weighted risk allele score created from all 33 variants to age, sex, and smoking (model 4) significantly increased the discriminatory capacity to 0.727 (95% CI: (0.6956–0.7584)) in the training set (pcompared to model 1 training set <0.001). However, the discriminatory capacity of model 4 in the test set was only 0.7047 (95% CI: (0.6718–0.7375)), and although it replicated the training set AUC (pcompared to model 4 training set = 0.3359), it was not significantly higher than that of model 1 (pcompared to model 1 test set = 0.1193). Adding the weighted risk allele score constructed from four variants, which increased prediction, to model 2 (model 5) increased the AUC to 0.7112 (95% CI: (0.679–0.7434)) in the training set and was replicated in the test set (AUC: 0.729, 95% CI: (0.6968–0.7611)), pcompared to model 5 training set = 0.4435). The discriminatory capacity of model 5 was significantly higher than that of model 1 (pcompared to model 1 training set <0.001, pcompared to model 1 test set <0.001). We then compared with model 2 to determine if the genetic risk score improved risk prediction in addition to family history. The discriminatory capacity of model 5 was significantly greater than that of model 2 in the training set and the test set (pcompared to model 2 training set = 0.003, pcompared to model 2 test set = 0.04).
Figure 2.

Receiver operating characteristic curves comparing model 1 with model 2, model 3, model 4, and model 5 in the (a) training and (b) test sets. AUC, area under the curve.
Sensitivity analysis
We performed a sensitivity analysis to evaluate the impact of including subjects with missing or unknown family history information. ORs for all covariates and AUCs in all models remained unchanged when those with missing family history (n = 107) were excluded from the training set (Supplementary Table S4 online).
DISCUSSION
In this study, we compared five models of PD risk prediction. All five models contained the covariates age, sex, and smoking. In model 3, a weighted risk allele score constructed from four variants in the SNCA, LRRK2, and MAPT genes was added to the baseline model, whereas model 4 included a weighted risk allele score constructed from a total of 33 SNPs in 25 genes. A larger risk allele score was associated with greater risk for PD for models 3 and 4. Although each one unit increase in risk allele score was associated with a nearly threefold greater risk of PD in either model, using the risk allele score in a model to predict risk for PD had a fairly low (0.69–0.73) discriminatory ability. Pepe and colleagues21 reported that ORs of this size, which are commonly observed in complex diseases, have little impact on the C-statistic. In addition, they estimate that the contribution of the predictors included in a model requires an OR of about 16 (corresponding to an AUC of 0.84) to achieve reasonable discrimination. Therefore, even though the OR for our combined genetic data was associated with a nearly threefold significant increase in risk, this OR was not strong enough to translate into significant discriminatory capacity for our genetic models. The common variants identified for almost all complex diseases, including PD, have very modest ORs. Identifying additional genetic variants with considerably larger effects will be necessary to achieve any substantial improvement in the AUC.
Several risk prediction studies have been published for various chronic diseases such as cardiovascular disease, diabetes, and breast and prostate cancer. The discriminatory ability of the models including genotype information ranges from 0.53 to 0.61.22–26 23andMe recently published a paper including several models of risk prediction for PD based on between 9 and 803 genetic variants, resulting in discriminatory ability ranging from 0.55 to 0.61,10 which is within the range reported for other complex diseases. In general, better discriminatory ability is seen from genetic variants included in risk prediction models for diseases with autoimmune etiology (e.g., age-related macular degeneration, psoriasis, and Crohn disease), ranging from 0.72 to 0.80.27–29
Generally, genetic risk prediction for common diseases has resulted in low discriminatory ability, which may, in part, be due to limitations in this method of risk prediction. Specifically, one limitation with many modeling approaches is that they may not accurately approximate the biological processes underlying the molecular pathogenesis of PD. Another limitation is the common practice of excluding highly correlated SNPs from analysis, as we have done, which might diminish predicted risk given that there is evidence that poorly performing but highly correlated markers can add substantially to model performance.30 Third, logistic regression analysis implicitly assumes a multiplicative risk model. But it is unclear whether a multiplicative model or an additive model best fits genetic risk data. The discriminative power (e.g., AUC) of the multiplicative risk model is greater and risks predicted under a multiplicative model are more extreme than those predicted under an additive risk model.31 Without knowing the mode of biological interaction underlying gene variant contribution to pathogenesis of disease, choosing the wrong model will cause overestimation or underestimation of risk and predictive ability of the risk model.31 Fourth, as mentioned earlier, combined risk from genetic variants must be quite large to observe any significant increase in discriminatory ability. Fifth, the sensitivity and specificity of a model are dependent on the subjects included for risk prediction.32,33 As such, misclassification due to heterogeneity in disease etiology, case–control status, or exposure variables will affect the model’s predictive ability.
The goal of this study was to compare the predictive ability of family history vs. specific genetic risk variants. Adding family history data to standard demographic risk factors for PD resulted in significantly better discriminatory ability than demographic risk factors alone. Adding a weighted risk allele score to family history information also significantly improved prediction, increasing discriminatory capacity from 0.71 to 0.73. Family history is a crude genomic measure, incorporating not only shared genetic variants but also shared environmental risk factors and interactions. Because both family history and the risk allele score were as strongly associated with PD in the model that contained both (model 5) as in the models that analyzed each separately (models 2 and 3), we hypothesize that family history incorporates genetic risk factors, including rare variants with larger effect sizes, that are different from genetic risk factors identified through genome-wide association studies. These differences may reflect different underlying etiology of patients with PD that could be used to identify more homogeneous subgroups for further study. However, self-reported family history is subject to recall bias, whereas assessment of genetic risk factors is an objective measure. Because of this, predicted risks derived from self-reported family history may be overestimated.
Practical genetic risk prediction for PD will require identification of more genetic risk factors, especially those contributing to familial disease. A recent study used genetic complex trait analysis to quantify the heritability of PD. Overall, the heritability of PD was estimated at 0.27; known GWAS SNPs in PD regions contributed 0.03 to the heritability estimate,34 indicating that a large proportion of the genetic variance is not yet accounted for. Furthermore, using this method, early onset cases had lower heritability than late onset cases, an unexpected result. The authors hypothesized that rare genetic variants contributing to early onset cases—generally understood to have a greater familial and genetic component—were poorly accounted for with standard genotyping platforms. In our study, we observed minimal improvement in risk prediction when we included a weighted genetic risk score. This reflects the observations that known variants account for only ~10% of the heritability of PD and that genes involved in early onset cases, which may have larger effect sizes, are not generally included on standard genotyping platforms and are difficult to impute. In addition to finding more genetic variants, better methods are needed to appropriately model genetic risk for disease, accounting for the molecular biology of associated genetic variants and allowing for interactions between both genes and environmental factors.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the Department of Veterans Affairs (1I01BX000531) and the National Institutes of Health (P50 NS062684, R01 NS065070, R01 NS036960).
Footnotes
DISCLOSURE The authors declare no conflict of interest.
SUPPLEMENTARY MATERIAL Supplementary material is linked to the online version of the paper at http://www.nature.com/gim
REFERENCES
- 1.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 2.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125(Pt 4):861–870. doi: 10.1093/brain/awf080. [DOI] [PubMed] [Google Scholar]
- 3.Gagne JJ, Power MC. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 2010;23(74):12. doi: 10.1212/WNL.0b013e3181d5a4a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23:228–242. doi: 10.1177/0891988710383572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesage S, Condroyer C, Klebe S, et al. Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology. 2012;78:1449–1450. doi: 10.1212/WNL.0b013e318253d5f2. [DOI] [PubMed] [Google Scholar]
- 6.Vilariño-Güell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:62–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimprich A, Benet-Pagès A, Struhal W, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:68–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.International Parkinson’s Disease Genomics Consortium Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.International Parkinson’s Disease Genomics Consortium (IPDGC) Wellcome Trust Case Control Consortium 2 (WTCCC2). A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genetics. 2011;7(8):e1002142. doi: 10.1371/journal.pgen.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Do CB, Tung JY, Dorfman E, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011;7:e1002141. doi: 10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pankratz N, Wilk JB, Latourelle JC, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saad M, Lesage S, Saint-Pierre A, et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum Mol Genet. 2011;20:615–627. doi: 10.1093/hmg/ddq497. [DOI] [PubMed] [Google Scholar]
- 14.Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 15.Simón-Sánchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simón-Sánchez J, van Hilten JJ, van de Warrenburg B, et al. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet. 2011;19:655–661. doi: 10.1038/ejhg.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatr. 1988;51:745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kay DM, Zabetian CP, Factor SA, et al. Parkinson’s disease and LRRK2: frequency of a common mutation in U.S. movement disorder clinics. Mov Disord. 2006;21:519–523. doi: 10.1002/mds.20751. [DOI] [PubMed] [Google Scholar]
- 20.Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pepe MS, Janes H, Longton G, Leisenring W, Newcomb P. Limitations of the odds ratio in gauging the performance of a diagnostic, prognostic, or screening marker. Am J Epidemiol. 2004;159:882–890. doi: 10.1093/aje/kwh101. [DOI] [PubMed] [Google Scholar]
- 22.Davies RW, Dandona S, Stewart AF, et al. Improved prediction of cardiovascular disease based on a panel of single nucleotide polymorphisms identified through genome-wide association studies. Circ Cardiovasc Genet. 2010;3:468–474. doi: 10.1161/CIRCGENETICS.110.946269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Machiela MJ, Chen CY, Chen C, Chanock SJ, Hunter DJ, Kraft P. Evaluation of polygenic risk scores for predicting breast and prostate cancer risk. Genet Epidemiol. 2011;35:506–514. doi: 10.1002/gepi.20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mealiffe ME, Stokowski RP, Rhees BK, Prentice RL, Pettinger M, Hinds DA. Assessment of clinical validity of a breast cancer risk model combining genetic and clinical information. J Natl Cancer Inst. 2010;102:1618–1627. doi: 10.1093/jnci/djq388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meigs JB, Shrader P, Sullivan LM, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359:2208–2219. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Hoek M, Dehghan A, Witteman JC, et al. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes. 2008;57:3122–3128. doi: 10.2337/db08-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen H, Poon A, Yeung C, et al. A genetic risk score combining ten psoriasis risk loci improves disease prediction. PLoS ONE. 2011;6:e19454. doi: 10.1371/journal.pone.0019454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hageman GS, Gehrs K, Lejnine S, et al. Clinical validation of a genetic model to estimate the risk of developing choroidal neovascular age-related macular degeneration. Hum Genomics. 2011;5:420–440. doi: 10.1186/1479-7364-5-5-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang J, Kugathasan S, Georges M, Zhao H, Cho JH, NIDDK IBD Genetics Consortium Improved risk prediction for Crohn’s disease with a multi-locus approach. Hum Mol Genet. 2011;20:2435–2442. doi: 10.1093/hmg/ddr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bansal A, Pepe MS. UW Biostatistics Working Paper Series. The Berkley Electronic Press; Berkley, CA: 2011. When does combining markers improve classification performance and what are the implications for practice. (Paper 378) [Google Scholar]
- 31.Moonesinghe R, Khoury MJ, Liu T, Janssens ACJW. Discriminative accuracy of genomic profiling comparing mulitplicative and additive risk models. Eur J Hum Genet. 2011;19:180–185. doi: 10.1038/ejhg.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brenner H, Gefeller O. Variation of sensitivity, specificity, likelihood ratios and predictive values with disease prevalence. Stat Med. 1997;16:981–991. doi: 10.1002/(sici)1097-0258(19970515)16:9<981::aid-sim510>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 33.Moons KG, van Es GA, Deckers JW, Habbema JD, Grobbee DE. Limitations of sensitivity, specificity, likelihood ratio, and bayes’ theorem in assessing diagnostic probabilities: a clinical example. Epidemiology. 1997;8:12–17. doi: 10.1097/00001648-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Keller MF, Saad M, Bras J, et al. Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum Mol Genet. 2012;21:4996–5009. doi: 10.1093/hmg/dds335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.