

Abstract
The radish displays great morphological variation but the genetic factors underlying this variability are mostly unknown. To identify quantitative trait loci (QTLs) controlling radish morphological traits, we cultivated 94 F4 and F5 recombinant inbred lines derived from a cross between the rat-tail radish and the Japanese radish cultivar ‘Harufuku’ inbred lines. Eight morphological traits (ovule and seed numbers per silique, plant shape, pubescence and root formation) were measured for investigation. We constructed a map composed of 322 markers with a total length of 673.6 cM. The linkage groups were assigned to the radish chromosomes using disomic rape-radish chromosome-addition lines. On the map, eight and 10 QTLs were identified in 2008 and 2009, respectively. The chromosome-linkage group correspondence, the sequence-specific markers and the QTLs detected here will provide useful information for further genetic studies and for selection during radish breeding programs.
Keywords: radish, STS, linkage map, chromosome assignment, QTL
Introduction
The radish, Raphanus sativus L. (2n = 18), is an important vegetable, especially in Asia (see Kaneko et al. 2007 for a review), and is also used as fodder and oil plants. Its hypertrophied root with hypocotyl is mainly used for human consumption. Young pods are also eaten as a vegetable in tropical Asia. Although nomenclature is complex, cultivated radishes are classified into three main types based on edible parts: fodder and oilseed radish (convar. oleifer Alef.), seed-pod or rat-tailed radish (convar. caudatus (L.f.) Pistr.) and small- and large-rooted radish (convar. sativus L.) (Crisp 1995). Wide morphological variations are observed in the roots of local and commercial radish cultivars. Despite such importance, few studies have been undertaken to analyze the genetic factors that determine the variability of agronomic traits in radish. The radish genome is closely related to the Brassica genomes carried by the vegetable and oilseed plants, Brassica rapa, B. nigra, B. oleracea and the amphidiploid species (B. juncea, B. napus and B. carinata). Linkage maps of Brassica species have been extensively studied and numerous genetic loci for qualitative and quantitative traits have been identified (see Schmidt and Bancroft 2011 for a review). The whole genome data are now available for B. rapa (Wang et al. 2011). In contrast, limited number of genetic maps has been constructed for radish (Bett and Lydiate 2003, Budahn et al. 2009, Kamei et al. 2010, Li et al. 2011, Nakatsuji et al. 2011, Shirasawa et al. 2011, Tsuro et al. 2005, 2008, Xu et al. 2012), few of which have included agronomic trait loci.
In addition to the molecular genetic map, cytogenetic study can provide information complementary to that from genetic and physical maps. A complete set of disomic rape-radish chromosome-addition lines has also been developed (Budahn et al. 2008) and the position of a locus for resistance against the beet cyst nematode has been determined using the chromosome-addition lines (Budahn et al. 2009). However, the cytogenetic data have not yet been integrated to sequence-specific molecular markers of radish.
Molecular markers provide a powerful tool for gene mapping and efficient selection of the genotypes having desired agronomic traits. This strategy is known as marker-assisted selection (MAS). Among the molecular markers, simple sequence repeats (SSRs), tandem repeats of short DNA sequences from one to six base pairs, are useful tools. Because of their high level of polymorphism, ubiquity and co-dominance, SSRs have become a valuable source of molecular markers in genetic analysis (Jones et al. 2009). SSRs have also been useful to detect the conserved order of genes among related genomes, referred to as “synteny”. Although we have previously developed 417 SSRs, including 293 genomic SSRs, from the rat-tailed radish, Japanese cultivar ‘Harufuku’ and their F1 plant (Nakatsuji et al. 2011), most of our SSRs have not yet been employed for mapping studies. The rat-tail radish and ‘Harufuku’ have several differences in vegetative and reproductive morphology traits (e.g. ovule and seed numbers, plant shape, leaf pubescence and root formation). Such the differences between the two radishes allowed us to investigate genetic loci controlling their morphologies.
The objective of this study was the construction of a radish linkage map containing radish SSR and sequence-tagged site (STS) markers, assignment of the linkage groups (LGs) to the chromosomes and mapping of quantitative trait loci (QTLs) for vegetative and generative characters.
Materials and Methods
Plant materials
A population of 94 F4 and F5 recombinant inbred lines (RILs) was developed from a cross between the rat-tail radish and the Japanese radish ‘Harufuku’ inbred lines (Saya-S and Haru-S, respectively) using a single-seed-descent of the F1 plant after four generations of selfings. The rat-tail radish is grown in Southeast Asia for its edible young seed-pods (Kitamura 1958). Instead of many seed-pods and larger plant top, the rat-tailed radish forms no fleshy root (Supplemental Fig. 1A). Its leaves are erect without pubescence on the surface. The ‘Harufuku’ is classified into Spring radish, which has a white, wedge-shaped root (Furusato and Miyazawa 1958) (Supplemental Fig. 1B). It has hairy, prostrate leaves. Although these two cultivars are classified as different convarieties (caudatus and sativus), they intercrossed freely. Both parental lines were supplied by the NARO Institute of Vegetable and Tea Science, Tsu, Japan. The parental lines Saya-S and Haru-S used in this study are highly homozygous by five or six successive selfings. A commercial rat-tail radish cultivar (Chia Tai Co. Ltd., Bangkok, Thailand) (Supplemental Fig. 1A) was used for field tests instead of the original parental line Saya-S because of the severe inbred depression in the Saya-S. Note that the ‘Harufuku’ inbred line Haru-S used in this study had shorter main root and more lateral roots than the original cultivar (Supplemental Fig. 1B), probably being an aspect of inbred depression.
Field tests
The population of 94 F4 or F5 families as well as their parental lines was used for investigation of eight agronomic traits (Table 1). For scoring the ovule (OV) and seed numbers (SE) per silique, each F4 RIL was grown in a heated greenhouse under a natural photoperiod until June 2008. For the other agronomic traits, F5 RILs were cultivated under natural conditions in an open field of the Kyoto Prefectural University Farm (Seika, Kyoto, Japan). Six plants per each RIL were cultivated from mid-September to mid-December. The field test was performed once in 2008 and 2009, respectively. Plant shape (PS) and pubescence (Pub) were categorized visually on 1–5 scales two months after sowing (Table 1 and Supplemental Fig. 2). Fresh weights for the following parts were measured at harvest (three months after sowing). After measurement of whole plant weight (WW), plant tops were removed to measure upper part weight (UW). The remainder (hypocotyls and roots) was taken as whole root weight (RW). The whole root was separated into main and lateral roots to measure main root weight (MW) and lateral root weight, respectively. The lateral root weight trait was excluded from further analysis because the variation was small (data not shown). After removal of extreme outliers, the mean score of the six F5 plants for each line was used as data for each F4 line.
Table 1.
List of traits analyzed and the phenotypic values of the F4 or F5 population and the parental lines
| Trait | Abbreviation | Year | Mean (±SD) in RIL | Range in RIL | Mean (±SD) in rat-tail radish | Mean (±SD) in Haru-S |
|---|---|---|---|---|---|---|
| Ovule number per silique | OV | 2008 | 9.93 ± 2.07 | 5.00–16.25 | 9.40 ± 0.80 | 6.60 ± 0.49 |
| Seed number per silique | SE | 2008 | 3.09 ± 1.00 | 1.14–5.55 | N.D.c | 3.04 ± 1.51 |
| Plant shape | PS | 2008 | 3.1 ± 0.8 | 1–5a | 5 | 1 |
| 2009 | 3.2 ± 0.7 | 1–5a | 5 | 1 | ||
| Pubescence | Pub | 2008 | 2.9 ± 1.3 | 1–5b | 1 | 5 |
| 2009 | 2.9 ± 1.3 | 1–5b | 1 | 5 | ||
| Whole plant weight (g) | WW | 2008 | 598 ± 236 | 142.6–1516 | 982 ± 311 | 429 ± 173 |
| 2009 | 1267 ± 458 | 426.7–2890 | 2486 ± 1140 | 1038 ± 252 | ||
| Upper part weight (g) | UW | 2008 | 416 ± 173 | 119.8–1142 | 879 ± 268 | 232 ± 86.9 |
| 2009 | 1005 ± 385 | 341.7–2518 | 2346 ± 1096 | 716 ± 174 | ||
| Whole root weight (g) | RW | 2008 | 180 ± 119 | 16.8–648 | 103 ± 46.5 | 197 ± 88.4 |
| 2009 | 249 ± 174 | 26.0–801 | 135 ± 63.9 | 320 ± 83.4 | ||
| Main root weight (g) | MW | 2008 | 148 ± 96.3 | 15.7–534 | 87.5 ± 43.3 | 175 ± 76.3 |
| 2009 | 239 ± 165 | 26.0–744 | 123 ± 63.3 | 314 ± 84.8 |
1 = prostrate; 2 = semi-prostrate; 3 = intermediate; 4 = open; 5 = erect, see Supplemental Fig. 2.
1 = no hair; 2 = sparse; 3 = intermediate; 4 = abundant; 5 = very abundant hair like Haru-S.
N.D., not determined.
Genotyping, linkage mapping, synteny detection and QTL analysis
Genomic DNA was extracted using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) from a bulk of leaf tissue consisting on average of 13 F5 plants for each line, because the initial DNA extracted from F4 plants by the CTAB method was not of sufficient quality for AFLP analysis. We confirmed no significant incongruence in genotypes between the F4 and the bulked F5 DNAs for each line with tested markers (data not shown). AFLP was performed according to method described by Tsuro et al. (2008). SSR primers from B. rapa (Suwabe et al. 2006) and radish (Nakatsuji et al. 2011, Wang et al. 2007, this study) were randomly picked up to screen polymorphic markers between the parental lines. PCR amplification and electrophoresis were carried out as described previously (Saito et al. 2006). The published STS markers for B. rapa and radish (Kamei et al. 2010, Li et al. 2011, Niikura and Matsuura 1998, Saito et al. 2006) were also used. New STS markers were developed from the sequences of Arabidopsis, B. rapa and radish at the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/nucleotide/) and the Radish-Database (Supplemental Fig. 3, http://radish.plantbiology.msu.edu/index.php/Sequences:All) (their accession nos. AB630374-AB630382 and AB611697-AB611700).
Segregation of each marker in the bulked F5 families was scored and treated as a genotype of the F4 line. Markers deviating significantly (P < 0.001) from the expected segregation ratio were excluded from the analysis. A linkage map was constructed using JoinMap ver. 3.0 (Van Ooijen and Voorrips 2001) as described previously (Saito et al. 2006). To detect potential synteny, nucleotide sequences of the mapped sequence-tagged markers were searched for the Arabidopsis genome data using a BlastN algorithm under the default parameter sets at the NCBI. QTL analysis was performed using MapQTL 6 software (Van Ooijen 2009) as described previously (Kubo et al. 2010). The significant logarithm of odds (LOD) thresholds of each QTL (α = 0.05) were estimated by 1000 permutations. They ranged from 2.75 (UW in 2009) to 3.00 (OV).
Chromosomal assignment
A complete set of disomic rape-radish chromosome-addition lines (Budahn et al. 2008) was used for assignment of the chromosomes corresponding to the radish LGs. Each disomic addition line has one of the nine pairs of the radish-derived chromosomes (pairs AA-II) on the synthetic B. napus background (Peterka et al. 2004). Thirty six markers (33 radish SSRs, one radish STS and two B. rapa SSRs) were screened for PCR, using the genomic DNAs of the nine disomic addition lines as well as their parental lines as templates. Loci which yielded radish-specific PCR products in the particular chromosome addition line (Supplemental Fig. 5, arrowheads) were assigned to one of the nine radish chromosomes (Supplemental Fig. 5, letters).
Results
Analysis of phenotypic variation
Phenotypic variation of eight agronomic traits examined in RILs and the parental lines are shown in Supplemental Fig. 4 and summarized in Table 1. The mean scores of the rat-tailed radish were larger than those of the Haru-S in OV, PS, WW and UW in each year (and presumably also in SE, see Discussion) (Table 1). It was vice versa in Pub, RW and MW. The majority of RILs were roughly segregated into two categories of pubescence (Pub), presence (categories 2–5) and absence (category 1), whose distribution was biased towards the presence of pubescence (Supplemental Fig. 4D). Since the segregation ratio, divided into the above two categories of Pub as a qualitative trait, was significantly deviated at P < 0.0001 from an expected segregation ratio for RIL population, we conducted the QTL analysis for Pub as below. For the other traits, RILs did not show a discrete segregation, suggesting that these traits are controlled by multiple genes. The distributions of RILs for weight traits (WW, UW, RW and MW) were shifted towards larger values in 2009 than seen in 2008 (Supplemental Fig. 4E–4H, gray and black bars, respectively) probably because of an environmental effect. In root weight traits (RW and MW), RILs with transgressive weight exceeding the values for the Haru-S (Supplemental Fig. 4G–4H, pink arrows) were found. In OV, transgression beyond the value for the rat-tailed radish also occurred (Supplemental Fig. 4A, green arrow).
Linkage mapping, LG assignment using the disomic chromosome addition lines and synteny to Arabidopsis
The map comprised nine LGs, including 163 radish AFLPs, 97 radish SSRs (75 genomic SSRs and 22 EST-SSRs), 37 radish STSs, 15 B. rapa SSRs, seven B. rapa STSs and three Arabidopsis STSs (Fig. 1). Of these, 10 SSR and 25 STS markers were newly developed or modified in this study (Table 2). The total map length was 673.6 cM, with an average marker interval of 2.1 cM. A total of 36 markers were tested for assignment of the present LGs to radish chromosomes using the disomic addition lines (Budahn et al. 2008) (Supplemental Fig. 5 for examples). Of these, 19 markers (18 radish SSRs and one radish STS) were assigned to the radish chromosomes (Fig. 1, gray boxes). There was no incongruence between the markers and the assigned chromosomes. Thus, we also refer each LG to our previous nomenclature (Chrs A–I) as well as LG numbers (R1–R9) given by Li et al. (2011) (Fig. 1, parentheses). Nucleotide sequences of 138 sequence-tagged markers showed homology to Arabidopsis (data not shown). Most of the homologies to the five Arabidopsis chromosomes were divided into many segments and dispersed throughout the present radish map. Relatively large clusters were found in all radish LGs, which may represent syntenies to Arabidopsis.
Fig. 1.
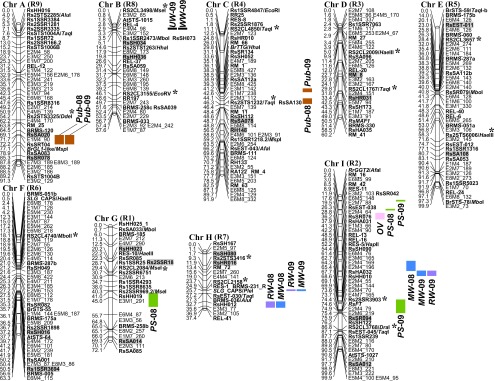
A linkage map of radish and the locations of QTLs for eight agronomic traits. Linkage groups were referred to chromosome numbers (Chrs A–I) (Budahn et al. 2008) and LG numbers (R1–R9) (Li et al. 2011). Marker names and genetic distances (cM) are shown on the right and left of each LG, respectively. SSR and STS markers are indicated with bold-face type. Markers used for the assignment to radish chromosomes and LG numbers are indicated with gray boxes and asterisks, respectively. Locations of QTLs are indicated with differentially-colored, vertical thick bars, the lengths of which correspond to the map positions in Table 3. The QTLs detected in 2008 and 2009 are represented with suffix numbers such as “−08” and “−09”, respectively.
Table 2.
List of SSR and STS markers developed and modified in this study
| Marker namea | Marker type | Repeat motif | Primer sequence (5′-3′) | Product size (bp)b | GenBank accession no. | |
|---|---|---|---|---|---|---|
| Radish genomic SSR markers | ||||||
| REL-4 | SSR | (CTT)6 | Forward Reverse |
GATACGGAGACAAGAAGAAG CGGACAGAAGAGAAAAAGAC |
237 | AB630374 |
| REL-12 | SSR | (GAA)25 | Forward Reverse |
CTTTAACACCAGGAATCC GCCTCTGAAATCCTCCAATC |
330 | AB630375 |
| REL-13 | SSR | (CA)13 | Forward Reverse |
CTAGCAATGCATACCAAACAG AACTTGGTCGTTGAGCAG |
166 | AB630376 |
| REL-16 | SSR | (TG)13 | Forward Reverse |
ACAGCAACGTTTTCAAGTGCTC CTCACATGCAATGCAATGCATAC |
197 | AB630377 |
| REL-20 | SSR | (TC)8 | Forward Reverse |
CCTTCATTCTTTGCTTCTTCC TTGTTTCTTCCCAGAAAGCC |
162 | AB630378 |
| REL-37 | SSR | (GA)16 | Forward Reverse |
GAAAACGGGAATCAGATACAAC CTCATTCACTTTCTTCGCCA |
94 | AB630379 |
| REL-40 | SSR | (AC)12 | Forward Reverse |
TGATAGCAAAGGCAGAGGTGGC CAAGAAACCTGTCGCAAGGTACAG |
176 | AB630380 |
| REL-41 | SSR | (CA)6 | Forward Reverse |
CGTTCATATCAACTTTTTGAGCGAG TTATGCCAGCCTTAGCTGC |
154 | AB630381 |
| SSR and STS markers developed from radish ESTs | ||||||
| RES-8 | SSR | (T)15GT C(A)21 |
Forward Reverse |
GGATAACAATTACCTCGATCA GGAAACCAAATACCACAAAAG |
206 | AF248491 |
| RES-10/HaeIII | STS (CAPS) | – | Forward Reverse |
CTTCCTAGTAGCTAACTC AACAGTCATGACACGTGG |
195 | AF263920 |
| RrGGT2/AfaI | STS (CAPS) | – | Forward Reverse |
CTATCTCCAGGCACTGGAATCG ATCGTCCCTCCGGCGATTGG |
431 | EX761507 |
| RsEST-011 | STS (indel) | – | Forward Reverse |
ATCATGTCTACGTGCGGAAACT ACCATTACATCACACACCATGC |
353 | AF051121 |
| RsEST-012 | STS (indel) | – | Forward Reverse |
GTACCAAGTTCCCCAGTTATGC GATGCCAACAAAGGACTTGTAA |
294 | AF051120 |
| RsEST-030/TaqI | STS (CAPS) | – | Forward Reverse |
CTATTCCTTTCACCCGTTTCT CTGACAGACTCTTGCTTCTCAG |
303 | T25178 |
| RsEST-038 | STS (indel) | – | Forward Reverse |
GATGTATCAGGTGGTGAAAT TACACCACCAGAAAGTCCAC |
192 | EL738632 |
| RsEST-043/AfaI | STS (CAPS) | – | Forward Reverse |
ATCATGGTCCGATACACAAATC TGCATTGAAACCAAAAGACC |
350 | EL738626 |
| RsEST-045/TaqI | STS (CAPS) | – | Forward Reverse |
GCAGAATTTTCCGCTGTAGTTC TGACAAAATACTTCCTGCAAGC |
275 | EL738624 |
| RsFT | STS (indel) | – | Forward Reverse |
TAGGTTTTCTGTTATATCACTGCT AAATCGAGTCTCTGAAAGTTTTAC |
103 | AB611697 |
| RsMAF1 | STS (indel) | – | Forward Reverse |
GTCAGGATCCTTGATCGATATGG GCTCAGAGATTTTGACTGAAGAT |
81 | FD958357 |
| RsSTS1004A/TaqI | STS (CAPS) | – | Forward Reverse |
ATTTCAATCCACTCAAGCTGAATCT TTTTATCTTGCATGGAACTCTAACC |
522 | AB611698 |
| RsSTS1004B | STS (indel) | – | Forward Reverse |
TCTAAGCCCATGGTTATGAGAC AAGGAAAGGCCCACTTGAATA |
154 | AB611699 |
| RsSTS1006B | STS (indel) | – | Forward Reverse |
CCCTAACGCACAAGTAATTA TAATCCGTGTAGCAGAACAATA |
191 | AB611700 |
| Rs1SSR7063 | SSR | (CT)7 | Forward Reverse |
GGACAAGTTTATACTTTAAGTG GATTTTTGCACAAGTTTTCCTAGAG |
223 | RS1CL7063Contig1c |
| Rs2STS1232/TaqI | STS (CAPS) | – | Forward Reverse |
AGCCTGGTAAGCATCTCCAA TGACCATGGTGGGAGTGAAT |
468 | RS2CL1232Contig1c |
| Rs2STS1263/HhaI | STS (CAPS) | – | Forward Reverse |
GATCCAAGGTTCCATGCCCATG GACCCGGCAGCATTCTGCTCTA |
366 | RS2CL1263Contig1c |
| Rs2STS2285/AluI | STS (CAPS) | – | Forward Reverse |
TGTTCTTCAGGGGAGTTTGATT CCTTGGTCTTCTAGTTGCGTTT |
489 | RS2CL2285Contig1c |
| Rs2STS3325/DdeI | STS (CAPS) | – | Forward Reverse |
CTAGCTCCAGGGAGTCAATTCA CCATCTGAGTAACGAGTGCG |
517 | RS2CL3325Contig1c |
| Rs2STS3416 | STS (indel) | – | Forward Reverse |
TACCAAAGAAGAGACCCTTATG TCTAAGAACCCTTGATCTTTGT |
451 | RS2CL3416Contig1 c |
| Rs2STS6006/HaeIII | STS (CAPS) | – | Forward Reverse |
CACAGCCATCACTTGCAGATCC ATGCTGCTACTGTACACCGGGT |
539 | RS2CL6006Contig1c |
| Markers developed from Brassica rapa genomic DNA | ||||||
| BrGL1-like/MspI | STS (CAPS) | – | Forward Reverse |
GAACAGATAACCAAGTCAAG TTAAACCAGAAAGCGTTACTGC |
310 | AC232566 |
| Marker modified from the previously reported B. rapa SSR | ||||||
| BRMS-036_R/AluI | STS (CAPS) | – | Forward Reverse |
GGTCCATTCCTTTTTGCATCTG GGGTAACAAACATTTCTTAACCTCTC |
141 | AB630382 |
| BRMS-231_R | STS (indel) | – | Forward Reverse |
CGGTGTATTTCCATTTATGCGTTT TTCTGGACTTCAATTAGAGATGGTC |
180 | CU695294 |
| Markers developed from Arabidopsis genome data | ||||||
| AtSTS-64 | STS (indel) | – | Forward Reverse |
CTCTCGGGTAGTTTACCTGACTTC GAACTTGGCCAGTTAGGTTATTGT |
640 | At3g05990d |
| AtSTS-1015 | STS (indel) | – | Forward Reverse |
GTTGGTTAGGCTTAAACAGATG TCCTACAATATCTTCAGCTTCTC |
850 | At1g74860d |
| AtSTS-1027 | STS (indel) | – | Forward Reverse |
TAAAACCTATCATCTCCGTACC GCACAACTTGAATCACACC |
880 | At1g71950d |
In CAPS markers, restriction enzymes used for polymorphism detection are indicated after slashes (/).
Sizes are estimated by agarose gel electrophoresis or predicted from the sequences used for primer design.
For markers designed from radish cDNA contig data, the cDNA contig nos. are shown instead of GenBank accession nos. (their sequences are shown in Supplemental Fig. 3).
For markers designed from Arabidopsis genome data, the locus tags are shown instead of GenBank accession nos.
QTL analysis
On the present map, QTLs were identified for five and six traits in 2008 and 2009, respectively (Table 3 and Fig. 1, thick bars). The phenotypic variations explained by each QTL varied from 9.3% to 37.7% (Table 3). A single OV QTL was detected on Chr I (Fig. 1, pink thick bar). The Saya-S allele resulted in an increase of the OV. PS QTLs were detected on Chr G in 2008 and on the lower part of Chr I in 2009 (Fig. 1, green thick bars). On the upper part of Chr I, PS QTL was located within the vicinity in each year. The QTLs on Chr G and on the upper part of Chr I had additive effects of the same direction; the Saya-S allele increased the PS scale, whereas the QTL on the lower part of Chr I had an opposite effect (Table 3). Pub QTLs were detected on Chrs A and C in both years (Fig. 1, brown thick bars). The QTLs on Chrs A and C explained a proportion of phenotypic variance of 34.4–37.7% and 24.2–25.3%, respectively and the Haru-S allele resulted in an increase of Pub. QTLs for WW and UW were identified at an overlapped region on Chr B in 2009 (Fig. 1, black thick bars). The QTL alleles affecting an increase of WW and UW would come from the Saya-S. RW QTLs were detected on Chr H in both years and on Chr I in 2009 (Fig. 1, purple thick bars). The Haru-S allele increased RW (Table 3). MW QTLs were found on Chrs H and I in both years (Fig. 1, blue thick bars). Three of the MW QTLs overlapped with the RW QTLs. These QTLs explained 14.6–33.9% of phenotypic variation. Like the case in RW, the Haru-S allele increased the MW.
Table 3.
Map positions and effect of QTLs detected in RILs
| Trait | Year | Chr | Map position in cM (peak)a | Marker intervalb | LODc | R2 (%)d | Additive effectc |
|---|---|---|---|---|---|---|---|
| OV | 2008 | I | 10.9–14.9 (11.9) | RES-11–E6M5_148 | 3.5 | 11.8 | 0.79 |
| PS | 2008 | G | 42.0–50.7 (45.0) | RsHH019–E6M4_87 | 3.8 | 10.8 | 0.31 |
| I | 8.5–8.9 (8.5) | RM_42–RES-11 | 6.4 | 20.9 | 0.40 | ||
| 2009 | I | 2.6–3.0 (2.6) | RM_16–E6M5_99 | 3.5 | 16.9 | 0.32 | |
| I | 67.4–75.9 (68.6) | E2M4_196–RsSR094 | 3.2 | 11.9 | −0.27 | ||
| Pub | 2008 | A | 72.2–78.1 (77.1) | RsSR104–RsSA083 | 13.6 | 37.7 | −0.86 |
| C | 42.6–46.1 (44.1) | E1M7_238–Rs2STS1232/TaqI | 9.7 | 24.2 | −0.68 | ||
| 2009 | A | 72.2–78.1 (77.1) | RsSR104–RsSA083 | 12.8 | 34.4 | −0.82 | |
| C | 36.1–38.3 (38.3) | RM_1–E5M8_427 | 10.1 | 25.3 | −0.69 | ||
| WW | 2009 | B | 1.8–2.8 (2.8) | AtSTS-1015–REL-4 | 3.8 | 18.7 | 213.34 |
| UW | 2009 | B | 1.8–2.8 (2.8) | AtSTS-1015–REL-4 | 3.6 | 17.8 | 175.33 |
| RW | 2008 | H | 34.7–35.6 (35.6) | RsHH012–E3M3_77 | 3.7 | 9.3 | −51.95 |
| 2009 | H | 24.9–25.7 (24.9) | RES-1–SLG_CAPS/PleI | 11.6 | 33.9 | −113.01 | |
| I | 48.5–51.9 (48.9) | REL-13–RES-5/HapII | 6.6 | 16.8 | −82.24 | ||
| MW | 2008 | H | 32.7–36.6 (35.6) | RsHH012–E3M2_105 | 8.4 | 25.1 | −53.52 |
| I | 42.2–48.5 (46.2) | E5M4_90–REL-13 | 5.4 | 14.6 | −42.84 | ||
| 2009 | H | 24.9–25.7 (24.9) | RES-1–SLG_CAPS/PleI | 11.7 | 33.9 | −107.27 | |
| I | 48.5–51.9 (48.9) | REL-13–RES-5/HapII | 6.9 | 17.3 | −79.06 |
Region above the LOD threshold, in which the peak position is in parentheses.
Smallest marker interval flanking a region above the LOD threshold.
Scores at the LOD peaks.
Phenotypic variation explained by each QTL at the LOD peak.
Discussion
Recently, we made radish linkage maps using F1 and F2 populations (Budahn et al. 2009, Kamei et al. 2010, Nakatsuji et al. 2011, Tsuro et al. 2005, 2008). These studies included limited number of specific markers derived from radish. Here we located 134 radish-specific markers (97 SSRs and 37 STSs) on the map using RILs. The map consisted of nine LGs, which corresponds to the haploid chromosome number of radish. Based on the anchor markers, correspondence of LGs between this study and Li et al. (2011) has been accomplished (Fig. 1, asterisks and parentheses). Through these maps, LGs from different studies (Kamei et al. 2010, Nakatsuji et al. 2011, Shirasawa et al. 2011) can also be assigned (data not shown). The total length of our map (673.6 cM) was shorter than those of Li et al. (2011) and Shirasawa et al. (2011). Assumed from the positions of anchor markers and the distributions of syntenic regions, the present map was roughly estimated to cover >70% of the above two maps (data not shown). Since our map still includes anonymous AFLPs and STSs that did not hit to Arabidopsis locus, the value might be underestimated. Thus, our map is considered to cover a significant fraction of the radish genome.
Two recent reports (Li et al. 2011, Shirasawa et al. 2011) involved many more markers and gave a more detailed picture of Arabidopsis synteny than this study. However, the present map has the following two advantages over the previous maps. First, each LG on the present map was assigned to a radish chromosome (Fig. 1, gray boxes). Previous work of chromosome-LG assignment was done with anonymous RAPD markers (Budahn et al. 2009). In this study, the assignment has been completed with radish-specific markers (Supplemental Fig. 5). This allows us to conduct easier and more concrete chromosome-LG assignment than in many other radish maps. Although Akaba et al. (2009) have made monosomic rape-radish addition lines, no assignment to radish linkage map has been reported. The chromosomal correspondence to the set of addition lines in Akaba et al. (2009) and ours should be considered in the future. Second, by using the present map, several QTLs for morphological traits were detected. Some of the QTLs showed low consistency between years probably because of an environmental effect. Indeed, the weight traits in 2008 were mostly smaller than those in 2009 (Supplemental Fig. 4E–4H). Low precipitation during the primary growth of radish could be a reason for such difference, because the monthly total precipitation in October, 2008 was lower than that of the climate normal (averages of 1997–2005) (data not shown). Unfortunately, the seeds of RILs were insufficient to assess the possibility of environmental effects and genotype-environment interaction by planting several fields. In contrast to the QTLs with low consistency, QTLs that were reproducible in the two years suggest the presence of loci with seemingly stable effects. The markers linked to the stable QTLs could provide practical tools for selecting radish lines. The details of QTLs are further described below.
The ovule and seed number traits in our radish population were quantitative. Although the SE remains to score in the rat-tailed radish inbred line Saya-S, presumably the Saya-S had more SE than the Haru-S, assumed from a report describing that seed numbers in rat-tailed radishes were ≥8 (Yazawa et al. 1953). In Arabidopsis, both of the OV and SE traits were also quantitative, in which QTLs overlapped on chrs 1 and 2 (Alonso-Blanco et al. 1999). In wild radish R. raphanistrum, the ovule number had a positive effect on the seed number (Conner et al. 1996). Unlike these cases, in this study, only an OV QTL has been found whereas no SE QTL has been detected. Such discrepancy might be because the number of ovules becoming seeds may be influenced by factors such as the ovule number itself, the quantity and quality of pollen transferred, nutrients and photosynthate (Lee 1988).
Two QTLs for pubescence were found in radish. There is no report of a pubescence locus on a radish map, whereas many reports of B. rapa studies (Ågren and Schemske 1992, Kubo et al. 2010, Li et al. 2009, Lou et al. 2007, Nozaki et al. 1997, Song et al. 1995, Teutonico and Osborn 1994, Zhang et al. 2009) have indicated that the pubescence is controlled by a single locus with the proposal of a few candidate genes. In the present case, a marker made from the B. rapa candidate gene (BrGL1) was mapped at the peak position of the Pub QTL on Chr A (Table 3 and Fig. 1). This gene is a good candidate for a Pub gene in our population; however, we have no information regarding the second Pub gene in the radish. The Pub QTLs found in this study are potentially used for breeding a pest-resistant radish line, as utilized in Brassica crops (Ågren and Schemske 1992).
The plant shape (e.g. leaf angle) is an important trait for cultivation because it largely affects planting density and effectiveness of receiving solar energy. This trait has extensively been studied in maize and rice (Tian et al. 2011 and references therein), whereas little is known for Brassicaceae crops. In the PS QTLs identified here, the rat-tail radish allele would influence the plant shape into an erect form (Table 3). Although this trait might be environmentally variable, these QTLs could be useful for the selection of radish lines with a more compact plant shape, thus improving canopy.
Root formation is one of the most important traits for the Asian radish cultivars, because a thick and long cylindrical root is indispensable for Asian cultivars. In the present population between rat-tail and Japanese radishes (Saya-S and Haru-S), most MW QTLs were associated with RW QTLs (Table 3 and Fig. 1). This clearly demonstrated that thickness of the main root contributed to the total root weight. In this study, we have found two potential QTL clusters for root formation. We previously identified four genomic regions of root-related QTLs using an F2 population between Chinese and Japanese radishes (Tsuro et al. 2008). No significant overlap was detectable in the positions of root QTLs between the previous and the present studies (data not shown). The root formation QTL might act in a cultivar-specific manner.
In conclusion, we have constructed a chromosome-assigned, sequence-tagged linkage map of the radish, and have identified several QTLs for morphological traits. The present data will provide useful information for further genetic studies and for selection during radish breeding programs. As an example, the QTL detected in radish could be introduced into Brassica crops via homoeologous recombination as in the case of the fertility restorer gene (Sakai et al. 1996). Genetic markers closely linked to the loci are powerful tools for detecting recombination in interspecific hybrids. For this purpose, the present chromosome-assigned, sequence-tagged linkage map of the radish would be useful for MAS and chromosome manipulation not only in the radish, but also in a wide range of Brassica crops.
Acknowledgments
We are grateful to Prof. T. Nishio and Dr. S. Matsumoto for providing the marker information. We thank Dr. N. Wang, Mr. M. Saito and Mr. A. Kamei for development of part of the markers and Ms. H. Kasaoka, Ms. A. Garve, Ms. K. Maier and Ms. E. Koblitz for technical assistance. This work was partly supported by the Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (BRAIN) to M.H. and JSPS Kakenhi 23510236 to N.K.
Literature Cited
- Ågren, J., and Schemske, D.W. (1992) Artificial selection on trichome number in Brassica rapa. Theor. Appl. Genet. 83: 673–678 [DOI] [PubMed] [Google Scholar]
- Akaba, M., Kaneko, Y., Ito, Y., Nakata, Y., Bang, S.W., and Matsuzawa, Y. (2009) Production and characterization of Brassica napus-Raphanus sativus monosomic addition lines mediated by the synthetic amphidiploid “Raphanobrassica”. Breed. Sci. 59: 109–118 [Google Scholar]
- Alonso-Blanco, C., Blankestijn-de Vries, H., Hanhart, C.J., and Koornneef, M. (1999) Natural allelic variation at seed size loci in relation to other life history traits of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 96: 4710–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett, K.E., and Lydiate, D.J. (2003) Genetic analysis and genome mapping in Raphanus. Genome 46: 423–430 [DOI] [PubMed] [Google Scholar]
- Budahn, H., Schrader, O., and Peterka, H. (2008) Development of a complete set of disomic rape-radish chromosome-addition lines. Euphytica 162: 117–128 [Google Scholar]
- Budahn, H., Peterka, H., Mousa, M.A.A., Ding, Y., Zhang, S., and Li, J. (2009) Molecular mapping in oil radish (Raphanus sativus L.) and QTL analysis of resistance against beet cyst nematode (Heterodera schachtii). Theor. Appl. Genet. 118: 775–782 [DOI] [PubMed] [Google Scholar]
- Conner, J.K., Rush, S., and Jennetten, P. (1996) Measurements of natural selection on floral traits in wild radish (Raphanus raphanistrum). I. Selection through lifetime female fitness. Evolution 50: 1127–1136 [DOI] [PubMed] [Google Scholar]
- Crisp, P. (1995) Radish, Raphanus sativus (Cruciferae). In: Smartt,, J., and Simmonds, N.W. (eds.) Evolution of Crop Plants, 2nd edn Longman Scientific & Technical, Harlow, pp. 86–89 [Google Scholar]
- Furusato, K., and Miyazawa, A. (1958) Japanese radish cultivars considered viewed from horticulture. In: Nishiyama, I. (ed.) Japanese Radish, Japan Society for the Promotion of Science, Tokyo, pp. 138–161 [Google Scholar]
- Jones, N., Ougham, H., Thomas, H., and Pašakinskienė, I. (2009) Markers and mapping revisited: finding your gene. New Phytol. 183: 935–966 [DOI] [PubMed] [Google Scholar]
- Kamei, A., Tsuro, M., Kubo, N., Hayashi, T., Wang, N., Fujimura, T., and Hirai, M. (2010) QTL mapping of clubroot resistance in radish (Raphanus sativus L.). Theor. Appl. Genet. 120: 1021–1027 [DOI] [PubMed] [Google Scholar]
- Kaneko, Y., Kimizuka-Takagi, C., Bang, S.W., and Matsuzawa, Y. (2007) Radish. In: Kole, C. (ed.) Genome Mapping and Molecular Breeding in Plants, vol. 5 Vegetables, Springer-Verlag, Heidelberg, pp. 141–160 [Google Scholar]
- Kitamura, S. (1958) Cultivars of radish and their change. In: Nishiyama,, I. (ed.) Japanese Radish, Japan Society for the Promotion of Science, Tokyo, pp. 1–19 [Google Scholar]
- Kubo, N., Saito, M., Tsukazaki, H., Kondo, T., Matsumoto, S., and Hirai, M. (2010) Detection of quantitative trait loci controlling morphological traits in Brassica rapa L. Breed. Sci. 60: 164–171 [Google Scholar]
- Lee, T.D. (1988) Patterns of fruit and seed production. In: Doust,, J.L., and Doust, L.L. (eds.) Plant Reproductive Ecology: Patterns and Strategies, Oxford University Press, New York, pp. 179–202 [Google Scholar]
- Li, F., Kitashiba, H., Inaba, K., and Nishio, T. (2009) A Brassica rapa linkage map of EST-based SNP markers for identification of candidate genes controlling flowering time and leaf morphological traits. DNA Res. 16: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F., Hasegawa, Y., Saito, M., Shirasawa, S., Fukushima, A., Ito, T., Fujii, H., Kishitani, S., Kitashiba, H., and Nishio, T. (2011) Extensive chromosome homoeology among Brassiceae species were revealed by comparative genetic mapping with high-density EST-based SNP markers in radish (Raphanus sativus L.). DNA Res. 18: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, P., Zhao, J., Kim, J.S., Shen, S., Del Carpio, D.P., Song, X., Jin, M., Vreugdenhil, D., Wang, X., Koornneef, M.et al. (2007) Quantitative trait loci for flowering time and morphological traits in multiple populations of Brassica rapa. J. Exp. Bot. 58: 4005–4016 [DOI] [PubMed] [Google Scholar]
- Nakatsuji, R., Hashida, T., Matsumoto, N., Tsuro, M., Kubo, N., and Hirai, M. (2011) Development of genomic and EST-SSR markers in radish (Raphanus sativus L.). Breed. Sci. 61: 413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura, S., and Matsuura, S. (1998) Identification of self-incompatibility alleles (S) by PCR-RFLP in radish (Raphanus sativus L.). Euphytica 102: 379–384 [Google Scholar]
- Nozaki, T., Kumazaki, A., Koba, T., Ishikawa, K., and Ikehashi, H. (1997) Linkage analysis among loci for RAPDs, isozymes and some agronomic traits in Brassica campestris L. Euphytica 95: 115–123 [Google Scholar]
- Peterka, H., Budahn, H., Schrader, O., Ahne, R., and Schütze, W. (2004) Transfer of resistance against the beet cyst nematode from radish (Raphanus sativus) to rape (Brassica napus) by monosomic chromosome addition. Theor. Appl. Genet. 109: 30–41 [DOI] [PubMed] [Google Scholar]
- Saito, M., Kubo, N., Matsumoto, S., Suwabe, K., Tsukada, M., and Hirai, M. (2006) Fine mapping of the clubroot resistance gene, Crr3, in Brassica rapa. Theor. Appl. Genet. 114: 81–91 [DOI] [PubMed] [Google Scholar]
- Sakai, T., Liu, H.J., Iwabuchi, M., and Kohno-Murase, J. (1996) Introduction of a gene from fertility restored radish (Raphanus sativus) into Brassica napus by fusion of X-irradiated protoplasts from a radish restorer line and iodacetoamide-treated protoplasts from a cytoplasmic male-sterile cybrid of B. napus. Theor. Appl. Genet. 93: 373–379 [DOI] [PubMed] [Google Scholar]
- Schmidt, R., and Bancroft, I. (2011) Plant genetics and genomics: crops and models, vol. 9, Genetics and genomics of the Brassicaceae. Springer Science and Business Media, New York [Google Scholar]
- Shirasawa, K., Oyama, M., Hirakawa, H., Sato, S., Tabata, S., Fujioka, T., Kimizuka-Takagi, C., Sasamoto, S., Watanabe, A., Kato, M.et al. (2011) An EST-SSR linkage map of Raphanus sativus and comparative genomics of the Brassicaceae. DNA Res. 18: 221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, K., Slocum, M.K., and Osborn, T.C. (1995) Molecular marker analysis of genes controlling morphological variation in Brassica rapa (syn. campestris). Theor. Appl. Genet. 90: 1–10 [DOI] [PubMed] [Google Scholar]
- Suwabe, K., Tsukazaki, H., Iketani, H., Hatakeyama, K., Kondo, M., Fujimura, M., Nunome, T., Fukuoka, H., Hirai, M., and Matsumoto, S. (2006) Simple sequence repeat-based comparative genomics between Brassica rapa and Arabidopsis thaliana: the genetic origin of clubroot resistance. Genetics 173: 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teutonico, R.A., and Osborn, T.C. (1994) Mapping of RFLP and qualitative trait loci in Brassica rapa and comparison to the linkage maps of B. napus, B. oleracea, and Arabidopsis thaliana. Theor. Appl. Genet. 89: 885–894 [DOI] [PubMed] [Google Scholar]
- Tian, F., Bradbury, P.J., Brown, P.J., Hung, H., Sun, Q., Flint-Garcia, S., Rocheford, T.R., McMullen, M.D., Holland, J.B., and Buckler, E.S. (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 43: 159–162 [DOI] [PubMed] [Google Scholar]
- Tsuro, M., Suwabe, K., Kubo, N., Matsumoto, S., and Hirai, M. (2005) Construction of a molecular linkage map of radish (Raphanus sativus L.), based on AFLP and Brassica-SSR markers. Breed. Sci. 55: 107–111 [Google Scholar]
- Tsuro, M., Suwabe, K., Kubo, N., Matsumoto, S., and Hirai, M. (2008) Mapping of QTLs controlling for root shape and red pigmentation in radish, Raphanus sativus L. Breed. Sci. 58: 55–61 [Google Scholar]
- Van Ooijen, J.W. (2009) MapQTL 6, software for the mapping of quantitative trait loci in experimental populations of diploid species. Kyazma BV, Wageningen [Google Scholar]
- Van Ooijen, J.W., and Voorrips, R.E. (2001) JoinMap Version 3.0, Software for the calculation of genetic linkage maps. Plant Research International, Wageningen [Google Scholar]
- Wang, N., Hu, J., Ohsawa, R., Ohta, M., and Fujimura, T. (2007) Identification and characterization of microsatellite markers derived from expressed sequence tags (ESTs) of radish (Raphanus sativus L.). Mol. Ecol. Notes 7: 503–506 [Google Scholar]
- Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., Bai, Y., Mun, J.H., Bancroft, I., Cheng, F.et al. (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43: 1035–1039 [DOI] [PubMed] [Google Scholar]
- Xu, L., Wang, L., Gong, Y., Dai, W., Wang, Y., Zhu, X., Wen, T., and Liu, L. (2012) Genetic linkage map construction and QTL mapping of cadmium accumulation in radish (Raphanus sativus L.). Theor. Appl. Genet. 125: 659–670 [DOI] [PubMed] [Google Scholar]
- Yazawa, S., Asahira, T., Matsumoto, Y., and Namiki, T. (1953) Morphological and physiological characteristics of rat-tailed radish introduced from India and Thailand. Agriculture and Horticulture 58: 704–706 [Google Scholar]
- Zhang, J., Lu, Y., Yuan, Y., Zhang, X., Geng, J., Chen, Y., Cloutier, S., McVetty, P.B.E., and Li, G. (2009) Map-based cloning and characterization of a gene controlling hairiness and seed coat color traits in Brassica rapa. Plant Mol. Biol. 69: 553–563 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.