

Abstract
Objective
To examine changes in the response properties of meningeal nociceptors that might lead to migraine pain and examine endogenous processes that could play a role in mediating them using a clinically relevant model of migraine triggering, namely infusion of the NO donor nitroglycerin (NTG).
Methods
Single unit recordings made in the trigeminal ganglion of rats were used to test changes in the activity and mechanosensitivity of meningeal nociceptors in response to administration of the migraine trigger NTG or another NO donor SNAP at doses relevant to the human model of migraine headache. Immunohistochemistry and pharmacological manipulations were used to investigate the possible role of meningeal vascular signaling in mediating the responses of meningeal nociceptors to NO.
Results
Infusion of NTG promoted a delayed and robust increase in the mechanosensitivity of meningeal nociceptors with a time course resembling the development of the delayed migraine headache. A similar sensitization was elicited by dural application of NTG and SNAP. NTG-evoked delayed meningeal nociceptor sensitization was associated with a robust ERK phosphorylation in meningeal arteries. Pharmacological blockade of meningeal ERK phosphorylation inhibited the development of NTG-evoked delayed meningeal nociceptor sensitization
Interpretation
The development of delayed mechanical sensitization evoked by the migraine trigger NTG is potentially of great importance as the first finding of a neurophysiological correlate of migraine headache in meningeal nociceptors. The arterial ERK phosphorylation and its involvement in mediating the NTG-evoked delayed sensitization points to an important, yet unappreciated, role of the meningeal vasculature in the genesis of migraine pain.
Introduction
Migraine is one of the most prevalent neurological disorders, affecting about 15% of the adult population worldwide 1. Despite this high prevalence, the mechanisms underlying the headache remain incompletely understood. Although migraine is not accompanied by any readily identified pathology, it is characterized by features that suggest the presence of an abnormal intracranial mechanosensitivity, in common with the headaches that accompany certain intracranial pathologies (i.e. worsening of the pain by coughing, straining, bending over, or rapid head movement) 2. A possible mechanism underlying this hypersensitivity could be an increased mechanosensitivity, or sensitization, of nociceptive neurons that innervate the intracranial meninges 3–5. Previous studies demonstrated the occurrence of such sensitization following the application of inflammatory mediators to the receptive fields of intracranial meningeal nociceptors 6–8. However the nature of the endogenous factors that contribute to the establishment of such sensitization during a clinically occurring migraine attack remain unknown. To begin exploring possible endogenous processes that could mediate the sensitization of meningeal nociceptors during migraine, in the present study we investigated for the first time changes in the response properties of meningeal nociceptors under a condition that is known to cause migraine headache in humans, namely systemic administration of the migraine trigger nitroglycerin (NTG).
NTG and other nitric oxide (NO) donor vasoactive compounds have the ability to evoke headache in human subjects when administered systemically. These compounds typically induce a brief, minor headache that occurs immediately after administration in all subjects, which is followed after a delay of 3–4 hrs by a sustained headache that generally occurs only in migraineurs, and reproduces the clinical characteristics of the subject’s spontaneously occurring attacks 9–11. A large body of evidence now supports the idea that the delayed headache is identical to a clinically occurring migraine attack. For example, as with spontaneous migraine, NTG-induced migraine headache is also relieved by sumatriptan 12. Furthermore, similar to spontaneous migraine, NTG-evoked migraine is associated with increased serum levels of CGRP during the headache phase 13 and is also accompanied by changes in blood levels of cytokines and other markers of inflammation that are also found during migraine 14. Consequently, systemic NTG administration and its delayed effects have been used as an experimental model of migraine headache in animals and humans 15, 16. The fact that the migrainous headache develops only after a delay, at a time when NTG is no longer present, implies that NTG (or NO) is not acting as a direct excitant of sensitizing agent on meningeal nociceptors, but rather serving as a trigger for a series of endogenous processes that in turn lead to nociceptor sensitization. This means that, while NTG itself is an exogenous agent, the NTG model can be used to investigate the endogenous processes that capable of producing migraineous headache.
The meningeal vasculature has been postulated to have a role in migraine pathogenesis 17, but the exact nature of this potential role is not yet clear. Existing evidence does not support a mechanism whereby meningeal vasodilatation produces a direct, immediate action of either nociceptor sensitization or activation 18–20. Nonetheless, the dense sensory innervation that terminates in the vicinity of the meningeal vasculature and the comorbidity of migraine with various cerebrovascular diseases strongly suggests a possible contribution of meningeal vascular factors to migraine pathophysiology 17. Thus, a more indirect role for the meningeal vasculature, for example as part of the triggering process, cannot be excluded. Recently we reported that the meningeal vasculature plays an important indirect role in mediating the sensitization of meningeal nociceptors in response to a local inflammatory stimulus 21. To further investigate a potential role of the meningeal vasculature in migraine pain, we examined whether the effect of NTG on meningeal nociceptors involves signaling processes in meningeal vascular cells.
Materials and methods
Animals
Sprague-Dawley male rats (250–350 g) were used in compliance with the experimental protocol approved by the institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center and Harvard Medical School and adhered to the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain 22.
Electrophysiological recordings
Rats were deeply anesthetized with Urethane (1.5 g/kg i.p.) and prepared for single-unit recordings of meningeal nociceptors in the trigeminal ganglion as previously described before, using either the exposed 23, 24 or non-exposed 25, 26 receptive field preparations. Single-unit activity was recorded using a platinum-coated tungsten microelectrode (impedance ~150 KΩ; FHC, Bowdoin, ME) advanced into the left trigeminal ganglion. In the exposed receptive field preparation, meningeal nociceptors were identified by stimulating electrically (0.5 ms pulse, 0.5–5 mA, 0.5 Hz) the transverse sinus. In the non-exposed (non-craniotomized) receptive field preparation, electrical stimulation was conducted through two burr holes located 1–2 mm caudal to the sinus, a site that is remote from the receptive field of the recorded neurons. Our previous study indicated that these burr holes are not associated with a marked mast cell degranulation at the receptive field of the recorded neurons. Conduction velocities were calculated based on a distance between the stimulation site on the dura and the recording site in the trigeminal ganglion of 12.5 mm and units were classified as either Aδ (CV >1.5 m/sec) or C (CV ≤1.5 m/sec). During the experiments, action potentials were acquired using a real-time waveform discriminator (Spike 2, CED, Cambridge, UK). Only one unit was tested in each animal. To prevent drying, the exposed dura was constantly bathed with modified synthetic interstitial fluid (SIF; 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 5 mM CaCl2, 10 mM glucose and 10 mM HEPES, pH 7.2).
Detection of mechanical sensitization
In the exposed dural preparation, the receptive field of each neuron was initially mapped using a series of calibrated von Frey monofilaments (range 38–443 kPa). Quantitative changes in the nociceptor mechanical responsiveness were then examined using a feedback-controlled mechanical stimulator (Series 300B, Aurora Scientific, Aurora, ON) fitted with a flat-ended plastic cylinder (0.5 or 0.8 mm). Stimulus trials for testing changes in mechanical sensitivity consisted of graded square-wave stimuli (100-msec rise time, 2-sec width, 60-sec inter-stimulusinterval) delivered in ascending order. Each trial included one threshold stimulus followed by 2 suprathreshold stimuli (usually X2 and X4 of the threshold). Neuronal responses were recorded every 15 min throughout the experiment. Ongoing activity was recorded between trials. Baseline ongoing discharge and responses to mechanical stimulation were determined during at least 3 consecutive trials prior to drug administration. Only units that exhibited consistent responses during baseline recordings were tested further.
Immunohistochemistry and labeled vessel counting
Following treatments, rats were anesthetized with an overdose of pentobarbital and perfused transcardially with cold heparinized solution of 0.1 M phosphate-buffered saline (PBS, pH 7.4), followed by freshly made made 4% paraformaldehyde in 0.1 M PBS. Dural tissues were dissected free and subjected to pERK immunohistochemistry. To block non-specific binding, tissues were first incubated for 2 h at room temperature in 0.1 M PBS containing 2% fetal bovine serum together with non-immune serum from the host species of the secondary antibody and 0.1% Triton-X. Specimens were then incubated for 48 h at 4 °C with a primary antibody against pERK (Mouse monoclonal anti-pERK, #4376, 1:5000, Cell signaling Technology, Danvers, MA). To determine arterial or venular localization of the vascular pERK, double labeling was conducted in conjunction with an antibody against the arterial marker Ephrin-B2 (Rabbit polyclonal, #SC-1010, Santa Cruz biotechnology, 1:500). For all indirect immunofluorescence, Alexa 594 and/or Alexa 488- conjugated secondary antibodies (1:200, Invitrogen,) were employed. For all immunohistochemical studies, controls for non-specific staining included omission of the primary antibody, which resulted in a complete lack of labeling. To examine potential changes in dural vascular pERK expression, the number of pERK-labeled vessels were counted in 15 separate randomized visual fields (regions of interest) using a Leica fluorescent microscope (Leica DM6000 with a motorized Z-focus) under X200 objective by an observer blinded to the treatment group as described previously 21.
Drug administration
Nitroglycerin (NTG, American Reagents Inc.) was mixed in 0.9% sterile saline and was slowly infused through a femoral catheter at a rate of 2 μg/kg/min for 30 min). This dose has been shown to produce a maximal headache in humans 27. At this dose and route of administration, NTG does not affect mean arterial pressure or heart rate 28, 29. To test for local action of NTG, it was diluted in SIF to a 10 μM concentration, which is the estimated blood plasma concentration achieved by the systemic infusion, and applied topically to the dura for 30 min. To further explore the local action of NO, another NO donor, S-Nitroso-N-acetyl-DL-penicillamine (SNAP, Sigma Aldrich, 10 μM) was applied topically. To block local meningeal ERK phosphorylation, we used the selective membrane-permeable MEK 1 (MAP kinase kinase 1) inhibitor PD98059 ([2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one] or the MEK 1/2 inhibitor U0126 ([1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] (Both from Sigma Aldrich). For their topical application, the inhibitors were dissolved in 100% DMSO and further diluted with SIF to a final concentration of 50 μM (<1% DMSO). The inhibitors were applied to the meninges 30 min after the end of the NTG infusion and left on the dura until the end of the recording period.
Statistical analyses
Data are presented as mean ± SEM. To test the effects of the NO donors and the MEK inhibitors on the response properties of meningeal nociceptors, two-way mixed factorial ANOVA was employed with time and drugs as factors. Additional analyses were conducted to compare average changes in neural responses at 180 and 360 min following the various treatments using the Mann- Whitney test. Changes in vascular pERK expression and the effect of the MEK inhibitors on meningeal nociceptors were compared using the Mann- Whitney test. The level of significance was set at p=0.05.
Results
Slow NTG infusion promotes delayed mechanical sensitization of meningeal nociceptors
The effect of a slow infusion of NTG or vehicle on the activity and mechanosensitivity of meningeal nociceptors was tested initially using the exposed receptive field preparation. Among the 16 units tested, only 1 C-unit showed an increase in ongoing discharge level, which started 185 minutes after the infusion and lasted for 90 minutes. As shown in Figures 1 and 2, despite the lack of activation seen in most of the recorded nociceptors, NTG infusion gave rise to a delayed and progressive increase in the mechanical responsiveness of 12/16 (75%) units tested (5 Aδ, 7 C-units). A slow infusion of the NTG vehicle (6 Aδ, 6 C-units) did not cause any significant increases in the mechanosensitivity or ongoing discharge levels in any of the units tested (Figure 2B). When comparing the overall changes in the neural responses to threshold stimuli observed in the NTG and vehicle treated animals, there were significant differences with regard to the effects of drugs (F(1,27)=24.27, p<0.0001) and time (F(1,24)=4.98, p<0.0001) as well their interaction (F(1,24)=6.47, p<0.0001). The mean onset latency for the sensitization was 175±15 min (range 120–300 minutes). At 180 min post NTG infusion, the average increase in threshold responses was 277±141%. The magnitude of the sensitization continued to increase over time and rose to 917±343% at 360 min post NTG infusion (p<0.05, 6 hrs vs. 3 hrs). NTG infusion also evoked statistically significant increases in suprathreshold responses with regard to drugs (F(1,27)=16.14, p<0.01), time (F(1,24)=1.61, p<0.05) and their interaction (F(1,24)=3.08, p<0.0001). This change however was less robust and reached its peak already at 180 min (160±26% vs. 169±25% at 360 min post NTG infusion).
FIGURE 1.
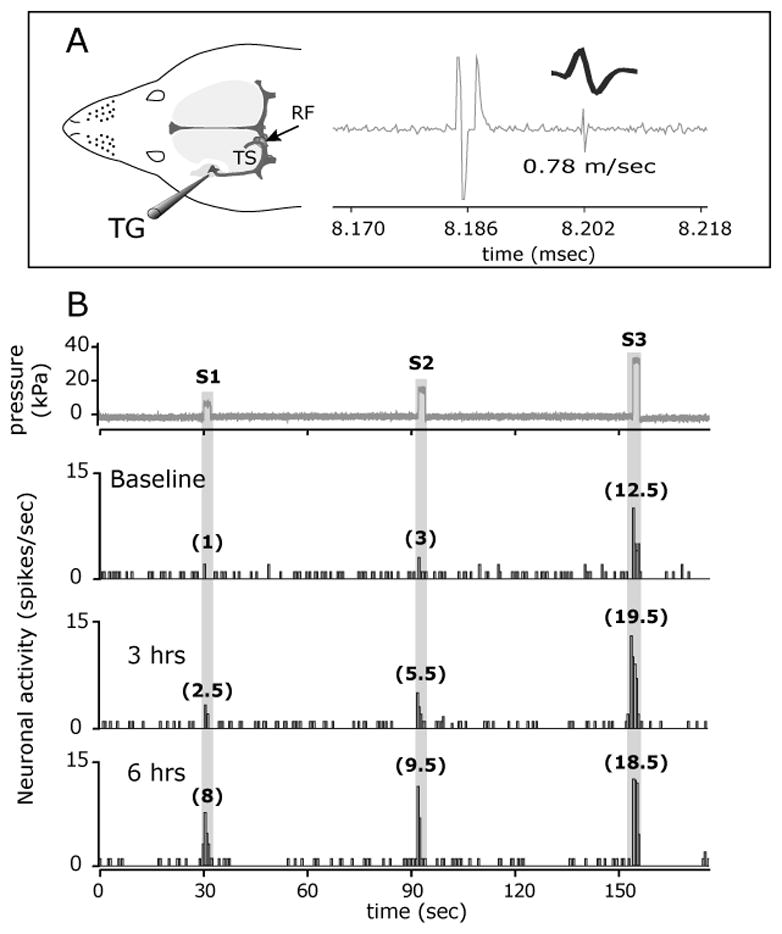
NTG infusion evokes a delayed mechanosensitization of a C-unit meningeal nociceptor. (A) Experimental setup (left) and the identification and isolation of a C-unit meningeal nociceptor based on the response latency (16 milliseconds) to electrical stimulation of the dura and a consistent waveform (right). (B) Peri-stimulus time histograms depicting the responses to threshold (S1) and suprathreshold (S2-3) mechanical stimulation of the dura at baseline, before NTG infusion, and at 3 and 6 hrs post infusion. The numbers in parenthesis indicate mean responses to mechanical stimuli in spikes/sec. Note the progressive increase in threshold (S1) responsiveness and the lack of increases in ongoing discharge (between trials) in response to NTG. RF, receptive field; TS, Transverse sinus; TG, Trigeminal ganglion.
FIGURE 2.

(A) Time course changes in the average responses of Aδ and C-units meningeal nociceptors in response to NTG infusion. Note that in response to NTG infusion, the Aδ and C-units had a similar delayed increase in both threshold and suprathreshold responses, but did not exhibit an increase in their ongoing discharge rate. (B) Time course changes in the responses of meningeal nociceptors following infusion of the NTG vehicle (6 Aδ, 6 C-units) against a combined re-plotting of the Aδ and C-units responses to NTG from the same 16 neurons plotted in Fig 2A.
NTG infusion in a non-craniotomized preparation has been shown to promote a delayed dural mast cell (MC) degranulation 29, a process that could lead to the activation of meningeal nociceptors 26. In the craniotomized preparation typically used for neurophysiological studies, the dural MCs are degranulated by the craniotomy 26. Because NTG infusion led to a negligible nociceptor activation in the exposed (i.e. craniotomized) dural preparation, we also tested whether NTG might activate meningeal nociceptors in the non-craniotomized preparation, in which the MC population around the meningeal receptive field of the neurons remains largely intact and thus more amenable to degranulation. In this preparation, the basal ongoing activity level is also reduced. Out of the 12 units (5 Aδ, 7 C) tested using this non-craniotomized receptive field preparation, there was an increase in ongoing discharge rate in 3 C-units (25%) that started between 120–150 min following NTG infusion. When compared to baseline, the average increase in ongoing discharge rate in these neurons was 240±108% at 180 min and 251±45% at 360 min.
NTG-evoked meningeal nociceptor sensitization is mediated by local meningeal NO action
NTG is believed to trigger migraine headache by releasing NO 9, but the anatomical site underlying this nociceptive action remains an open question. To investigate whether the delayed nociceptor sensitization evoked by NTG infusion could be due to a local meningeal action, we studied nociceptor responses following meningeal application of NTG. To examine whether this local action is NO-related, we further tested the effect of another NO donor, SNAP. Similar to the effect of its systemic administration, when compared to the effect of vehicle treatment (4 Aδ, 5 C units), meningeal NTG application promoted a significant mechanical sensitization in 3/5 Aδ and 5/6 C units (Figure 3). Statistical analyses revealed significant differences in the responsiveness to threshold stimuli with regard to the effects of drug (F(1,19)=6.32, p<0.01), time (F(1,24)=8.93, p<0.0001) and their interaction (F(1,24)=3.09, p<0.0001). The mean onset latency for the sensitization was also similar to that seen after systemic infusion of NTG at 195±65 min (range 120–300 min). The magnitude of the increase in threshold responsiveness was also comparable to that seen after NTG infusion with 219±110% at 180 min, which increased to 899±272% at 360 min (p<0.01 180 vs. 360 min). Responses to suprathreshold stimuli were also statistically different with regard to the effect of drug (F(1,19)=4.02, p<0.01), time (F(1,24)=1.87, p<0.01) and their interaction (F(1,24)=1.65, p<0.01). The magnitude of the increases in suprathreshold responses were also comparable to that seen after NTG infusion and were similar at 180 min (145±34%) and 360 min (182±35%). Only 1 C-unit displayed a mild increase in ongoing activity 145–225 min following treatment. Local application of the second NO donor SNAP also promoted a delayed sensitization in 3/6 of the Aδ and 5/7 C-units with mean onset latency of 185±45 min (range 120–240 min). When compared to the effect of vehicle treatment, SNAP elicited statistically significant increases in threshold responsiveness with regard to the effects of drugs (F(1,21)=22.18, p<0.001), time (F(1,24)=6.23, p<0.0001) and their interaction (F(1,24)=8.09, p<0.0001). Similar differences were noted in the responsiveness to suprathreshold stimuli with regard to the effects of drugs (F(1,21)=4.788, p<0.05), time (F(1,24)=1.836, p<0.05) and their interaction (F(1,24)=2.37, p<0.0001). The mean increase in threshold responses at 180 minutes was 300±98%, which was further increased to 634±162% at 360 minutes (p<0.01 180 vs. 360 min). Suprathreshold responses also increased mildly to 123±21% at 180 min and 135±18% at 360 min. In none of the units tested did SNAP treatment promote an increase in ongoing activity. Topical application of the NO-donors’ vehicle also did not affect the ongoing activity in any of the units tested.
FIGURE 3.
Time course changes in threshold and suprathreshold responses following topical meningeal application of NTG (5 Aδ, 6 C), SNAP (6 A-δ, 7 C) or their vehicle (4 Aδ, 5 C). Note the development of the delayed sensitization similar to that seen following NTG infusion.
NTG evokes meningeal vascular pERK expression
NO activates the MAP kinase ERK in vascular cells 30, 31. ERK activation in the meningeal vasculature leads to increased expression and release of proinflammatory sensitizing mediators 32–34. To begin examining whether meningeal vascular ERK activation might play a role in mediating the NTG-evoked sensitization of meningeal nociceptors, we tested the effect of NTG on pERK expression in dural vessels. Infusion of NTG, but not its vehicle, led to increased pERK immunofluorescence in the walls of meningeal vessels and a time-dependent increase in the overall number of pERK-labeled dural blood vessels (Figure 4A–C). When compared to vehicle treatment, the number of pERK-positive blood vessels was higher at 2 and 4 hrs post-NTG infusion (both p<0.05, Figure 4G). At all the time points tested following NTG or vehicle treatments, no pERK immunereactivity was detected in dural axons, indicating a lack of neural expression. Following NTG infusion, pERK labeling was confined to vessels that also expressed Eph-B2 (Figure 4D–G), suggesting ERK activation in dural arteries 35. Topical administration of the MEK inhibitor U0126 onto the dura blocked the increase in vascular pERK expression at 2 (not shown) and 4 hrs (Figure 4G).
FIGURE 4.

NTG infusion promotes ERK activation in dural arteries. Immunohistochemical localization of pERK-immunoreactivity in the dura mater of animals infused with vehicle (A) or NTG (B,C). Note the increased expression of pERK in the walls of the middle meningeal artery (MMA) and the second and third-order arterioles that arise from it (B). Increases in the number of medium size vessels expressing pERK was also found in dural territories remote from the MMA (C). (D) An example of a dural vessel showing pERK-immunoreactivity, which is also labeled with antibody against the arterial marker Ephrin B2 (E). (F) Co-localization (yellow) is shown in (F). The Average±SEM number of pERK labeled dural blood vessels observed per visual field at 1, 2 and 4 hrs following NTG infusion. Note the inhibitory effect induced by local application of the MEK inhibitor U0126 (* p < 0.05, Mann Whitney test NTG vs. vehicle).
NTG-evoked delayed sensitization of meningeal nociceptors is mediated by local ERK activation
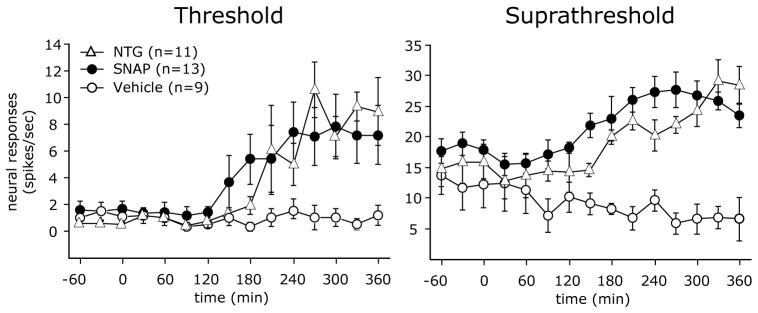
To test whether the NTG-evoked vascular ERK activation (i.e. phosphorylation) contributes to the delayed sensitization of meningeal nociceptors, we examined the effect of MEK inhibition using PD98059 and U0126 (Figure 5). Local application of PD98059 (3 Aδ, 5 C units) suppressed the NTG-evoked increases in threshold responses (F(1,23)=5.73, p<0.01 for the effect of drugs, F(1,24)=7.09, p<0.0001 for the effect of time, and F(1,24)=5.06, p<0.0001 for the interaction between drug and time). A similar inhibition was noted for the suprathreshold responses (F(1,23) =12.64, p<0.01 for the effect of drugs, F(1,24) =1.56, p<0.05 for the effect of time, and F(1,24)=4.47, p<0.0001 for the interaction between drug and time). Local administration of U0126 (6 Aδ, 6 C units) had a similar suppressive effect on the development of the NTG-evoked delayed increases in threshold (F(1,27)=23.60, p<0.0001 for the effect of drugs, F(1,24)=9.06, p<0.0001 for time effect, and F(1,24)=10.05, p<0.0001 for the interaction between drugs and time) and suprathreshold responses (F(1,27)=10.83, p<0.01 for the effect of drugs, F(1,24) =1.80, p<0.01 for the effect of time, and F(1,24)=8.92, p<0.0001 for the interaction between drug and time). When compared to treatment with the NTG vehicle, the application of PD98059 (4 Aδ, 5 C units) or U0126 (4 Aδ, 5 C units) alone did not reduce the activity or responses to threshold or suprathreshold stimuli of the neurons tested, indicating that the suppressive effects of the MEK inhibitors on the increased responsiveness following NTG infusion were not due to a direct inhibition of the basal mechanosensitivity of the meningeal nociceptors
FIGURE 5.

NTG-evoked delayed sensitization of meningeal nociceptors sensitization involves pERK. Changes in threshold (A) and suprathreshold (B) responses of meningeal nociceptors to mechanical stimulation of the dura 180 and 360 min following infusion with NTG alone (7 Aδ, 9 C), NTG infusion followed by local MEK inhibition using U0126 (6 A-δ, 6 C) or PD98059 (3 A-δ, 5 C) or local treatment with U0126 (4 A-δ 5C) or PD98059 (4 A-δ 5C) alone. (*p < 0.05, Mann Whitney test NTG alone vs NTG+MEK inhibitors).
Discussion
In this study we examined for the first time changes in the responses of meningeal nociceptors under a condition known to trigger migraine headache in humans, namely infusion of NTG, at a dose relevant to the human experimental model of NTG-triggered migraine headache. Our data provides evidence for a delayed and robust increase in the discharge of meningeal nociceptors in response to mechanical stimulation (i.e mechanosensitization) with a time course resembling the development of the delayed migraine headache. This finding provides experimental evidence that substantiates the role of meningeal nociceptors in mediating the headache during a migraine attack and further points to an increase in their mechanosensitivity as a key process underlying the headache.
The critical question of what endogenous physiological processes promote the increased discharge of meningeal nociceptors during migraine remains open. In a recent study, Asghar and colleagues 36 demonstrated that systemic infusion of the vasodilator CGRP to migraineurs causes a delayed migrainous headache, which coincides with about a 10% increase in the diameter of intracranial meningeal arteries. Given this relatively small vasomotor change, the authors suggested that it contributes to the headache by activating meningeal nociceptors whose mechanical threshold was lowered (i.e. sensitized), presumably through a delayed action of CGRP. Similar to CGRP, NTG infusion also promotes an acute meningeal vasodilation 37, a phenomenon seen also in animal models 38. However, unlike CGRP, a recent imaging study could not detect a delayed meningeal vasodilatation concomitant with the delayed migrainous headache evoked by NTG infusion 11. While the induction of a delayed meningeal vasodilatation in response to NTG remains questionable 11, 36, the notion that the initial meningeal vascular response, which occurs during the infusion of NTG, serves as an initiating factor that contributes to the development of the delayed sensitization of meningeal nociceptors cannot be excluded. Nonetheless, the findings that administration of the vasodilator peptide VIP does not trigger migraine headache in migraineurs, despite its ability to evoke a marked acute cephalic vasodilatation 39, suggests that intracranial meningeal vasodilatation alone may not be sufficient to drive the nociceptor sensitization and consequently the migrainous headache. Future studies will be needed to examine whether other vasodilators can induce a delayed sensitization of meningeal nociceptors. Of additional interest will be to determine whether meningeal vasodilatation can promote the activation of mechanosensitive meningeal nociceptors under a sensitized condition such as the one elicited by NTG.
Whereas the direct contribution of meningeal vasodilatation to migraine pain remains dubious, the meningeal vasculature could play an indirect role in headache pathogenesis 17. Vascular cells contribute to inflammatory responses by releasing pro-inflammatory factors 40. In previous animal studies, infusion of NTG has been shown to promote a delayed meningeal inflammation, attendant by increased local expression of pro-inflammatory cytokines 29, 41. We have shown recently that the pro-inflammatory cytokine TNF-α promotes a robust sensitization of meningeal nociceptors through a mechanism that involves p38 MAP kinase signaling in meningeal vascular cells and the release of COX-derived prostaglandins 21. Our present data further supports the notion that vascular signaling processes contribute to migraine headache by showing that NTG infusion evokes a delayed activation of another MAP signaling cascade, ERK phosphorylation, in meningeal arterial cells and the contribution of this vascular signaling event to the delayed sensitization of meningeal nociceptors. That vascular ERK signaling contributes to the elaboration of proinflammatory nociceptive molecules within the intracranial meninges 32, 34 further points to the involvement of a vascular inflammatory response in migraine pain. Further studies will be required to elucidate the exact nature of the endogenous process that promotes this vascular response in migraineurs and the identity of the vascular factors that interact with meningeal nociceptors to promote their sensitization. Of particular interest will be to examine the possibility that other migraine triggering factors promote the headache by eliciting a similar vascular response.
The present study found only an increase in mechanosensitivity, with no significant increase in the ongoing discharge of primary afferent meningeal nociceptors, when using the standard, craniotomized preparation that is typically used in neurophysiological studies of the meningeal sensory system. It should be noted that in this preparation, meningeal nociceptors have a baseline level of ongoing discharge, likely as a result of the tissue injury that is produced by the surgical exposure of the dural receptive field 26. In contrast, prior studies did find an increase in ongoing discharge in trigeminal dorsal horn neurons following systemic administration of NTG and other NO donors, using the same craniotomized preparation 42–44. If local (or systemic) actions of NO do not activate meningeal nociceptors, what might be the source of the increased discharge of meningeal-sensitive dorsal horn neurons evoked by NO? One possibility is that NO also acts locally within the trigeminal dorsal horn and directly promotes the activation of dorsal horn neurons 43, 45. Another possibility is that NO enhances the excitatory effect of the ongoing primary afferent input on the dorsal horn neurons 42, 44. Such an effect would be expected to be present only in the craniotomized preparation, since meningeal nociceptors have essentially no ongoing discharge in the non-craniotomized preparation 26. Trigeminal dorsal horn neural discharge has not been studied in a non-craniotomized preparation, but studies of c-fos expression (a marker of central neuron activation) in the non-craniotomized preparation found no increase in the dorsal horn labeling following systemic administration of the same dose of NTG as used in our study 46, 47. This suggests that NO does not activate dorsal horn neurons directly but rather promotes a process that enhances their response to peripheral nociceptive input, possibly in a manner resembling central sensitization 48. The present work thus supports previous suggestions that the NO-related increased activity of dorsal horn neurons that receive input from meningeal nociceptors 43, 44 is mediated by central facilitation of the neuronal response to afferent traffic that originates in the meninges as well as other tissues that become injured in this preparation 49.
One fundamental question related to the clinical relevance of the effect of NTG on meningeal nociceptors is what type of intracranial mechanical forces can promote the activation of meningeal nociceptors under the sensitized state induced by NTG. Our earlier studies of meningeal nociceptors indicate that at baseline their mechanical thresholds are distributed over a wide range but the lower end of this range corresponds to the upper limit of normal intracranial pressures in humans (about 20 mmHg or 2.6 kPa at the peak of the arterial pulsation) 4. In a previous study we have shown that the sensitization induced by local application of inflammatory mediators leads to about 50% reduction in the response thresholds of meningeal nociceptors 7. In the current study, the mechanical sensitization induced by NTG was even greater than induced by local application of inflammatory mediators. Therefore, the cascade of events that occur as a result of NTG infusion is expected to render a larger proportion of the meningeal nociceptors sensitive to the forces evoked by normal changes in ICP. Furthermore, under such a sensitized condition, additional recruitment of afferents (and likely headache exacerbation) are expected to be induced in response to conditions that promote transient increases in ICP such as straining 50 and coughing 51.
Another important question related to our study is why in humans an equivalent dosing regimen of NTG triggers the delayed headache only in migraineurs? One possibility is that the delayed mechanical sensitization and its related afferent response evoked by NTG is not by itself sufficient to cause the migraine pain, but that migraineurs may have an additional level of susceptibility such that this response does lead to headache. For example, in individuals not suffering from migraine, the afferent volleys related to the activation of the sensitized meningeal nociceptors by the mechanical forces within the intracranial space may not be sufficient to activate the second-order dorsal horn neurons and thus the migraine pain pathway. In migraineurs however, the central transmission of the same afferent traffic could be facilitated downstream, potentially at the first synapse, a process that could lead to the headache. Such facilitation may be due to neuroplastic changes, such as an upregulation of sensory neurotransmitters 52–54 or their receptors 55. Relevant to this concept is the finding that the development of a delayed pain behavior reminiscent of migraine headache (cephalic allodynia) is exhibited in animals with increased expression of the neurotransmitter CGRP in meningeal nociceptors 53.
Another conceivable mechanism that might be involved in promoting specifically the headache in migraineurs in response to NTG, and possibly to other triggering factors, is the induction of a higher level of ongoing discharge in meningeal nociceptors. How NTG infusion could promote meningeal nociceptors’ excitation is, as yet, unclear. Using a non-craniotomized preparation in naïve rats, Reuter et al. 29 have shown that NTG infusion promotes a moderate level of degranulation of meningeal MCs. We have previously shown that robust MC degranulation following administration of the powerful degranulating agent 48/80 can produce a pronounced increased in ongoing discharge in nociceptors, in a non-craniotomized preparation 26. However, our current data shows only a slight increase in the nociceptors’ discharge level following NTG in the non-craniotomized preparation, which suggests that the degree of MC degranulation evoked by NTG is not sufficient to induce substantial discharge in naïve animals. One possible mechanism that might enhance the nociceptive response to NTG in migraineurs is an increase in the density of meningeal MCs. Recent data from our lab suggest that changes in estrogen levels, which are associated with the increased propensity of migraine in women 56, promote enhanced trafficking of mast cells to the meninges, leading to an increase in their density 57. A rise in the propensity of meningeal MCs to become activated by NO and possibly other migraine triggering factors 58 may also play a role in mediating the increased sensitivity of migraineurs.
Conclusions
The present study provides data that suggest that NO donors such as NTG could contribute to the genesis of migraine headache by promoting a massive increase in the mechanosensitivity of meningeal nociceptors through their local action in the meninges. The finding that meningeal arterial ERK activation mediates this nociceptive response suggests that meningeal vascular processes occurring within the meningeal arterial wall might play a role in mediating the sensitization of meningeal nociceptors in response to local meningeal NO action. We propose that signaling processes in meningeal arterial cells could play a key role in the pathogenesis of migraine pain through an inflammatory-related mechanism.
Acknowledgments
We would like to thank Rami Burstein for sharing the motorized Z-focus fluorescent microscope. This work was supported in part by NIH grants (NINDS) NS061116, NS077882 and (NIAAA) AA020305 (D.L.); (NINDS) NS032534 (A.M.S) and the National Headache Foundation (D.L).
References
- 1.Stovner L, Hagen K, Jensen R, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia. 2007 Mar;27(3):193–210. doi: 10.1111/j.1468-2982.2007.01288.x. [DOI] [PubMed] [Google Scholar]
- 2.Blau JN, Dexter SL. The site of pain origin during migraine attacks. Cephalalgia. 1981;1(3):143–7. doi: 10.1046/j.1468-2982.1981.0103143.x. [DOI] [PubMed] [Google Scholar]
- 3.Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet neurology. 2009 Jul;8(7):679–90. doi: 10.1016/S1474-4422(09)70090-0. [DOI] [PubMed] [Google Scholar]
- 4.Strassman AM, Levy D. Response properties of dural nociceptors in relation to headache. Journal of neurophysiology. 2006 Mar;95(3):1298–306. doi: 10.1152/jn.01293.2005. [DOI] [PubMed] [Google Scholar]
- 5.Messlinger K. Migraine: where and how does the pain originate? Experimental brain research Experimentelle Hirnforschung. 2009 Jun;196(1):179–93. doi: 10.1007/s00221-009-1756-y. [DOI] [PubMed] [Google Scholar]
- 6.Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996 Dec 12;384(6609):560–4. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- 7.Levy D, Strassman AM. Distinct sensitizing effects of the cAMP-PKA second messenger cascade on rat dural mechanonociceptors. J Physiol (Lond) 2002. 2002;538(2):483–93. doi: 10.1113/jphysiol.2001.013175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy D, Jakubowski M, Burstein R. Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. Proceedings of the National Academy of Sciences of the United States of America. 2004 Mar 23;101(12):4274–9. doi: 10.1073/pnas.0306147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olesen J, Thomsen LL, Iversen H. Nitric oxide is a key molecule in migraine and other vascular headaches. Trends Pharmacol Sci. 1994 May;15(5):149–53. doi: 10.1016/0165-6147(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 10.Sances G, Tassorelli C, Pucci E, Ghiotto N, Sandrini G, Nappi G. Reliability of the nitroglycerin provocative test in the diagnosis of neurovascular headaches. Cephalalgia. 2004 Feb;24(2):110–9. doi: 10.1111/j.1468-2982.2004.00639.x. [DOI] [PubMed] [Google Scholar]
- 11.Schoonman GG, van der Grond J, Kortmann C, van der Geest RJ, Terwindt GM, Ferrari MD. Migraine headache is not associated with cerebral or meningeal vasodilatation--a 3T magnetic resonance angiography study. Brain. 2008 May 23; doi: 10.1093/brain/awn094. [DOI] [PubMed] [Google Scholar]
- 12.Iversen HK, Olesen J. Headache induced by a nitric oxide donor (nitroglycerin) responds to sumatriptan. A human model for development of migraine drugs. Cephalalgia. 1996 Oct;16(6):412–8. doi: 10.1046/j.1468-2982.1996.1606412.x. [DOI] [PubMed] [Google Scholar]
- 13.Juhasz G, Zsombok T, Modos EA, et al. NO-induced migraine attack: strong increase in plasma calcitonin gene-related peptide (CGRP) concentration and negative correlation with platelet serotonin release. Pain. 2003 Dec;106(3):461–70. doi: 10.1016/j.pain.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Martelletti P, Stirparo G, Morrone S, Rinaldi C, Giacovazzo M. Inhibition of intercellular adhesion molecule-1 (ICAM-1), soluble ICAM-1 and interleukin-4 by nitric oxide expression in migraine patients. J Mol Med. 1997 Jun;75(6):448–53. doi: 10.1007/s001090050130. [DOI] [PubMed] [Google Scholar]
- 15.Bergerot A, Holland PR, Akerman S, et al. Animal models of migraine: looking at the component parts of a complex disorder. The European journal of neuroscience. 2006 Sep;24(6):1517–34. doi: 10.1111/j.1460-9568.2006.05036.x. [DOI] [PubMed] [Google Scholar]
- 16.Magis D, Bendtsen L, Goadsby PJ, et al. Evaluation and proposal for optimization of neurophysiological tests in migraine: Part 2-neuroimaging and the nitroglycerin test. Cephalalgia. 2007 Dec;27(12):1339–59. doi: 10.1111/j.1468-2982.2007.01435.x. [DOI] [PubMed] [Google Scholar]
- 17.Dodick DW. Examining the essence of migraine--is it the blood vessel or the brain? A debate. Headache. 2008 Apr;48(4):661–7. doi: 10.1111/j.1526-4610.2008.01079.x. [DOI] [PubMed] [Google Scholar]
- 18.Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: Implications for the pathophysiology of migraine. Annals of neurology. 2005 Nov;58(5):698–705. doi: 10.1002/ana.20619. [DOI] [PubMed] [Google Scholar]
- 19.Levy D, Burstein R. The vascular theory of migraine: leave it or love it? Annals of neurology. 2011 Apr;69(4):600–1. doi: 10.1002/ana.22422. [DOI] [PubMed] [Google Scholar]
- 20.Charles A. Migraine is not primarily a vascular disorder. Cephalalgia: an international journal of headache. 2012 Apr;32(5):431–2. doi: 10.1177/0333102412441717. [DOI] [PubMed] [Google Scholar]
- 21.Zhang XC, Kainz V, Burstein R, Levy D. Tumor necrosis factor-alpha induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions. Pain. 2010 Oct 29; doi: 10.1016/j.pain.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983 Jan;16(2):109–10. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Strassman AM, Burstein R, Levy D. Sensitization and activation of intracranial meningeal nociceptors by mast cell mediators. J Pharmacol Exp Ther. 2007 May 4;322(2):806–12. doi: 10.1124/jpet.107.123745. [DOI] [PubMed] [Google Scholar]
- 24.Levy D, Zhang XC, Jakubowski M, Burstein R. Sensitization of meningeal nociceptors: inhibition by naproxen. Eur J Neurosci. 2008 Feb;27(4):917–22. doi: 10.1111/j.1460-9568.2008.06068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy D, Burstein R, Strassman AM. Mast Cell Involvement in the Pathophysiology of Migraine Headache: A Hypothesis. Headache. 2006;46(S1):S13–S8. doi: 10.1111/j.1526-4610.2006.00485.x. [DOI] [PubMed] [Google Scholar]
- 26.Levy D, Burstein R, Kainz V, Jakubowski M, Strassman AM. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain. 2007 Jul;130(1–2):166–76. doi: 10.1016/j.pain.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iversen HK, Olesen J, Tfelt-Hansen P. Intravenous nitroglycerin as an experimental model of vascular headache. Basic characteristics. Pain. 1989 Jul;38(1):17–24. doi: 10.1016/0304-3959(89)90067-5. [DOI] [PubMed] [Google Scholar]
- 28.Read SJ, Manning P, McNeil CJ, Hunter AJ, Parsons AA. Effects of sumatriptan on nitric oxide and superoxide balance during glyceryl trinitrate infusion in the rat. Implications for antimigraine mechanisms. Brain research. 1999 Nov 13;847(1):1–8. doi: 10.1016/s0006-8993(99)01985-x. [DOI] [PubMed] [Google Scholar]
- 29.Reuter U, Bolay H, Jansen-Olesen I, et al. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain. 2001;124(Pt 12):2490–502. doi: 10.1093/brain/124.12.2490. [DOI] [PubMed] [Google Scholar]
- 30.Komalavilas P, Shah PK, Jo H, Lincoln TM. Activation of mitogen-activated protein kinase pathways by cyclic GMP and cyclic GMP-dependent protein kinase in contractile vascular smooth muscle cells. The Journal of biological chemistry. 1999;274(48):34301–9. doi: 10.1074/jbc.274.48.34301. [DOI] [PubMed] [Google Scholar]
- 31.Doronzo G, Viretto M, Russo I, et al. Nitric oxide activates PI3-K and MAPK signalling pathways in human and rat vascular smooth muscle cells: influence of insulin resistance and oxidative stress. Atherosclerosis. 2011 May;216(1):44–53. doi: 10.1016/j.atherosclerosis.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 32.Maddahi A, Ansar S, Chen Q, Edvinsson L. Blockade of the MEK/ERK pathway with a raf inhibitor prevents activation of pro-inflammatory mediators in cerebral arteries and reduction in cerebral blood flow after subarachnoid hemorrhage in a rat model. J Cereb Blood Flow Metab. 2010 Apr 28; doi: 10.1038/jcbfm.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maddahi A, Edvinsson L. Enhanced expressions of microvascular smooth muscle receptors after focal cerebral ischemia occur via the MAPK MEK/ERK pathway. BMC neuroscience. 2008;9:85. doi: 10.1186/1471-2202-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maddahi A, Edvinsson L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. Journal of neuroinflammation. 2010;7:14. doi: 10.1186/1742-2094-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000 Sep 14;407(6801):242–8. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 36.Asghar MS, Hansen AE, Amin FM, et al. Evidence for a vascular factor in migraine. Annals of neurology. 2011 Apr;69(4):635–45. doi: 10.1002/ana.22292. [DOI] [PubMed] [Google Scholar]
- 37.Hansen JM, Pedersen D, Larsen VA, et al. Magnetic resonance angiography shows dilatation of the middle cerebral artery after infusion of glyceryl trinitrate in healthy volunteers. Cephalalgia: an international journal of headache. 2007 Feb;27(2):118–27. doi: 10.1111/j.1468-2982.2006.01257.x. [DOI] [PubMed] [Google Scholar]
- 38.Gozalov A, Jansen-Olesen I, Klaerke D, Olesen J. Role of BK(Ca) channels in cephalic vasodilation induced by CGRP, NO and transcranial electrical stimulation in the rat. Cephalalgia: an international journal of headache. 2007 Oct;27(10):1120–7. doi: 10.1111/j.1468-2982.2007.01409.x. [DOI] [PubMed] [Google Scholar]
- 39.Rahmann A, Wienecke T, Hansen JM, Fahrenkrug J, Olesen J, Ashina M. Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia. 2008 Mar;28(3):226–36. doi: 10.1111/j.1468-2982.2007.01497.x. [DOI] [PubMed] [Google Scholar]
- 40.Loppnow H, Buerke M, Werdan K, Rose-John S. Contribution of vascular cell-derived cytokines to innate and inflammatory pathways in atherogenesis. J Cell Mol Med. 2011 Mar;15(3):484–500. doi: 10.1111/j.1582-4934.2010.01245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reuter U, Chiarugi A, Bolay H, Moskowitz MA. Nuclear factor-kappaB as a molecular target for migraine therapy. Annals of neurology. 2002;51(4):507–16. doi: 10.1002/ana.10159. [DOI] [PubMed] [Google Scholar]
- 42.Jones MG, Lever I, Bingham S, Read S, McMahon SB, Parsons A. Nitric oxide potentiates response of trigeminal neurones to dural or facial stimulation in the rat. Cephalalgia. 2001;21(6):643–55. doi: 10.1046/j.1468-2982.2001.00213.x. [DOI] [PubMed] [Google Scholar]
- 43.Koulchitsky S, Fischer MJ, De Col R, Schlechtweg PM, Messlinger K. Biphasic response to nitric oxide of spinal trigeminal neurons with meningeal input in rat--possible implications for the pathophysiology of headaches. Journal of neurophysiology. 2004 Sep;92(3):1320–8. doi: 10.1152/jn.01210.2003. [DOI] [PubMed] [Google Scholar]
- 44.Koulchitsky S, Fischer MJ, Messlinger K. Calcitonin gene-related peptide receptor inhibition reduces neuronal activity induced by prolonged increase in nitric oxide in the rat spinal trigeminal nucleus. Cephalalgia: an international journal of headache. 2009 Apr;29(4):408–17. doi: 10.1111/j.1468-2982.2008.01745.x. [DOI] [PubMed] [Google Scholar]
- 45.De Col R, Koulchitsky SV, Messlinger KB. Nitric oxide synthase inhibition lowers activity of neurons with meningeal input in the rat spinal trigeminal nucleus. Neuroreport. 2003;14(2):229–32. doi: 10.1097/00001756-200302100-00014. [DOI] [PubMed] [Google Scholar]
- 46.Martin RS, Martin GR. Investigations into migraine pathogenesis: time course for effects of m-CPP, BW723C86 or glyceryl trinitrate on appearance of Fos-like immunoreactivity in rat trigeminal nucleus caudalis (TNC) Cephalalgia. 2001 Feb;21(1):46–52. doi: 10.1046/j.1468-2982.2001.00157.x. [DOI] [PubMed] [Google Scholar]
- 47.Offenhauser N, Zinck T, Hoffmann J, et al. CGRP release and c-fos expression within trigeminal nucleus caudalis of the rat following glyceryltrinitrate infusion. Cephalalgia. 2005 Mar;25(3):225–36. doi: 10.1111/j.1468-2982.2004.00845.x. [DOI] [PubMed] [Google Scholar]
- 48.Lin Q, Palecek J, Paleckova V, et al. Nitric oxide mediates the central sensitization of primate spinothalamic tract neurons. Journal of neurophysiology. 1999 Mar;81(3):1075–85. doi: 10.1152/jn.1999.81.3.1075. [DOI] [PubMed] [Google Scholar]
- 49.Roch M, Messlinger K, Kulchitsky V, Tichonovich O, Azev O, Koulchitsky S. Ongoing activity in trigeminal wide-dynamic range neurons is driven from the periphery. Neuroscience. 2007 Dec 12;150(3):681–91. doi: 10.1016/j.neuroscience.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 50.Greenfield JC, Jr, Rembert JC, Tindall GT. Transient changes in cerebral vascular resistance during the Valsalva maneuver in man. Stroke. 1984 Jan-Feb;15(1):76–9. doi: 10.1161/01.str.15.1.76. [DOI] [PubMed] [Google Scholar]
- 51.Williams B. Cerebrospinal fluid pressure changes in response to coughing. Brain. 1976;99(2):331–46. doi: 10.1093/brain/99.2.331. [DOI] [PubMed] [Google Scholar]
- 52.De Felice M, Ossipov MH, Wang R, et al. Triptan-induced enhancement of neuronal nitric oxide synthase in trigeminal ganglion dural afferents underlies increased responsiveness to potential migraine triggers. Brain: a journal of neurology. 2010 Aug;133(Pt 8):2475–88. doi: 10.1093/brain/awq159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Felice M, Ossipov MH, Wang R, et al. Triptan-induced latent sensitization: a possible basis for medication overuse headache. Annals of neurology. 2010 Mar;67(3):325–37. doi: 10.1002/ana.21897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Felice M, Porreca F. Opiate-induced persistent pronociceptive trigeminal neural adaptations: potential relevance to opiate-induced medication overuse headache. Cephalalgia: an international journal of headache. 2009 Dec;29(12):1277–84. doi: 10.1111/j.1468-2982.2009.01873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raddant AC, Russo AF. Calcitonin gene-related peptide in migraine: intersection of peripheral inflammation and central modulation. Expert Rev Mol Med. 2011;13:e36. doi: 10.1017/S1462399411002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.MacGregor EA. Oestrogen and attacks of migraine with and without aura. Lancet neurology. 2004 Jun;3(6):354–61. doi: 10.1016/S1474-4422(04)00768-9. [DOI] [PubMed] [Google Scholar]
- 57.Boes T, Levy D. Influence of sex, estrous cycle and estrogen on intracranial dural mast cells. Cephalalgia. 2012 doi: 10.1177/0333102412454947. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baun M, Pedersen MH, Olesen J, Jansen-Olesen I. Dural mast cell degranulation is a putative mechanism for headache induced by PACAP-38. Cephalalgia: an international journal of headache. 2012 Mar;32(4):337–45. doi: 10.1177/0333102412439354. [DOI] [PubMed] [Google Scholar]