

Abstract
Lead (Pb), one of the most toxic heavy metals, can be absorbed and accumulated by plant roots and then enter the food chain resulting in potential health risks for human beings. The radish (Raphanus sativus L.) is an important root vegetable crop with fleshy taproots as the edible parts. Little is known about the mechanism by which radishes respond to Pb stress at the molecular level. In this study, Next Generation Sequencing (NGS)–based RNA-seq technology was employed to characterize the de novo transcriptome of radish roots and identify differentially expressed genes (DEGs) during Pb stress. A total of 68,940 assembled unique transcripts including 33,337 unigenes were obtained from radish root cDNA samples. Based on the assembled de novo transcriptome, 4,614 DEGs were detected between the two libraries of untreated (CK) and Pb-treated (Pb1000) roots. Gene Ontology (GO) and pathway enrichment analysis revealed that upregulated DEGs under Pb stress are predominately involved in defense responses in cell walls and glutathione metabolism-related processes, while downregulated DEGs were mainly involved in carbohydrate metabolism-related pathways. The expression patterns of 22 selected genes were validated by quantitative real-time PCR, and the results were highly accordant with the Solexa analysis. Furthermore, many candidate genes, which were involved in defense and detoxification mechanisms including signaling protein kinases, transcription factors, metal transporters and chelate compound biosynthesis related enzymes, were successfully identified in response to heavy metal Pb. Identification of potential DEGs involved in responses to Pb stress significantly reflected alterations in major biological processes and metabolic pathways. The molecular basis of the response to Pb stress in radishes was comprehensively characterized. Useful information and new insights were provided for investigating the molecular regulation mechanism of heavy metal Pb accumulation and tolerance in root vegetable crops.
Introduction
The problem of heavy metal pollution is becoming a major worldwide ecological concern due to its potential adverse impacts on human health through the food chain [1], [2]. Lead (Pb) is one of the most toxic heavy metals and can be easily taken up by plants and accumulated in different parts [3], [4]. The sources of Pb contamination not only include roots in natural processes but also derive anthropogenic activities, such as exhaust fumes of automobiles, emissions from industrial factories and land applications of agrochemicals including insecticides, herbicides and fertilizers [3], [5].
Lead is absorbed by plants mainly through the roots, and in most plant species the Pb content in organs tends to decrease in the order of root, leaves, stem, inflorescence and seeds [6]–[9]. Many previous studies reported that Pb overload in plants causes a range of negative effects, and the visually nonspecific symptoms of Pb toxicity are rapid inhibition of seed germination, stunted growth of the plant and chlorosis [1], [4]. Additionally, Pb can cause oxidative damage by stimulating the formation of free radicals and reactive oxygen species, resulting in oxidative stress and DNA damage [10]–[12].
Radish (Raphanus sativus L.), belonging to the family Brassicaceae, is an economically important vegetable crop with an edible taproot [13]. It was reported that radish roots could accumulate large amounts of lead (Pb), which could cause a potential health risk in polluted conditions [6], [14]. Considerable efforts have been invested into investigating Pb stress in radishes, especially its accumulation, translocation, physiological and metabolic variations, and cell deposition [6], [15]–[16]. However, little is known about the mechanism of radish plant responses to Pb stress at the molecular level,i.e.gene expression variations under Pb stress.
The next-generation sequencing (NGS) technologies including Roche/454’s sequencing [17],Illumina/Solexa’s sequencing technology (San Diego, California, USA), and Applied Biosystems’ SOLiD sequencing technology have provided novel opportunities for accelerating genome projects such as whole-genome re-sequencing for variation analysis, RNA sequencing (RNA-seq) for transcriptome and non-coding RNAome analysis [18], [19]. Among them, NGS-based RNA-seq for transcriptome methods can permit simultaneous acquisition of sequences and can be used to characterize genes, detect gene expression, identify and quantify rare transcripts without prior knowledge of a particular gene, and provide information regarding alternative splicing and sequence variations in identified genes [20]. In recent years, RNA-seq has been proven as a powerful method for understanding the complexity of gene expression and regulation networks in several plant species responding to many kinds of biotic and abiotic stresses, such as cotton [21], chinese cabbage [22], chickpea [23], maize [24], potato [25], Ammopiptanthus mongolicus [26] and soybean [27].
Previous studies indicated that plant in different stress responses is controlled by a range of gene regulatory mechanisms which could act together in various response and defense systems. Transcription factors, transport proteins and some other critical genes involved in certain signal transduction and secondary metabolite pathways were considered to be common stress-related transcripts activated in both biotic and abiotic stresses[27]–[29]. However, there were some unique genes involved in response to a specific stress [28]. For example, plants counteract the heavy metal stress by adopting both common and some specific means to defense and detoxifying the excessive concentration occurring in their surroundings, which includes generating metal ions signal sensing and transduction related pathways, activating numerous metal transporters and modulating related transcription factors[30]–[33]. It was found that the major activated genes not only involved in common stress-induced responses but also in some specific pathways including sulfur assimilation, glutathione (GSH) metabolism, IAA and jasmonic acid (JA) biosynthesis in A. thaliana under Pb treatments [34].
Although the molecular regulation mechanisms and massive candidate genes involved in various stress conditions have been successfully revealed by employing NGS technologies based on two main platforms including Roche/454 and Solexa/Illumina, their applications in investigating the dynamically transcriptional changes in response to heavy metal Pb stress in radishes has not been reported. The present study is aimed to elucidate the molecular regulation mechanisms and the critical genes involved in regulating radish responses to Pb stress. NGS-based Illumina paired-end Solexa sequencing platform was employed to characterize the fleshy taproot de novo transcriptome of two radish root cDNA samples, one untreated control (CK) and one Pb-stressed with Pb(NO3)2 at 1000 mg L−1(Pb1000). Furthermore, an overall gene expression analysis was compared between CK and Pb1000 based on the assembled de novo transcriptomes. From that, a large set of radish transcript sequences that could be used to discover root-specific functions were generated, an abundance of differentially expressed Pb-responsive genes were quantified, and the enriched networks for regulating heavy metal Pb stress were acquired in radishes. Additionally, expression profiling of some differentially regulated genes were validated by qRT-PCR. The molecular basis of the response to Pb stress was first comprehensively characterized in radish, and the resulting information would facilitate further investigation of the mechanisms of Pb accumulation/tolerance in plants and the effective management of Pb contamination in vegetable crops by genetic manipulation.
Results
Illumina Sequencing and de novo Transcriptome Assembly of Radish Roots
A large volume data was generated with the Illumina HiSeq 2000 sequencing the two radish cDNA libraries, CK, an untreated control and Pb1000, a Pb-stressed with Pb(NO3)2 at 1000 mg L−1. According to Illumina’s RNA transcriptome sequencing, a sequence can generate 2×101 basepairs (bps) from each Paired-End (PE) of a cDNA fragment. After removing the low quality reads and trimming off the adapter sequences, 26,381,880 (CK) and 22,535,988 (Pb1000) high-quality, clean paired-end sequencing reads with a total of 2,636,208,012 and 2,251,686,397 nucleotides were obtained, respectively, for the two pools. Because no appropriate reference genome sequence was available for radishes, a de novo assembly program Trinity [35] was selected for de novo assembly of all the 48,918,868 clean reads. Then, we combined all the assembling transcripts with sequencing of 31, 4823 radish expressed sequence tags (ESTs) and 17,187 unigenes from the NCBI database (http://www.ncbi.nlm.nih.gov/). Finally, a total of 68,940 assembled transcripts including 33,337 unigenes were obtained with the contig size ranging from 306 to 16,101 bp in transcript length and the average size of 1,226.63 bp (Table 1). The size distribution of the assembled transcripts is shown in Figure 1.
Table 1. Overview of the sequencing and assembly.
| Items | Number |
| Total genes | 33,337 |
| Max contig length(bp) | 16.101 |
| Min contig length(bp) | 306 |
| Whole dataset lengths(bp) | 84,563,748 |
| Average contig lengths(bp) | 1,226.63 |
| Total isogenes | 68,940 |
| N50 | 1,262 |
| N90 | 437 |
Figure 1. The length distribution of the assembled transcripts.

Function Annotation and Classification of the Assembled Transcripts
Gene Ontology (GO) assignments were used to classify the functions of the predicted radish root genes under the heavy metal lead (Pb) stress. Based on sequence homology, 46,966 transcript sequences (68.12% of all assembled sequences) were assigned at least one GO term, including 57 functional groups at the second level (Figure 2). Among which, ‘cell’ (34,290 sequences, 73.01%), ‘cell part’ (36,289 sequences, 73.01%) and ‘organelle’ (23,439 sequences, 49.91%) terms were dominant in the cellular component, while ‘cellular process’ (30533 sequences, 65.01%),‘metabolic process’ (27,531 sequences, 58.62%) and ‘response to stimulus’ (14,392 sequences, 30.64%) were the most highly represented under biological process, and ‘binding’ (28856 sequences, 61.44%) and ‘catalytic activity’ (21742 sequences, 46.29%) were the most abundant subcategories for the molecular function. Furthermore, only a few transcripts were clustered in terms of ‘virion part’, ‘protein tag’, ‘translation regulator activity’, ‘metallochaperone activity’, ‘channel regulator activity’ and ‘sulfur utilization’.
Figure 2. Gene ontology classification of assembled transcripts.

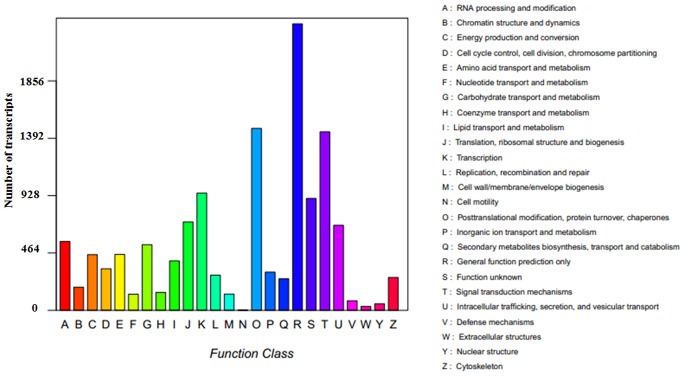
To further evaluate the completeness of the de novo assembly transcriptome and predict possible functions, all assembled transcripts were compared against the Cluster of Orthologous Groups (COG database) for the analysis of phylogenetically widespread domain families. The results revealed 22,518 sequences with significant homology and assigned them into the appropriate COG clusters. These COG classifications were grouped into 23 functional categories (Figure 3). The five largest categories were ‘general function prediction only’ (30.05%), ‘signal transduction mechanisms’ (15.89%), ‘transcription’ (14.70%), ‘post translational modification, protein turnover, chaperones’ (13.59%), and ‘function unknown (12.69%).
Figure 3. COG function classification of assembled transcripts.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database resource for the systematic understanding of high-level gene functions in terms of networks of the biological system, such as the cell, organism and ecosystem, from molecular-level information (http://www.genome.jp/kegg/). The assembled transcript sequences were searched against the KEGG database using BLASTX with a cut-off E-value of 10−5 to identify the biological pathways activated in the roots of radishes in response to heavy metal Pb stress. Of a total of 48,864 annotated sequences, 19,480 had a significant match in the database and were assigned to 296 KEGG pathways (Table S1). The major pathways were ‘metabolic pathways [ko01100]’, ‘biosynthesis of secondary metabolites [ko01110]’, ‘microbial metabolism in diverse environments [ko01120]’, ‘ribosome [ko03010]’ and ‘plant hormone signal transduction [ko04075]; the sequence numbers and percentage of sequences assigned to each pathway were 4,269 (28.20%), 1,954 (12.91%), 1,059 (7.21%), 839 (5.54%) and 629 (4.15%), respectively.
Differentially Expressed Genes Involved in the Response to Pb Stress in Radish Roots
To investigate the changes in gene expression and understand the critical genes in radish roots responding to the stress of heavy metal Pb, clean reads of the Pb1000 and the CK libraries were respectively mapped to the de novo assembly transcriptome reference sequences with Bowtie [36]–[37] and were assigned to unigenes and isoforms with the RSEM (RNA-Seq by Expectation Maximization) software [38]. The assigned unigene and isoform expression levels were calculated using a normalizing statistic called FPKM (fragments mapped per kilobase of exon per million reads mapped), which provides a measure of expression level that accounts for variation in gene length [39]. The differentially expressed genes (DEGs) (including unigene and isoform) were determined with a log-fold expression change (log FC) greater than 2 or less than –2 using a greater statistically significant value (P<0.005) as well as false discovery rates (FDR <0.001). From that, a total of 4,614 DEGs (1,398 unigenes) were detected between the two libraries, and these DEGs included both upregulated (2,154 transcripts) and downregulated genes (2,460 transcripts) under the Pb treatment (Figure 4).
Figure 4. Volcano plot of gene expression difference between Pb1000 and CK samples.

Gene Ontology (GO) and Pathway Functional Enrichment Analysis of the DEGs
All of the DEGs were performed on GO function and pathway enrichment analysis with a hypergeometric test and Bonferroni Correction [40]. GO terms significantly enriched in the DEGs were identified using goatools (https://github.com/tanghaibao/goatools). According to GO functional enrichment analysis, a total of 270 terms for the upregulated transcripts were significantly enriched at a Bonferroni-corrected P-value ≤0.05, while for downregulated ones only 107 terms were enriched. The GO terms predominantly enriched for the upregulated transcripts were related to biological processes including ‘defense response by callose deposition in cell wall’ (GO:0052544), ‘defense response by cell wall thickening’ (GO:0052482), ‘response to oxygen levels’ (GO:0070482), ‘glycoside catabolic process’ (GO:0016139), and ‘glutathione conjugation reaction’ (GO:0006803), and those related to molecular function include ‘glutathione transferase activity’(GO:0004364) (Table S2). The predominant GO terms identified as enriched among the downregulated transcripts related to ‘catalytic activity’ (GO: 0003824) in molecular functions, such as ‘cellulose synthase (GDP-forming) activity’ (GO:0016761), ‘carbon-oxygen lyase activity, acting on polysaccharides’ (GO: 0016837), and ‘cell wall organization or biogenesis’ (GO:0071554) in biological processes such as ‘cellular cell wall organization (GO:0007047) and cell wall modification (GO:0042545) (Table S3). The KEGG Orthology-Based Annotation System (KOBAS) was employed to find most statistically significantly enriched pathways for all the DEGs, which revealed that 13 and six pathways were observed significantly enriched for up- and down-regulated DEGs, respectively. As shown in Table 2, the pathways predominantly enriched for the upregulated transcripts were related to xenobiotics biodegradation and metabolism, such as drug metabolism-cytochrome P450 [ko00982], metabolism of xenobiotics by cytochrome P450 [ko00980], polycyclic aromatic hydrocarbon degradation [ko00624], aminobenzoate degradation [ko00627] and bisphenol degradation [ko00363], and metabolism of other amino acids such as glutathione metabolism [ko00480]. The central pathways identified as enriched for the downregulated transcripts were involved in carbohydrate metabolism including pentose and glucuronate interconversions [ko00400], starch and sucrose metabolism [ko00500], amino sugar and nucleotide sugar metabolism [ko00520] (Table 3).
Table 2. Enriched pathways of the up-regulated DEGs.
| Pathway | Id | Sample number | Background number | P-Value | Corrected P-Value |
| Drug metabolism - cytochrome P450 | ko00982 | 16 | 65 | 4.98E-10 | 1.29E-07 |
| Metabolism of xenobiotics by cytochrome P450 | ko00980 | 15 | 64 | 3.61E-09 | 4.66E-07 |
| Glutathione metabolism | ko00480 | 20 | 163 | 1.01E-06 | 6.49E-05 |
| Bisphenol degradation | ko00363 | 13 | 84 | 6.10E-06 | 0.000225 |
| Polycyclic aromatic hydrocarbon degradation | ko00624 | 14 | 99 | 7.99E-06 | 0.000258 |
| Aminobenzoate degradation | ko00627 | 14 | 102 | 1.14E-05 | 0.000326 |
| Limonene and pinene degradation | ko00903 | 15 | 122 | 2.15E-05 | 0.000556 |
| Tryptophan metabolism | ko00380 | 14 | 119 | 6.56E-05 | 0.001539 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | ko00945 | 12 | 104 | 0.000257 | 0.004741 |
| Methane metabolism | ko00680 | 19 | 230 | 0.000443 | 0.007622 |
| Glucosinolate biosynthesis | ko00966 | 6 | 36 | 0.001357 | 0.020595 |
| Sphingolipid metabolism | ko00600 | 7 | 50 | 0.00159 | 0.022791 |
| Galactose metabolism | ko00052 | 11 | 114 | 0.002032 | 0.027596 |
Table 3. Enriched pathways of the down-regulated DEGs.
| Pathway | Id | Sample number | Background number | P-Value | CorrectedP-Value |
| Pentose and glucuronate interconversions | ko00040 | 26 | 104 | 1.34E-14 | 4.49E-12 |
| Starch and sucrose metabolism | ko00500 | 37 | 369 | 1.07E-07 | 1.79E-05 |
| Gap junction | ko04540 | 10 | 44 | 4.85E-06 | 0.000405 |
| Pathogenic Escherichia coli infection | ko05130 | 11 | 66 | 3.78E-05 | 0.002524 |
| Glucosinolate biosynthesis | ko00966 | 8 | 36 | 5.17E-05 | 0.002879 |
| Amino sugar and nucleotide sugar metabolism | ko00520 | 27 | 355 | 0.000594 | 0.022046 |
Validation of Illumina Expression Patterns by qRT-PCR Analysis
To confirm the reliability of the Solexa analysis, 22 candidate DEGs were selected and their expression was detected by qRT-PCR. The expression patterns from qRT-PCR showed general agreement with those from the Solexa sequencing (Table 4). The discrepancies with respect to ratio should be attributed to the essentially different algorithms and sensitivity between the two techniques [41]–[42]. In the analysis of gene expression profiling, the deep-sequencing method generated absolute rather than relative expression measurements.
Table 4. Validation of the RNA-Seq expression profiles of selected DEGs by qRT-PCR.
| Transcript ID | Description | RNA-Seq(FPKM) | qRT-PCR | ||
| CK | Pb1000 | Log FC | Pb1000/CK | ||
| comp7581_c0_seq1 | HP4 no annotation | 0 | 77.93 | 36.999341 | 2.62 |
| comp7904_c0_seq1 | NAC29 NAC domain-containing protein 29 | 0 | 33.42 | 35.500309 | 222.86 |
| comp13297_c0_seq1 | EFR088-2 ethylene-responsive transcription factor | 0 | 26.67 | 34.839795 | 56.49 |
| comp12162_c0_seq1 | LAA long-chain acyl-CoA synthetase | 0 | 15.31 | 34.74358 | 97.68 |
| comp25182_c0_seq1 | LEAD-1 late embryogenesis abundant hydroxyproline-rich glycoprotei | 0 | 10.05 | 33.74358 | 33.13 |
| comp2239_c0_seq1 | GSTU11 glutathione-s-transferase | 0.67 | 193.06 | 8.1793185 | 56.49 |
| comp5087_c0_seq1 | NDR1 NDR1/HIN1-like 25salicylic acid | 0.23 | 52.2 | 7.8221293 | 113.77 |
| comp5862_c0_seq1 | GST1-1 glutathione S-transferase | 0.89 | 44.35 | 5.6316074 | 10.85 |
| comp1331_c0_seq1 | GST3-1glutathione-s-transferase 3 | 4.86 | 114.66 | 4.5584999 | 10.13 |
| comp2192_c0_seq1 | GGT1 gamma-glutamyl transpeptidase 1 | 4.82 | 107.13 | 4.4724431 | 8.28 |
| comp9962_c0_seq1 | ASA Ascorbate Peroxidase | 2.89 | 38.79 | 3.7457784 | 5.24 |
| comp932_c0_seq1 | GST1-2glutathione S-transferase | 7.61 | 100.68 | 3.7247168 | 7.62 |
| comp2560_c0_seq1 | GSTL1 glutathione transferase lambda 1 | 6.33 | 82.32 | 3.7008077 | 776.05 |
| comp942_c0_seq1 | GST3-2 glutathione-s-transferase 3 | 6.36 | 75.85 | 3.5758534 | 17.15 |
| comp674_c0_seq1 | GSHB1 glutathione synthetase | 51.37 | 454.81 | 3.1451889 | 3.25 |
| comp630_c0_seq1 | LAX2-1 auxin transporter-like protein 2 | 327.47 | 45.41 | −2.85144 | 0.37 |
| comp5751_c0_seq1 | SPDS spermine synthase | 54.48 | 4.7 | −3.535811 | 0.02 |
| comp8656_c0_seq1 | LAX2-2 auxin transporter-like protein 2 | 60.46 | 1.83 | −5.049527 | 0.03 |
| comp3052_c0_seq1 | LAX3 auxin transporter-like protein 3 | 199.38 | 4.81 | −5.375419 | 0.13 |
| comp4226_c0_seq1 | GSTF11 glutathione S-transferase F1 | 94.71 | 0 | −36.5882 | 0.01 |
| comp5860_c0_seq1 | SSF sodium symporter family protein | 79.66 | 0 | −37.17514 | 0.05 |
| comp1679_c0_seq1 | CCD8 carotenoid cleavage dioxygenase 8 | 180.13 | 0 | −38.81567 | 0.01 |
For further investigating and verifying the expression variations of the DEGs, transcriptional qRT-PCR analysis of six selected genes including three upregulated (GST3-1, GGT1 and GSHB1) and three downregulated (SPDS, LAX2-1 and LAX2-2), were performed for detailed analysis of different concentrations of Pb(NO3)2 (0, 200, 500 and 1000 mg L−1) after 72 h treatment and temporal duration (0, 24, 48 and 72 h) of a fixed concentration of Pb(NO3)2 at 1000 mg L−1. As shown in Figure 5, three downregulated DEGs, including SPDS, LAX2-1 and LAX2-2, were downregulated under all the concentrations of Pb(NO3)2 treatments after 72 h, however, their expression exhibited variations among different time durations, which showed an upregulation for SPDS after 24 and 48 h, and LAX2-1 after 24 h, while the remaining expressions were all downregulated. With an increase in Pb treatment duration, the levels of SPDS, LAX2-1 and LAX2-2 were gradually brought down, except for LAX2-1 within 48 and 72 h where there was a slight increase. On the other hand, for glutathione metabolism upregulated genes, GST3-1 and GSHB1 showed downregulation to Pb(NO3)2 exposure for 200 and 500 mg L−1, but upregulation for 1000 mg L−1, whereas GGT1 showed upregulation among all concentration treatments after 72 h. Meanwhile, the expression of these three genes showed significant changes at different duration treatments. GST3-1and GSHB1 showed an upregulation at 24 and 72 h, respectively, while GGT1 showed an increase at every exposure time point.
Figure 5. qRT-PCR analysis of six selected DEGs with different concentrations and temporal treatments of Pb(NO3)2.

A-F represent different concentrations of Pb(NO3)2 after 72 h treatment and G-L represent different temporal duration of a fixed concentration of Pb(NO3)2 at 1000 mg L−1.
Identification of DEGs Involved in Signal Sensing and Transduction Proteins
The main pathway by which heavy metal can penetrate into the plants is through root uptake from soils, and the root cell wall is directly in contact with metals in the soil solution [31], [43]. When encounter an extra-cellular stimuli, the cell walls could activate a variety of specific stress-responsive signaling proteins to protect the cell from susceptible sites into the protoplast, such as Mitogen-activated protein kinases (MAPKs) and calcium-binding related protein kinase. In eukaryotes, MAPKs were consisted of three sequentially activated protein kinases including MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK, all of which were involved in response to a variety of environmental, hormonal and developmental stimuli [31]. In this study, four DEGs were identified that were highly homologous to genes encoding MAPKs such as MAPKKK7, MAPK6, MAPK18 and MPAK20, and 14 DEGs were similar to calcium-binding protein genes including CDPK9, CML45, CML22 and EICBP5, and most of these genes were upregulated under Pb stress (Table S4).
Identification of DEGs Involved in Transcription Factors
Transcription factors (TFs) could regulate corresponding transcriptional processes when plants were treated with toxic heavy metals [30]. Numerous TFs such as WRKY, basic leucine zipper (bZIP), ethylene-responsive factor (ERF) and myeloblastosis protein (MYB) have been verified to play a significant role in controlling the expression of specific stress-related genes [44]–[45]. In this study, there were 58 DGEs including up- and down-regulated genes that were involved in different kinds of TFs such as the WRKY family (i.e., WRKY4, 6, 28, 32, 33 and 75), ERF family (i.e. ERF1B, 018, 060, 070, 088 and 114), MYB family (i.e., MYB 1, 28, 29, 46, 51, 59, 95, 116 and 122), and dehydration-responsive element-binding protein-type transcription factors (DREB) (Table S4).
Identification of DEGs Involved in Metal Transporters
Metal transporters could play a vital role in alleviating heavy metal toxicity by transporting metal ions out of the cell or sequestering it into the vacuole. It was reported that a wide range of transporter families including ATP binding cassette (ABC), Natural resistance-associated macrophage proteins (Nramps), ZRT, IRT-like proteins (ZIPs) and the Cation diffusion facilitators (CDFs), may contribute to heavy metal resistance [46]–[47]. In this study, 30 DGEs were identified as candidate genes involved as members of different metal transporter families, which were mainly related to ABC and ZIP families (Table S4).
Identification of Candidate Genes Involved in Biosynthesis of Chelating Compounds and Glutathione Metabolism
Synthesis of Metal-chelating compounds is another mechanism used by plants to combat heavy metal stress, which can sequester and ultimately detoxify the excess metal ions [48]. Metallothioneins (MTs) are low-molecular-weight cysteine-rich metal binding peptides, which were usually classified into four groups (MT1–4). Currently, MT genes have been identified in a number of higher plants such as Arabidopsis and Brassica juncea [48]–[49]. In the present study, three DEGs were found homologs with genes encoding MT3. Phytochelatins (PCs) is another important class of heavy-metal-binding ligands, which can bind metal ions via thiolate coordination. PCs are not formed as a direct result of expression of a metal tolerance gene, but rather as a product of a biosynthetic pathway [50].Numerous physiological, biochemical and genetic studies have confirmed that glutathione (GSH) is the substrate for PC biosynthesis [51]. The conversion of GSH to PCs can be catalyzed by a special γ-glutamyl cysteine dipeptidyl transpeptidase (EC 2.3.2.15) called Phytochelatin sythase (PCS), and many genes involved in the GSH metabolism pathway [ko00480] have been found crucial in regulating GSH and PCs levels [52].
In the present study, only one DEG sequence was discovered encoding PCS. Based on the KEGG pathway assignment, 163 transcript sequences were found to encode 18 putative enzymes involved in glutathione metabolism from the present assembled de novo transcriptome background (Table 5, S1). More than one transcript sequences were annotated as the same enzyme, implying that such transcript sequences may represent different fragments of a single transcript, or different members of a gene family [53]. Comparative analysis of sequences from the two cDNA libraries during lead stress revealed that 25 transcript sequences were significantly differentially expressed. These sequences encoded five enzymes including four upregulated, L-ascorbate peroxidase (APX, EC 1.11.1.11), glutathione S-transferase (GST, EC 2.5.1.18), glutathione synthase (GSHB, EC 6.3.2.3) and gamma-glutamyltranspeptidase (GGT, EC 2.3.2.2), and one downregulated spermidine synthase (SPDS, EC 2.5.1.16) (Figure 6).
Table 5. The identified genes involved in glutathione metabolism of the de novo transcriptome.
| KO No. | Gene description | Gene Name | EC Number |
| K00434 | L-ascorbate peroxidase | APX | 1.11.1.11 |
| K00799 | glutathione S-transferase | GST | 2.5.1.18 |
| K01920 | glutathione synthase | GSHB | 6.3.2.3 |
| K00681 | gamma-glutamyltranspeptidase | GGT | 2.3.2.2 |
| K00797 | spermidine synthase | SPDS | 2.5.1.16 |
| K01469 | 5-oxoprolinase (ATP-hydrolysing) | OPLAH | 3.5.2.9 |
| K11205 | glutamate–cysteine ligase regulatory subunit | GCLM | N/A |
| K01919 | glutamate–cysteine ligase | GSHA | 6.3.2.2 |
| K01255 | leucyl aminopeptidase | PEPA | 3.4.11.1 |
| K01256 | aminopeptidase N | PEPN | 3.4.11.2 |
| K15428 | Cys-Gly metallodipeptidase DUG1 | DUG1 | 3.4.13.- |
| K00383 | glutathione reductase (NADPH) | GSR | 1.8.1.7 |
| K00031 | isocitrate dehydrogenase | IDH1, | 1.1.1.42 |
| K00033 | 6-phosphogluconate dehydrogenase | PGD | 1.1.1.44 |
| K00036 | glucose-6-phosphate 1-dehydrogenase | G6PD | 1.1.1.49 |
| K00432 | glutathione peroxidase | GPX | 1.11.1.9 |
| K10807 | ribonucleoside-diphosphate reductase subunit M1 | RRM1 | 1.17.4.1 |
| K10808 | ribonucleoside-diphosphate reductase subunit M2 | RRM2 | 1.17.4.1 |
KO (KEGG Orthology), a classification of ortholog and paralog groups based on highly confident sequence similarity scores, and the reaction classification system for biochemical reaction classification, along with other classifications for compounds and drug.
Figure 6. Expression pattern of genes involved in glutathione metabolism.

The red and green color in each column indicated up- and down-regulation enzymes. The heatmaps generated were colored to the corresponding DEGs from the KEGG database (map 00480).
Discussion
Lead (Pb) is one of the most abundant, ubiquitous, toxic heavy metal elements posing a critical concern to human and environmental health [54]–[56]. Therefore, how to control Pb pollution and reduce Pb risks to human health has been an important health issue all over the world. Understanding the molecular mechanisms of plants responding to heavy metal stresses to control its uptake, translocation, and accumulation has been a focus work for plant genetic manipulation and biotechnology research [30], [57].
Radish (Raphanus sativus L.) is a major root vegetable crop worldwide, which can accumulate a relatively high concentration of toxic heavy metal elements, such as lead (Pb) and cadmium (Cd) in roots when grown in heavy metal polluted conditions [14]–[16]. However, few works have been reported about the Pb-responsive genes and their regulation networks in radishes under Pb stress.
Because the genome sequence of the radish is still unavailable, expressed sequence tag (EST) analysis has become one of the most valuable tools to discover and identify novel genes. However, to date, only 31, 4823 radish ESTs can be retrieved from the GenBank of NCBI, which has a limited coverage of the transcriptome, and only a few candidate genes involved in complex biosynthetic pathways can be identified. The arrival of Next-Generation Sequencing (NGS) technologies could overcome the limitations and allow investigators to simultaneously measure the changes and regulation at a genome-wide level under certain biological conditions for non-model plants for which the background genomic information is not available [39], [58]–[59]. In recent years,the newly developed NGS-based RNA-seq technique has been widely used for de novo transcriptome sequencing assembly, discovery of novel genes and investigation of gene expression in many non-model plants such as rice [60], peanuts [61], purple sweet potato [62], medicinal plant Polygonum cuspidatum [63] and floral plant Dendrocalamus latiflorus [64].
In this study, based on de novo transcriptome sequencing and assembly, a total of 68,940 assembled transcripts including 33,337 unigenes were obtained from two radish root cDNA libraries of untreated control (CK) and Pb-treated with Pb(NO3)2 at 1000 mg L−1 (Pb1000). Comparison of the assembled transcripts to gene catalogs of other species, 48,864 transcript sequences (70.88% of 68,940 transcripts) were annotated by BLAST analysis and functional bioinformatics analyses including the GO, COG and KEGG databases. From the annotation results, it could be found that the main species with BLAST hits to annotated transcripts were A. thaliana, A. lyrata, B. napus, B. oleracea and B. rapa, which indicated that the sequences of the radish transcripts obtained in the present study were assembled and annotated correctly [65]. However, an abundance of unannotated transcripts were generated in the present study, which may represent a specific gene pool for radish root studies. The gene catalog provided a comprehensive understanding of the gene transcription profiles of radishes, and laid a solid foundation for further investigation of Pb-stress mechanisms and identification of novel genes in this species.
Gene expression analysis is often employed in exploring changes among different plant tissues, different developmental stages, or plants under different treatments [20], [66]–[68]. In this study, a total of 4,614 transcripts were identified as differentially expressed genes (DEGs). By analysis of the statistically enriched GO terms and pathways related to the DEGs, it was revealed that the most significant and high frequently enriched terms and pathways for upregulated transcripts during Pb stress were predominately involved in the defense response in cell walls (GO:0052544 and GO:0052482) and glutathione (GSH) metabolism-related (i.e., GO:0006803, GO:0004364, and ko00480) processes, while for downregulated genes, they were mainly related to carbohydrate metabolism-related pathways (i.e., ko00400, ko00500 and ko00520) (Table 3, S2 and S3). This information could offer more direct biological insights to elucidate the response mechanism of Pb stress in radishes, which was in high accordance with previous studies of heavy metal stresses in plants [69]–[71].
The plant cell wall played an important role for heavy metal defense and detoxification, which could act as a barrier limiting metal uptake and penetration into the protoplast [43]. With electron microscopy observation, Inoue et al [15] reported that the presence of Pb in the cell wall of radish roots could form extremely large crystalline-like deposits, which can completely saturate the cell wall and need special accommodation space or thickening of the cell wall at the site. These findings supported the roles of cell wall-related terms ‘defense response by callose deposition in cell wall’ (GO: 0052544) and ‘defense response by cell wall thickening’ (GO: 0052482) identified in the present study following Pb stress in radishes. Additionally, plant cell walls have been consided as active metabolic sites, where a variety of specific stress-responsive signaling molecules such as mitogen-activated protein kinases (MAPKs) and calcium-binding related protein kinase could be generated in response to extra-cellular stimuli [31], [72]. Many studies from different plant species indicated that several MAPK pathways were activated in response to heavy metal stress. A previous screen in Arabidopsis for cadmium-responsive genes identification revealed that a related MAPK named MAPKKKMEK K1 could be induced by high concentrations of CdCl2 at transcriptional level [73]. Jonak et al (2004) reported that exposure of alfalfa (Medicago sativa) seedlings to excess copper or cadmium ions could activated four distinct MAPKs [74]. Currently, four DEGs were identified as MAPKs including MAPKKK7, MAPK6, MAPK18 and MPAK20 in response to Pb treatment in radish. Because the activation of MAPK cascade is plant-species and metal variety-dependent, the role of distinct MAPK modules in radish responding to Pb stress still to be verified for further study. Calmodulin system was reported to be involved in sensing heavy metal stress such as Cd, Ni and Pb, which could regulate ion transport, gene expression and stress tolerance [31]. The different gene expression pattern in our study showed that 14 DEGs were found homologous encoding calmodulin-like protein including both up and down regulated following Pb stress.
It was reported that GSH played a pivotal role in protecting plants from lead stress by quenching lead-induced reactive oxygen species (ROS). Liu et al. [34] by genome-wide microarray expression profiling analysis revealed that lead treatment could induce different GSH genes in Arabidopsis thaliana. Estrella-Gomez et al. indicated that lead accumulation in Salvinia minima could increase the accumulation of GSH, the enzymatic activity of glutathione synthase (GS), and cause expression changes of SmGS genes in both leaf and root tissues [75]. In addition Gupta et al. also reported the role of GSH in lead detoxification in S. alfredii [76]. In the present study, 18 GSH-related enzymes were found based on assembled radish transcriptome background and five of them were identified as DEGs including four upregulated and one downregulated under Pb stress (Table 5, Figure 6). The different gene expression showed a similar result to the previous reports. Moreover, many kinds of transcription factors (TFs), metal transporters, and metal-binding ligands have been found to be responsive and defensive to Pb stress in many other species [43]. As expected, these related gene families were also successfully identified in our study.
In summary, Next Generation Sequencing (NGS)-based RNA-seq technology was firstly employed in this study to characterize the roots de novo transcriptome of the radish plant under Pb stress, and a total of 68,940 assembled unique transcripts representing 33,337 unigenes were obtained. Based on the assembled de novo transcriptome, 4,614 differentially expressed genes (DEGs) that play significant roles in the response to Pb stress were identified. Gene Ontology (GO) and pathway enrichment analysis revealed that the upregulated DEGs under Pb stress were predominately involved in defense responses in the cell wall and glutathione metabolism-related processes, while downregulated DEGs were mainly related to carbohydrate metabolism pathways. Multiple candidate genes involved in defense and detoxification were successfully identified in response to Pb stress, and expression patterns of selected DEGs were further validated with qRT-PCR, which reflected significant alterations in major biological processes and metabolic pathways during Pb stress. The molecular basis of the response to Pb stress in radishes was first comprehensively characterized in this study, which resulted in useful information and laid a solid foundation for future investigating the molecular regulation mechanism of heavy metal Pb accumulation and tolerance in root vegetable crops.
Materials and Methods
Plant Materials
The radish (Raphanus sativus L.) advanced inbred line, ‘NAU-RG’, was used in this study. The surface-sterilized seeds were sown into soil in plastic pots and the seedlings were cultured in a growth chamber with 14 h light at 25°C and 10 h dark at 18°C for 20 days. Seedlings with similar sizes were transplanted into a 20-L plastic container with modified half-strength Hoagland’s nutrient solution (pH 5.4). One week later, the plants were treated with Pb(NO3)2 at 0 and 1000 mg L−1, respectively, and cultured under glasshouse conditions with natural light and day/night temperature of 28/18°C. For Solexa analysis and qRT-PCR verification, plants were harvested when they were treated with Pb(NO3)2 at 0 and 1000 mg L−1after 72 h. For qRT-PCR analysis of the different concentrations and temporal variation of Pb treatments, plants were collected after 72 h with different concentrations of Pb(NO3)2 at 0, 200 and 500 mg L−1, and for the concentration at 1000 mg L−1 were collected after 0, 24, 48, and 72 h, respectively. All samples were washed with deionized water and immediately frozen in liquid nitrogen and stored at −80°C for RNA extraction.
RNA Isolation and Illumina Sequencing
Total RNA from different samples was isolated using the RNAprep pure Plant Kit according to the manufacturer’s protocol (Tiangen Biotech Co., Ltd., China). To avoid genomic DNA contamination, RNA samples were treated with RNase-free DNase I (Takara, Japan). Two radish root cDNA libraries were constructed using an mRNA-seq assay for paired-end transcriptome sequencing, which was performed by the Shanghai Majorbio Biopharm Technology Co., Ltd. (Shanghai, China). Poly(A) mRNA was enriched from total RNA by using Sera-mag Magnetic Oligo (dT) Beads (Thermo Fisher Scientific, USA) and then mRNA-enriched RNAs were chemically fragmented to short pieces using 1× fragmentation solution (Ambion, USA) for 2.5 min at 94°C. Double-stranded cDNA was generated using the SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen, USA). After that, the Illumina Paired End Sample Prep kit was used for RNA-seq library construction and then was sequenced using Illumina HiSeq™ 2000. The sequencing data were deposited in NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/Traces/sra) with accession numbers SRX256970 (CK) and SRX263753 (Pb1000).
Raw Sequence Processing and de novo Assembly
Raw reads generated by Illumina HiseqTM2000 were initially processed to get clean reads by removing the adapter sequences and low quality bases at the 3' end. Then, transcriptome de novo assembly was carried out using the short-read assembling program Trinity [35]. First, clean reads with a certain length of overlap were combined to form longer contiguous sequences (contigs), and then these reads were mapped back onto the contigs. The distance and relation among these contigs could be calculated based on paired-end reads, which enabled the detection of contigs from the same transcript and also calculation of the distances among these contigs. Finally, contigs were further assembled using Trinity, and the contigs that could not be extended on either end were defined as unique transcripts. Additionally, the unique assembled transcripts were further subjected to the process of sequence-splicing redundancy removal with sequence clustering software to acquire non-redundant transcripts called unigenes.
Functional Annotation of the Assembled Transcripts
All the assembled transcripts were searched against the NCBI non-redundant (nr) Swiss-Prot, COG and KEGG databases using BLASTX to identify the proteins that had the highest sequence similarity with the given transcripts to retrieve their function annotations and a typical cut-off E-value of <10−5 was set. For the nr annotations, the BLAST2GO program was used to get GO annotations of unique assembled transcripts for describing biological processes, molecular functions and cellular components [77] After getting GO annotations for each transcript, WEGO software [78] was used to conduct GO functional classification for understanding the distribution of gene functions at the macroscopic level.
Identification and Functional Enrichment Analysis of the Differentially Expressed Genes (DEGs)
To identify DEGs between the two different treatments, the expression level for each transcript was calculated using the fragments per kilobase of exon per million mapped reads (FRKM) method. R statistical package software edgeR (Empirical analysis of Digital Gene Expression in R) was employed to quantify differential gene expression [79]. In addition, functional-enrichment analysis including GO and KEGG were performed to identify which DEGs were significantly enriched in GO terms and metabolic pathways at Bonferroni-corrected P-value ≤0.05 compared with the whole-transcriptome background using the formula described in the previous studies [24], [42].
Validation of DEG Expression with Quantitative Real-time PCR (qRT-PCR)
Quantitative real-time PCR was performed on a MyiQ Real-Time PCR Detection System (Bio-Rad) platform using the SYBR Green Master ROX (Roche, Japan) following the manufacturer’s instructions. Primers were designed using Beacon Designer 7.0 software, and Actin2/7 (ACT) (Table S5) was selected as the internal control gene according to a previous report [80]. The amplification was achieved by the following PCR program of first denaturation 95°C for 3 min, then 40 cycles of denaturation at 95°C for 5 s, annealing and extension at 58°C [62], [80]. The relative expression levels of the selected transcripts normalized to ACT were calculated using the 2−ΔΔCt method. All reactions were performed with three replicates, and the data were analyzed using Bio-Rad CFX Manager software.
Supporting Information
KEGG pathways of the assembled transcripts.
(XLS)
The enriched GO terms of up-regulated DEGs.
(XLS)
The enriched GO terms of down-regulated DEGs.
(XLS)
Candidate genes involved in defense and detoxification mechanisms of radish roots in repose to heavy metal Pb.
(DOC)
Primers used for qRT-PCR.
(XLS)
Acknowledgments
We thank Zhu X. at North Dakota State University, ND, USA for his critical review and helpful comments during the preparation of this manuscript.
Funding Statement
This work was supported by grants from the NSFC (31171956), the National Key Technologies R&D Program of China (2012BAD02B01,2011GB23600006), the Key Technologies R&D Program of Jiangsu Province, China (BE2010328), the Program for NCET, MOE (NCET-10-0473), the PAPD of Jiangsu Higher Education Institutions, and the Fundamental Research Funds for the Central Universities (KYZ201209). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Li X, Bu N, Li Y, Ma L, Xin S, et al. (2012) Growth, photosynthesis and antioxidant responses of endophyte infected and non-infected rice under lead stress conditions. J Hazard Mater 213–214: 55–61. [DOI] [PubMed] [Google Scholar]
- 2. Gupta N, Khan DK, Santra SC (2012) Heavy metal accumulation in vegetables grown in a long-term wastewater-irrigated agricultural land of tropical India. Environ Monit Assess 184: 6673–6682. [DOI] [PubMed] [Google Scholar]
- 3.Huang ZY, Chen T, Yu J, Qin DP, Chen L (2012) Lead contamination and its potential sources in vegetables and soils of Fujian, China. Environ Geochem Health: 1–11. [DOI] [PubMed]
- 4. Sharma P, Dubey RS (2005) Lead toxicity in plants. Brazil J Plant Physiol 17: 35–52. [Google Scholar]
- 5. Liu J, Li K, Xu J, Zhang Z, Ma T, et al. (2003) Lead toxicity, uptake, and translocation in different rice cultivars. Plant Sci 165: 793–802. [Google Scholar]
- 6. Lane S, Martin E (1977) A histochemical investigation of lead uptake in Raphanus sativus . New Phytol 79: 281–286. [Google Scholar]
- 7. Chen M, Li XM, Yang Q, Zeng GM, Zhang Y, et al. (2008) Total concentrations and speciation of heavy metals in municipal sludge from Changsha, Zhuzhou and Xiangtan in middle-south region of China. J Hazard Mater 160: 324–329. [DOI] [PubMed] [Google Scholar]
- 8. Eun SO, Shik Youn H, Lee Y (2008) Lead disturbs microtubule organization in the root meristem of Zea mays . Physiol Plant 110: 357–365. [Google Scholar]
- 9. Liu W, Zhou Q, Zhang Y, Wei S (2010) Lead accumulation in different Chinese cabbage cultivars and screening for pollution-safe cultivars. J Environ Manage 91: 781–788. [DOI] [PubMed] [Google Scholar]
- 10. Shahid M, Pinelli E, Dumat C (2012) Review of Pb availability and toxicity to plants in relation with metal speciation; role of synthetic and natural organic ligands. J Hazard Mater 219: 1–12. [DOI] [PubMed] [Google Scholar]
- 11. Shu X, Yin LY, Zhang QF, Wang WB (2012) Effect of Pb toxicity on leaf growth, antioxidant enzyme activities, and photosynthesis in cuttings and seedlings of Jatropha curcas L. Environ Sci Pollut Res. 19: 893–902. [DOI] [PubMed] [Google Scholar]
- 12. Schutzendubel A, Polle A (2002) Plant responses to abiotic stresses: heavy metal-induced oxidative stress and protection by mycorrhization. J Exp Bot 53: 1351–1365. [PubMed] [Google Scholar]
- 13.Wang L, He Q (2005) Chinese radish. Beijing: Scientific and Technical Documents Publishing House.
- 14. He L, Liu L, Gong Y, Hou X, Zhu X, et al. (2008) Physiological responses of radish (Raphanus Sativus L.) to lead stress. ICBBE 2008: 4602–4605. [Google Scholar]
- 15. Inoue H, Fukuoka D, Tatai Y, Kamachi H, Hayatsu M, et al. (2013) Properties of lead deposits in cell walls of radish (Raphanus sativus L.) roots. J Plant Res 126: 51–61. [DOI] [PubMed] [Google Scholar]
- 16. El-Beltagi HS, Mohamed AA (2010) Changes in non protein thiols, some antioxidant enzymes activity and ultrastructural alteration in radish plant (Raphanus sativus L.) grown under lead toxicity. Not Bot Hort Agrobot Cluj 38: 76–85. [Google Scholar]
- 17. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, et al. (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lister R, Gregory BD, Ecker JR (2009) Next is now: new technologies for sequencing of genomes, transcriptomes, and beyond. Curr Opin Plant Biol 12: 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mochida K, Shinozaki K (2011) Advances in omics and bioinformatics tools for systems analyses of plant functions. Plant Cell Physiol 52: 2017–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Strickler SR, Bombarely A, Mueller LA (2012) Designing a transcriptome next-generation sequencing project for a nonmodel plant species. Am J Bot 99: 257–266. [DOI] [PubMed] [Google Scholar]
- 21. Wang G, Zhu Q, Meng Q, Wu C (2012) Transcript profiling during salt stress of young cotton (Gossypium hirsutum) seedlings via Solexa sequencing. Acta Physiol Plant 34: 107–115. [Google Scholar]
- 22. Yu S, Zhang F, Yu Y, Zhang D, Zhao X, et al. (2012) Transcriptome profiling of dehydration stress in the Chinese cabbage (Brassica rapa L. ssp. pekinensis) by tag sequencing. Plant Mol Biol Rep 30: 17–28. [Google Scholar]
- 23. Wang XS, Liu Y, Jia YY, Gu HY, Ma HY, et al. (2012) Transcriptional responses to drought stress in root and leaf of chickpea seedling. Mol Biol Rep 39: 8147–8158. [DOI] [PubMed] [Google Scholar]
- 24. Shen Y, Zhang Y, Chen J, Lin H, Zhao M, et al. (2013) Genome expression profile analysis reveals important transcripts in maize roots responding to the stress of heavy metal Pb. Physiol Plant. 147: 270–282. [DOI] [PubMed] [Google Scholar]
- 25. Nachappa P, Levy J, Tamborindeguy C (2012) Transcriptome analyses of Bactericera cockerelli adults in response to “Candidatus Liberibacter solanacearum” infection. Mol Genet Genomics 287: 803–817. [DOI] [PubMed] [Google Scholar]
- 26. Zhou Y, Gao F, Liu R, Feng J, Li H (2012) De novo sequencing and analysis of root transcriptome using 454 pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus . BMC Genomics 13: 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fan XD, Wang JQ, Yang N, Dong YY, Liu L, et al. (2013) Gene expression profiling of soybean leaves and roots under salt, saline-alkali and drought stress by high-throughput Illumina sequencing. Gene 512: 392–402. [DOI] [PubMed] [Google Scholar]
- 28. Atkinson NJ, Urwin PE (2012) The interaction of plant biotic and abiotic stresses: from genes to the field. J Exp Bot 63: 3523–3543. [DOI] [PubMed] [Google Scholar]
- 29. dos Reis SP, Lima AM, de Souza CRB (2012) Recent Molecular Advances on Downstream Plant Responses to Abiotic Stress. Int J Mol Sci 13: 8628–8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thapa G, Sadhukhan A, Panda SK, Sahoo L (2012) Molecular mechanistic model of plant heavy metal tolerance. Biometals 25: 489–505. [DOI] [PubMed] [Google Scholar]
- 31. DalCorso G, Farinati S, Furini A (2010) Regulatory networks of cadmium stress in plants. Plant Signal Behav 5: 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aery N (2012) Plant defence against heavy metal stress. In: Mérillon JM, Romawat KG, editors.Plant Defence: Biological Control. 241–269.
- 33.Sharma J, Chakraverty N (2013) Mechanism of Plant Tolerance in Response to Heavy Metals. Molecular Stress Physiology of Plants: Springer. 289–308.
- 34. Liu T, Liu S, Guan H, Ma L, Chen Z, et al. (2009) Transcriptional profiling of Arabidopsis seedlings in response to heavy metal lead (Pb). Environ Exp Bot 67: 377–386. [Google Scholar]
- 35. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang W, Wei Z, Lam TW, Wang J (2011) Next generation sequencing has lower sequence coverage and poorer SNP-detection capability in the regulatory regions. Sci Rep 1: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ward JA, Ponnala L, Weber CA (2012) Strategies for transcriptome analysis in nonmodel plants. Am J Bot 99: 267–276. [DOI] [PubMed] [Google Scholar]
- 40.Zhao Y, Yao B, Zhang M, Wang S, Zhang H, et al.. (2012) Comparative analysis of differentially expressed genes in Sika deer antler at different stages. Mol Biol Rep: 1–12. [DOI] [PubMed]
- 41. Li P, Ponnala L, Gandotra N, Wang L, Si Y, et al. (2010) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42: 1060–1067. [DOI] [PubMed] [Google Scholar]
- 42. Shi T, Gao Z, Wang L, Zhang Z, Zhuang W, et al. (2012) Identification of differentially-expressed genes associated with pistil abortion in Japanese apricot by genome-wide transcriptional analysis. PloS one 7: e47810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pourrut B, Shahid M, Dumat C, Winterton P, Pinelli E (2011) Lead uptake, toxicity, and detoxification in plants. Reviews of Environmental Contamination and Toxicology Volume 213: Springer. 113–136. [DOI] [PubMed]
- 44. Jakoby M, Weisshaar B, Dröge-Laser W, Vicente-Carbajosa J, Tiedemann J, et al. (2002) bZIP transcription factors in Arabidopsis . Trends Plant Sci 7: 106–111. [DOI] [PubMed] [Google Scholar]
- 45. Singh KB, Foley RC, Oñate-Sánchez L (2002) Transcription factors in plant defense and stress responses. Curr Opin Plant Biol 5: 430–436. [DOI] [PubMed] [Google Scholar]
- 46. Hall J (2002) Cellular mechanisms for heavy metal detoxification and tolerance. J Exp Bot 53: 1–11. [PubMed] [Google Scholar]
- 47.Lan HX, Wang ZF, Wang QH, Wang MM, Bao YM, et al. (2012) Characterization of a vacuolar zinc transporter OZT1 in rice (Oryza sativa L.). Mol Biol Rep: 1–10. [DOI] [PubMed]
- 48. Lin Y-F, Aarts MG (2012) The molecular mechanism of zinc and cadmium stress response in plants. Cell Mol Life Sci 69: 3187–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cobbett C, Goldsbrough P (2002) Phytochelatins and metallothioneins: roles in heavy metal detoxification and homeostasis. Annu Rev Plant Biol 53: 159–182. [DOI] [PubMed] [Google Scholar]
- 50. Cobbett CS (2000) Phytochelatin biosynthesis and function in heavy-metal detoxification. Curr Opin Plant Biol 3: 211–216. [PubMed] [Google Scholar]
- 51. Anjum NA, Ahmad I, Mohmood I, Pacheco M, Duarte AC, et al. (2012) Modulation of glutathione and its related enzymes in plants’ responses to toxic metals and metalloids–a review. Environ Exp Bot 75: 307–324. [Google Scholar]
- 52. Yadav S (2010) Heavy metals toxicity in plants: An overview on the role of glutathione and phytochelatins in heavy metal stress tolerance of plants. S Afr J Bot 76: 167–179. [Google Scholar]
- 53. Hyun TK, Rim Y, Jang HJ, Kim CH, Park J, et al. (2012) De novo transcriptome sequencing of Momordica cochinchinensis to identify genes involved in the carotenoid biosynthesis. Plant Mol Biol 79: 413–427. [DOI] [PubMed] [Google Scholar]
- 54. Jayalakshmi N, Venkatachalam P (2011) Biochemical and molecular effects of lead heavy metal toxicity in plants: A phytoremediation approach. Plant Cell Biotechnol Mol Biol 12: 1–14. [Google Scholar]
- 55. Chen Y, Wang J, Shi G, Sun X, Chen Z, et al. (2011) Human health risk assessment of lead pollution in atmospheric deposition in Baoshan District, Shanghai. Environ Geochem Health 33: 515–523. [DOI] [PubMed] [Google Scholar]
- 56. Rossato LV, Nicoloso FT, Farias JG, Cargnelluti D, Tabaldi LA, et al. (2012) Effects of lead on the growth, lead accumulation and physiological responses of Pluchea sagittalis . Ecotoxicology 21: 111–123. [DOI] [PubMed] [Google Scholar]
- 57.Seth CS (2012) A review on mechanisms of plant tolerance and role of transgenic plants in environmental clean-up. Bot Rev : 1–31.
- 58. Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Varshney RK, Nayak SN, May GD, Jackson SA (2009) Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol 27: 522–530. [DOI] [PubMed] [Google Scholar]
- 60. Jung K-H, Ko H-J, Minh Xuan N, Kim S-R, Ronald P, et al. (2012) Genome-wide identification and analysis of early heat stress responsive genes in rice. J Plant Biol 55: 458–468. [Google Scholar]
- 61. Chen X, Zhu W, Azam S, Li H, Zhu F, et al. (2013) Deep sequencing analysis of the transcriptomes of peanut aerial and subterranean young pods identifies candidate genes related to early embryo abortion. Plant Biotechnol J 11: 115–127. [DOI] [PubMed] [Google Scholar]
- 62.Xie F, Burklew CE, Yang Y, Liu M, Xiao P, et al. (2012) De novo sequencing and a comprehensive analysis of purple sweet potato (Impomoea batatas L.) transcriptome. Planta: 1–13. [DOI] [PubMed]
- 63. Hao DC, Chen SL, Xiao PG, Liu M (2012) Application of high-throughput sequencing in medicinal plant transcriptome studies. Drug Dev Res 73: 487–498. [Google Scholar]
- 64. Zhang XM, Zhao L, Larson-Rabin Z, Li DZ, Guo ZH (2012) De Novo Sequencing and Characterization of the Floral Transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PloS one 7: e42082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dang ZH, Zheng LL, Wang J, Gao Z, Wu SB, et al. (2013) Transcriptomic profiling of the salt-stress response in the wild recretohalophyte Reaumuria trigyna . BMC genomics 14: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li P, Ponnala L, Gandotra N, Wang L, Si Y, et al. (2010) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42: 1060–1067. [DOI] [PubMed] [Google Scholar]
- 67. Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, et al. (2011) Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC genomics 12: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, et al. (2010) Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol 152: 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Douchiche O, Driouich A, Morvan C (2010) Spatial regulation of cell-wall structure in response to heavy metal stress: cadmium-induced alteration of the methyl-esterification pattern of homogalacturonans. Ann Bot 105: 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Glaeser H, Coblenz A, Kruczek R, Ruttke I, Ebert-Jung A, et al. (1991) Glutathione metabolism and heavy metal detoxification in Schizosaccharomyces pombe . Curr Genet 19: 207–213. [Google Scholar]
- 71. Krzesłowska M (2011) The cell wall in plant cell response to trace metals: polysaccharide remodeling and its role in defense strategy. Acta Physiol Plant 33: 35–51. [Google Scholar]
- 72. Opdenakker K, Remans T, Vangronsveld J, Cuypers A (2012) Mitogen-activated protein (MAP) kinases in plant metal stress: regulation and responses in comparison to other biotic and abiotic stresses. Int J Mol Sci 13: 7828–7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Suzuki N, Koizumi N, Sano H (2001) Screening of cadmium-responsive genes in Arabidopsis thaliana. Plant Cell Environ 24: 1177–1188. [Google Scholar]
- 74. Jonak C, Nakagami H, Hirt H (2004) Heavy metal stress. Activation of distinct mitogen-activated protein kinase pathways by copper and cadmium. Plant Physiol 136: 3276–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Estrella-Gomez NE, Sauri-Duch E, Zapata-Perez O, Santamaria JM (2012) Glutathione plays a role in protecting leaves of Salvinia minima from Pb2+ damage associated with changes in the expression of SmGS genes and increased activity of GS. Environ Exp Bot 75: 188–194. [Google Scholar]
- 76. Gupta D, Huang H, Yang X, Razafindrabe B, Inouhe M (2010) The detoxification of lead in Sedum alfredii H is not related to phytochelatins but the glutathione. J Hazard Mater 177: 437–444. [DOI] [PubMed] [Google Scholar]
- 77. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 78. Ye J, Fang L, Zheng H, Zhang Y, Chen J, et al. (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34: 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xu Y, Zhu X, Gong Y, Xu L, Wang Y, et al. (2012) Evaluation of reference genes for gene expression studies in radish (Raphanus sativus L.) using quantitative real-time PCR. Biochem Biophys Res Commun 424: 398–403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
KEGG pathways of the assembled transcripts.
(XLS)
The enriched GO terms of up-regulated DEGs.
(XLS)
The enriched GO terms of down-regulated DEGs.
(XLS)
Candidate genes involved in defense and detoxification mechanisms of radish roots in repose to heavy metal Pb.
(DOC)
Primers used for qRT-PCR.
(XLS)