

Abstract
The use of thrombolytic agents has greatly improved patient outcomes, but the prothrombotic response to these drugs in vivo is unknown. Approximately 24 h after we induced thrombosis in male Sprague–Dawley rats, we placed an infusion line in the inferior vena cava and administered either saline or a thrombolytic agent (tissue plasminogen activator [tPA] or plasmin) for 30 min. Blood was drawn immediately after infusion; rats were euthanized 24 h after infusion for collection of blood and tissue (inferior vena cava and thrombus). Thrombus size was decreased in the tPA-treated rats but not in those that received saline or plasmin; this change correlated with the significant rise in D-dimer levels noted immediately after infusion in the tPA-treated rats. Plasma soluble P-selectin, a prothrombotic marker, was elevated at 24 h in the plasmin group compared with the other treatment groups. There were no significant differences in plasma C3a, C5a, or C5b9 levels or in thrombus C3 levels between groups. According to ultrastructural analysis, thrombus structure and vein wall effects did not differ between groups. Local tPA did not induce a prothrombotic state during acute DVT or after thrombolytic therapy in a rodent model of venous thrombolysis. Conversely, levels of the prothrombotic marker plasma soluble P-selectin increased when plasmin was administered.
Abbreviations: DVT, deep vein thrombosis; IVC, inferior vena cava; tPA, tissue plasminogen activator
Deep vein thrombosis (DVT) is part of a disease condition known as venous thromboembolism. Approximately 360,000 new cases of DVT occur annually7,36 in the United States, and this number has not changed appreciably over the years.33 DVT can lead to serious sequelae such as chronic venous insufficiency and postthrombotic syndrome,4,13 and recurrence of thrombosis is a significant risk.12 To prevent these complications, the goals of treatment are to restore vessel patency and prevent valvular damage.24 Standard-of-care anticoagulants do not always meet both of these goals, and catheter-directed thrombolysis has arisen as an alternative treatment method.24 This technique uses placement of a multiport catheter at the site of the thrombus, with subsequent infusion of a thrombolytic agent.24 The thrombolytic agents most commonly used are plasminogen activators, such as tissue-plasminogen activator (tPA), urokinase plasminogen activator (uPA), and streptokinase. These agents work on the fibrinolytic system and catalyze the conversion of plasminogen to plasmin.6 Plasmin then binds to fibrin, which leads to breakdown of fibrin and initiates the formation of fibrin degradation products, such as D-dimer.6,10 Recently, there has been increased interest in the use of plasmin,20 due to its direct thrombolytic ability (already in the active form to break down fibrin) and enhanced safety profile when compared with plasminogen activators.21
Inflammation plays an important role in the development and progression of DVT27 and therefore has become a target for treatment of this disease. Soluble plasma P-selectin has been documented to be a biomarker of thrombosis, and increased concentrations are associated with a prothrombotic state.2,25 Several components of the innate immune system, specifically the complement system, are upregulated in the presence of thrombolytics.1,3 In addition, the presence of certain complement components (for example, C3) within the thrombus may lead to states of hypofibrinolysis.14,16 Taken together, these factors may impede successful therapy via catheter-directed thrombolysis and lead to a poorer patient outcome.
Therefore, we set out to determine whether thrombolytic agents create a prothrombotic environment during acute DVT, especially at the site of treatment with catheter-directed thrombolysis. Our hypothesis was that local and systemic prothrombotic indicators would be activated after therapy with thrombolytic agents with different mechanisms of action (indirect, recombinant tPA; direct thrombolytic, plasmin) in a rat model of venous thrombolysis.
Materials and Methods
Animal use.
For this study, male (n = 96; mean body weight, 360.3 g) Sprague–Dawley rats (Charles River Laboratories, Portage, MI) were housed 3 per cage in static microisolation cages (8 in. × 17 in. × 7 in.; Allentown Caging, Allentown, NJ) in a temperature-controlled room on a 12:12-h light:dark cycle. All environmental parameters within the room complied with the recommendations in the Guide for the Care and Use of Laboratory Animals.18 Rats were fed a commercial diet ad libitum (Lab Diet 5053, PMI Nutrition International, Brentwood, MO), and provided filtered city water ad libitum by an automated watering system. Colony health was monitored by use of a semiannual dirty-bedding sentinel system. Three sentinel rats (Sprague–Dawley) were used for every 50 to 70 cages, and these sentinels remained negative for the following infectious agents during the time period that this study was conducted: sialodacryoadenitis virus, rat parvovirus, Kilham rat virus, Toolan H1 virus, rat minute virus, Syphacia spp., Aspiculuris tetraptera, Sendai virus, pneumonia virus of mice, Theiler murine encephalomyelitis virus strain GDVII, reovirus type 3, Mycoplasma pulmonis, lymphocytic choriomeningitis virus, and mouse adenovirus. This study was part of an IACUC-approved protocol, and rats were housed in an AAALAC-accredited research facility. Rats were randomly assigned to one of the following groups: controls (that is, surgically naïve animals), n = 21; saline treatment, n = 22; tPA treatment, n = 22; plasmin treatment, n = 22; and DVT only, n = 9. Due to insufficient sample collection from some rats, some of the test parameters have fewer than the number of samples listed here.
Animal welfare.
Because the current study investigated inflammatory parameters, the use of systemic analgesic agents was contraindicated. Furthermore, multiple major survival surgeries were a component of this particular animal model, so we sought alternatives to minimize pain and distress to the animals. Bupivacaine was used along the incision site before closure after every surgical procedure. This analgesic has been shown to be effective for postoperative pain,5,40 and because it was administered locally, it likely had minimal effects on systemic inflammatory parameters.31 In addition various nursing care measures were employed to improve animal comfort including: long sipper tubes on the water bottles, rodent chow placed on the floor, warmed subcutaneous fluids given daily, and a nutritionally complete soft diet (DietGel 76A, Clear H2O, Portland, ME) administered daily. None of these treatments were used on the surgically naïve control rats.
Animal model.
The modified inferior vena cava (IVC) ligation stasis model was used to induce thrombogenesis.28 Rats were anesthetized by using 2% isoflurane gas in 100% oxygen. A midline laparotomy was performed and the IVC exposed by blunt dissection. The back branches and the IVC itself, just caudal to the renal veins, were ligated by using 6-0 polypropylene suture (Prolene, Ethicon, Somerville, NJ). The abdominal muscle and skin were closed in 2 layers. Prior to closure of the skin, bupivacaine (1 mg/kg; Hospira, Lake Forest, IL) was injected into the abdominal musculature along the incision line. The rat then was allowed to recover from anesthesia.
Twenty-four hours later, rats were reanesthetized by using the same mixture of isoflurane gas and oxygen as previously mentioned. The incision site was opened and the IVC exposed via blunt dissection. The suture on the IVC was removed, and an infusion line (27-gauge needle; Baxter, Deerfield, IL) connected to a syringe pump (Braintree Scientific, Braintree, MA) was inserted directly into the IVC at the level of the thrombus. Recombinant tPA (5 mg/kg, in sterile water; Alteplase, Genentech, San Francisco, CA), plasmin (8 mg/kg; human; plasminogen activated to plasmin by using streptokinase; lyophilized from 20 mM NaPO4 with 10 mg/mL d-mannitol and 10 mg/mL NaCl, pH 7.4; Athens Research and Technology, Athens, GA), or 0.9% saline (equivalent volume) was infused for a total of 30 min. These dosages were based on allometric scaling using previously published information21 or on previous experience within the laboratory. Surgically naïve control rats (C) were also used for baseline measurements along with rats that only underwent the first surgical procedure to induce thrombosis (DVT). After the infusion, the needle was removed and hemostasis achieved by applying pressure to the site with a sterile cotton-tipped applicator and/or use of sterile hemostatic foam (Surgifoam, Ethicon). The site was observed for at least 5 min after needle removal to ensure adequate hemostasis. Blood (1 mL) was drawn from an area on the IVC, cranial to the thrombus for analysis of D-dimer and complement (C5a, C5b9, C3a) levels (0-h time point). The abdominal muscle and skin were closed. Prior to closure of the skin, bupivacaine (1 mg/kg; Hospira) again was injected into the abdominal musculature along the incision line, to provide analgesia. Rats then recovered from anesthesia.
Twenty-four hours after infusion, rats were reanesthetized and blood was collected via cardiocentesis (24-h time point) for analysis of the following parameters: C3a, C5a, C5b9, soluble P-selectin, CBC, fibrinogen, D-dimers, thrombin clotting time, and activated partial thromboplastin time. Rats were euthanized (removal of the heart under anesthesia) and the IVC and thrombus were harvested for analysis of the following parameters: weight and length of IVC and thrombus, C5b9 levels in the vein wall, C3 levels in the thrombus, and thrombus composition via scanning electron microscopy and transmission electron microscopy.
IVC+thrombus weights.
In brief, treatment groups were analyzed for wet thrombus weight (in grams) and length (in centimeters) at the time of euthanasia, when the IVC thrombus and its associated vein wall were removed. The weights were normalized to length to ensure uniform analysis across groups.
Soluble P-selectin and plasma complement levels.
After hematologic analyses were run, the same EDTA sample was processed for ELISA assays according to the manufacturer's instructions for analysis of circulating C5a, C5b9 (NovaTeinBio, Cambridge, MA), soluble P-selectin, and C3a (Kamiya Biomedical, Seattle, WA). All samples were run in duplicate, a positive control was run for each plate, and the results were normalized to total protein by using the standard bicinchoninic acid assay (Pierce, Rockford, IL). Blood collected via direct IVC draw was placed in EDTA microtainer tubes (Becton Dickinson, Franklin Lakes, NJ) and analyzed for C5a (sensitivity,1.0 ng/mL), C5b9 (sensitivity, 1.0 ng/mL), soluble P-selectin (sensitivity, 0.1 ng/mL), and C3a (minimal detectable dose, less than 0.225 ng/mL) levels. Plasma was immediately frozen in liquid nitrogen (−196 °C) and stored at −70 °C prior to processing.
C5b9 level in vein wall.
The vein wall was separated from the thrombus, immediately frozen in liquid nitrogen (−196 °C), and stored at −70 °C prior to processing. The vein wall was homogenized in 1 mL Complete Lysis Buffer (Roche, Indianapolis, IN), sonicated for 20 s, and centrifuged at 15,000 × g for 30 min. The supernatant was processed according to the manufacturer's suggestions for the C5b9 direct sandwich ELISA kit (NovaTeinBio) to measure the amount of C5b9 contained within the vein wall (lower limit of detection, 3.12 ng/mL). All samples were run in duplicate, a positive control was run for each plate, and the results were normalized to total protein by using the standard bicinchoninic acid assay (Pierce).
C3 level in thrombus.
The thrombus was separated from the vein wall, immediately frozen in liquid nitrogen (−196 °C), and stored at −70 °C prior to processing. The thrombus was homogenized in 1 mL of Complete Lysis Buffer (Roche), sonicated for 20 seconds, and centrifuged at 15,000 × g for 30 min. The supernatant was processed according to the manufacturer's suggestions for the C3 (Kamiya Biomedical) direct sandwich ELISA kit, to measure the amount of C3 contained within the thrombus (assay range is 12.5 to 800 ng/mL). All samples were run in duplicate, a positive control was run for each plate, and the results were normalized to total protein by using the standard bicinchoninic acid assay (Pierce).
Coagulation parameters.
Blood (1.8 mL) was drawn via cardiocenesis at 24 h after thrombolysis and placed in sodium citrate vacuum phlebotomy tubes (Becton Dickinson) for analysis of thrombin clotting time, activated partial thromboplastin time, and fibrinogen by using the BBL Fibrosystem (Becton Dickinson). After these samples were run, the same sodium citrate sample was processed according to the manufacturer's suggestions for the D-dimer direct sandwich ELISA kit (Diagnostica Stago, Parsippany, NJ), to measure circulating D-dimer levels (lower limit of detection, 10 ng/mL). In addition, blood samples were collected via direct IVC draw immediately after infusion and placed in sodium citrate. Plasma was immediately frozen by using liquid nitrogen (−196 °C) and stored at −70 °C prior to processing. All samples were run in duplicate, and the results were normalized to total protein using the standard bicinchoninic acid assay (Pierce).
Hematology.
Blood was drawn via cardiocentesis and placed in EDTA microtainer tubes (Becton Dickinson) for complete blood analysis by using an automated analyzer (CDC Technologies Hemavet Multispecies Hematology Analyzer, Drew Scientific, Oxford, CT).
Transmission electron microscopy.
Segments of the IVC containing the thrombus were excised and immersion-fixed by incubation in 2.5% glutaraldehyde in 0.1 M Sorensen buffer overnight at 4°C. After several buffer rinses, the tissue was postfixed in 1% osmium tetroxide in the same buffer. Samples were rinsed in buffer and then dehydrated in ascending concentrations of ethanol, treated with propylene oxide, and embedded in epoxy resin (Epon, Momentive Specialty Chemicals, Houston, TX). Semithin sections (thickness, 5 µm) were stained with toluidine blue for tissue identification. Selected regions of interest were sectioned (thickness, 70 nm), poststained with uranyl acetate and lead citrate, and examined at 60 kV (Philips CM100, FEI Company, Hillsboro, OR). Images were recorded digitally (ORCA-HR system, Hamamatsu Photonics, Hamamatsu, Japan) operated with AMT software (Advanced Microscopy Techniques, Danvers, MA). Digital images of thrombus and vein wall were analyzed by 2 board-certified veterinary pathologists blinded to the treatment groups.
Scanning electron microscopy.
Segments of the inferior vena cava with thrombus were excised and then fixed overnight in 2.5% glutaraldehyde in 0.1 M Sorensen buffer. After a buffer rinse, samples were postfixed for 1 h in 1% osmium tetroxide in the same buffer, rinsed with buffer, dehydrated in ascending concentrations of ethanol, treated with hexamethyldisilazane, and allowed to dry. Samples were mounted on specimen mounts, sputter-coated with gold, and viewed on a field-emission scanning electron microscope at 5 kV (Amray 1910 FE, SEMTech Solutions, Billerica, MA). Digital images were collected with a SEMTech X-Stream Imaging System (SEMTech Solutions). Digital images of thrombus and vein wall were analyzed by 2 board-certified veterinary pathologists blinded to the treatment groups.
Statistics.
All statistical analyses were performed by using Prism (version 5.0c, GraphPad Software, La Jolla, CA) or SAS (version 9.2, SAS Institute, Cary, NC). All parameters underwent one-way or repeated-measures ANOVA, depending on the data set (single time point compared with 2 time points, respectively). Bonferroni and Tukey posthoc tests were used to adjust for multiple comparisons among experimental treatment groups. For all analyses, a P value of less than 0.05 was considered statistically significant. Data are presented as mean ± SEM.
Results
tPA, but not plasmin, increased venous thrombolysis.
No thrombus was present in the nonsurgical control group, and their IVC+thrombus weight (representing vein wall only) was less (P < 0.05) than those of all other groups. The DVT group had a greater (P < 0.05) IVC+thrombus weight than those of the tPA and saline groups. The IVC+thrombus weight of the tPA group was less (P < 0.05) than those of the saline and plasmin groups, and the saline group had a lower (P < 0.05) IVC+thrombus weight than that of the plasmin group. These data suggest that tPA was an effective thrombolytic agent but plasmin was not. The gross appearance of a representative thrombus in each treatment group is presented in Figure 1.
Figure 1.
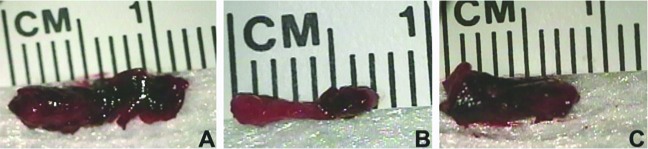
Gross appearance of the thrombus from a representative rat in the (A) saline group, (B) tPA group, and (C) plasmin group. The tPA thrombus is visibly the smallest in both length and width.
Soluble P-selectin and plasma complement levels.
In both rodents26 and humans,29 elevated plasma P-selectin levels have been associated with a prothrombotic state. Soluble P-selectin levels at the 24 h time point were significantly higher in the plasmin group compared with the control group (P = 0.0084), saline (P = 0.0354) and tPA (P = 0.0109) groups (Figure 2). Therefore, use of plasmin for treatment of DVT may lead to the creation of a prothrombotic environment. Plasma complement levels (C5a, C3a, C5b9) did not differ between any of the groups at either of the time points (0 or 24 h), except that at the 24-h time point, the saline group had a higher (P = 0.0094) C3a level than did the plasmin group (Figure 3).
Figure 2.
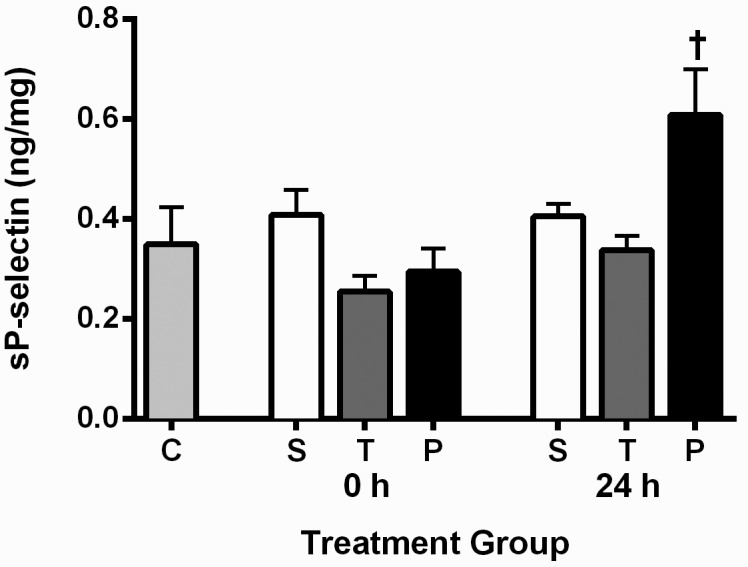
Plasma soluble P-selectin levels in all groups. Data are presented as mean ± SEM (n = 9 for all groups). Plasma samples were taken immediately (0 h) and 24 h after infusion. C, surgically naïve control; S, saline; T, tPA; and P, plasmin. †, Soluble P-selectin levels were significantly elevated in the plasmin group at the 24 h time point compared with the surgically naïve control (P = 0.0084), saline (P = 0.0354), and tPA (P = 0.0109) groups. The increased soluble P-selectin seen in the plasmin group at the 24 h time point is indicative of a prothrombotic environment.
Figure 3.

Plasma (A) C3a, (B) C5a, and (C) C5b9 levels in all groups. Data are presented as mean ± SEM (n = 11 for all groups except the saline groups were n = 12). Plasma samples were taken immediately (0 h) and 24 h after infusion. C, surgically naïve control; S, saline; T, tPA; and P, plasmin. †, saline group had a higher (P = 0.0094) C3a level at the 24 h time point than did the plasmin group.
Complement levels in thrombus and vein wall.
There were no statistically significant differences in thrombus C3 levels between the DVT only, saline, tPA, and plasmin groups (Figure 4). The DVT group had higher (P < 0.05) C5b9 levels in the vein wall than did all other groups; this unexpected finding may reflect an acute increase due to manipulation of the tissue for collection (the vessels from the saline, tPA, and plasmin groups had already been extensively manipulated).
Figure 4.
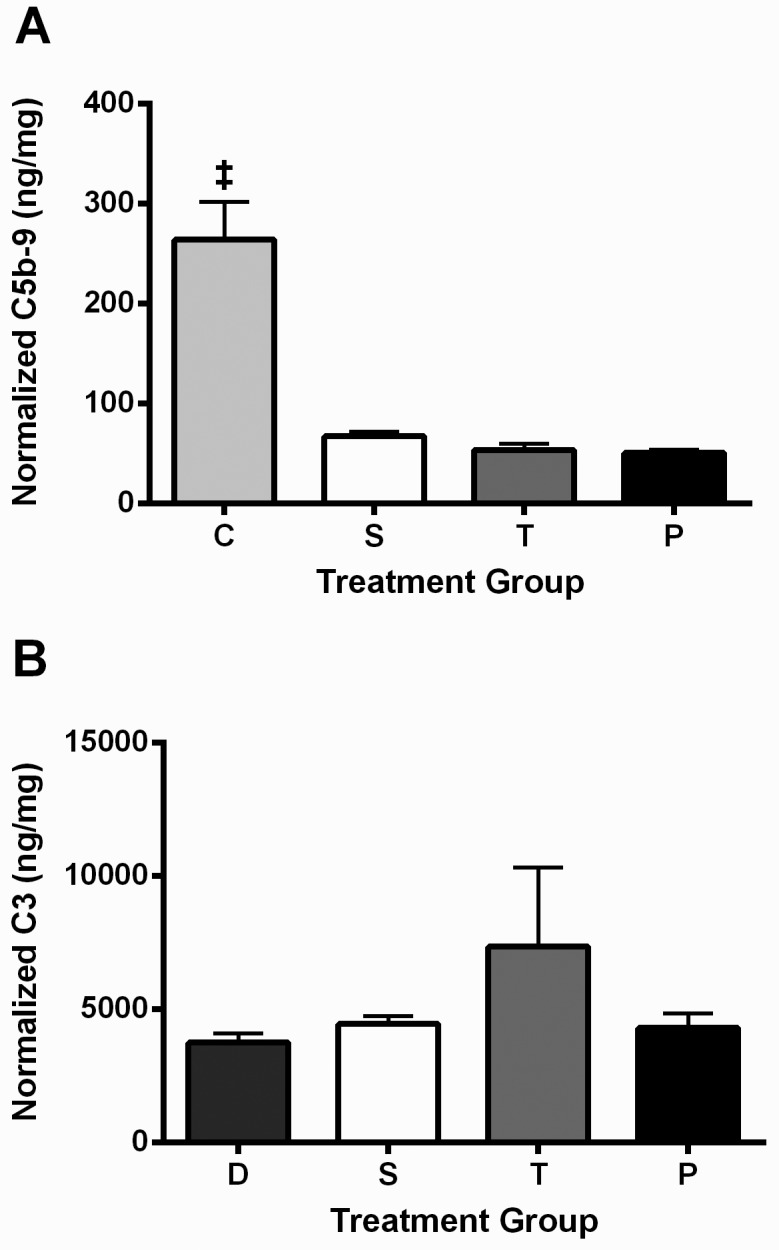
(A) Vein wall C5b9 levels and (B) thrombus C3 levels. Data are presented as mean ± SEM (n = 9 for all groups except n = 8 for the tPA and plasmin groups for thrombus C3 levels). C, surgically naïve control; S, saline; T, tPA; and P, plasmin. ‡, C5b9 level in vein wall of the surgically naïve control group was higher than those of other the treatment groups. The D group was used as the control for the thrombus C3 ELISA because the surgically naïve rats, due to their lack of a thrombus, were not an appropriate control for this particular assay.
Plasma D-dimer levels.
Derived from the interaction between plasmin and fibrin, plasma D-dimer levels are an indication of active thrombolysis.10 Plasma D-dimer levels were elevated in the tPA group immediately (0 h) after infusion compared with the control and other treatment groups at the same time point (P < 0.0001 in all cases; Figure 5), indicating that the tPA group was the only one in which active thrombolysis occurred.
Figure 5.
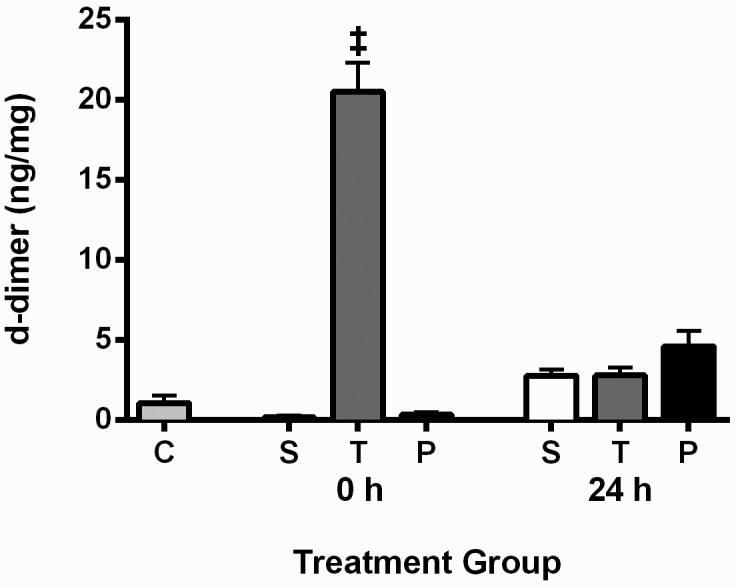
Plasma D-dimer levels for all groups. Data presented as mean ± SEM (n = 9 for all groups). Plasma samples were taken immediately (0 h) and 24 h after infusion. C, surgically naïve control; S, saline; T, tPA; and P, plasmin. ‡, D-dimer levels for the tPA treatment group at the 0 h time point were higher (P < 0.0001) than those of all other groups, indicating that tPA was causing thrombolysis.
Coagulation parameters.
Fibrinogen levels were significantly (P < 0.05) elevated in all of the treatment groups compared with the control group, indicating active thrombosis, and that of the saline group was significantly (P < 0.05) higher than that of the tPA group. Thrombin clotting time was elevated (P < 0.05) in all of the treatment groups compared with the control group, most likely because of the presence of fibrin degradation products in the treatment groups. Thrombin clotting time did not differ between treatment groups, which indicates that there were no abnormalities in the intrinsic (contact activation) or common coagulation pathways after treatment (Table 1).
Table 1.
Coagulation values
| Group | Fibrinogen (mg/dL) | TCT (s) | aPTTa (s) |
| Control | 211.9 ± 5.9b | 22.08 ± 0.3b | 15.73 ± 0.9 |
| Saline | 381.1 ± 10.7c | 25.21 ± 0.4 | 16.36 ± 0.3 |
| tPA | 330.6 ± 17.3 | 23.97 ± 0.5 | 16.42 ± 0.5 |
| Plasmin | 350.1 ± 10.1 | 24.89 ± 0.5 | 16.04 ± 0.6 |
Data presented as mean ± SEM (n = 18 for all groups except for fibrinogen measurement, where n = 9 for all groups).
aPTT, activated partial thromboplastin time, TCT, thrombin clotting time.
Not significantly different among groups.
Control value is lower (P < 0.05) than those of other groups.
Level for saline group is greater (P < 0.05) than that of the tPA group.
Hematology.
Neutrophil levels were elevated (P < 0.05) and hematocrit levels were decreased (P < 0.05) in all treatment groups compared to the control group. There were no differences in the remaining hematology values (monocytes, platelets, lymphocytes, total white blood cells) between groups (Table 2). These findings were expected given that DVT is an inflammatory process and that RBC are a large component of a thrombus.
Table 2.
Hematology values
| Group | WBC (×103/μL) | Neutrophils (×103/μL) | Monocytesa (×103/μL) | Lymphocytesa (×103/μL) | Plateletsa (×103/μL) | Hct (%) |
| Control | 9.85 ± 0.54 | 1.76 ± 0.15b | 0.50 ± 0.03 | 7.32 ± 0.37 | 687.20 ± 38.6 | 39.8 ± 0.5c |
| Saline | 10.47 ± 0.64 | 4.01 ± 0.26 | 0.38 ± 0.04 | 6.04 ± 0.44 | 593.70 ± 10.62 | 33.3 ± 0.6 |
| tPA | 10.54 ± 0.52 | 3.77 ± 0.24 | 0.39 ± 0.06 | 6.59 ± 0.45 | 607.30 ± 31.97 | 34.5 ± 0.9 |
| Plasmin | 11.05 ± 0.45 | 3.90 ± 0.19 | 0.48 ± 0.04 | 6.63 ± 0.41 | 608.70 ± 32.43 | 32.8 ± 1.1 |
Data are presented as mean ± SEM (n = 18 for all groups).
Not statistically different between groups.
Control value is lower (P < 0.05) than those for other groups.
Control value is greater (P < 0.05) those for other groups.
Ultrastructural imaging of thrombi.
There were no major differences between groups noted on transmission or scanning electron microscopy for either thrombus composition or its effect on vein wall. The main components in all thrombi were neutrophils, fibrin, and RBC (data not shown). There was also a large amount of cellular debris present, as well as scant monocytes, lymphocytes, and platelets. These findings were expected in light of the inflammatory nature of DVT.
Discussion
We used a stasis model of venous thrombosis to investigate the effects of 2 thrombolytic agents (tPA and plasmin) and their ability to produce a prothrombotic state. Elucidating the inflammatory response to thrombolytic agents is crucial to improve patient outcome by preventing serious sequelae (chronic venous insufficiency, postthrombotic syndrome)4,13 and recurrence of thrombosis.12 Our data showed that although the model worked well in regard to induction of thrombosis and administration of thrombolytic agents, there were minimal effects on the complement system at the time points evaluated. Plasma soluble P-selectin levels were significantly elevated in the plasmin group compared with other groups at 24 h after thrombolysis, a finding that may explain why the IVC+thrombus weights of the plasmin group were increased. In addition, the moderate alterations noted in various coagulation and hematologic parameters were more indicative of a state of thrombosis rather than of problems with the thrombolytic or excessive stimulation of the complement cascade. These findings prompt additional questions into the role of innate immunity in DVT as well as the differences that can be seen between the venous and arterial systems.
On activation after vascular injury, the soluble P-selectin glycoprotein relocates from secretory granules to the surfaces of platelets and endothelial cells.29 Soluble P-selectin is indicative of a prothrombotic state25,26 and is being investigated as a potential biomarker for DVT in humans.29 In addition, P-selectin inhibitors are being evaluated as treatment options for patients with DVT.23,30 Therefore, a successful DVT therapeutic agent would be one that prevents an elevation in soluble P-selectin and, ideally, decreases circulating levels of P-selectin. Plasmin does not cause systemic activation of plasminogen and is rapidly inactivated beyond the thrombus by α2-antiplasmin;8 plasmin therefore has a much wider margin of safety than that of tPA and other plasminogen activators in regard to bleeding risk.19,21,22 Therefore, plasmin seems like an ideal thrombolytic because it is safe for patients19,20 and because it would bind to and begin degrading fibrin immediately on delivery and without the need for activation.6 In addition, plasmin is an effective thrombolytic in a rabbit model of abdominal aortic thrombosis.21 However, in the current study, plasmin significantly elevated soluble P-selectin levels at the 24 h time point, whereas tPA did not at either time point. Of interest, the plasmin dose used was not effective as a thrombolytic, and these rats had a higher IVC+thrombus weight than did other groups. If plasmin initiates a systemic prothrombotic environment, this effect may negate any of the useful benefits of this compound as a therapeutic agent for DVT. In addition, P-selectin can activate the complement system by acting as a C3b-binding protein.9 This activity could further contribute to a prothrombotic state by inducing a localized inflammatory response at the thrombus site. In other studies, elevations in endogenous plasmin levels induced a proinflammatory environment by stimulating the production of cytokines, such as IL6, and the recruitment of monocytes to the site of inflammation.38,39 Therefore, plasmin may have limited usefulness as a thrombolytic agent for the treatment of DVT.
Plasmin and, by extension, tPA stimulates the complement system by cleaving C3 and C5 independently of any of the traditional complement pathways.1,3,32 This outcome is unwanted because the complement system propagates the inflammatory response and causes other local alterations,17,34,41 leading to a prothrombotic environment. All of these changes can result in restenosis or valvular damage. The presence of various complement components has been demonstrated in atherosclerosis, and higher levels of these components, especially of C5b9 (membrane attack complex), have been associated with poorer patient outcomes.11,35,42 Therefore, we considered that investigating the role of the complement system in DVT and after treatment with thrombolytic agents was a logical next step. We did not find any biologically significant activation of the complement system in any of the treatment groups. However, one of the limitations of the present study is the small number of time points that was assessed. In the future, analyzing additional acute (for example, 3 h, 6 h) time points would be helpful to better characterize the role complement plays after thrombolytic therapy. Other possibilities for the observed results are that the complement system does not play a role in venous disease, given that this environment is very different from the arterial side (lower flow, thinner vessel walls, less smooth muscle present in walls), or that the low-flow environment may promote local activation of the complement cascade and therefore render changes in plasma levels undetectable. Additional assessments of the thrombus and vein wall for other complement system components may be helpful to determine whether this effect is, in fact, local.
Although significant differences between groups were not seen, it was interesting to note that C3 was detected within the venous thrombi of all groups. Localized increases of C3 could be due to the fact that the α granules of unstimulated platelets contain large amounts of C3 that can be released into the area of thrombogenesis when platelets are activated.9 In addition, this component has been found in the thrombi of patients with various chronic diseases (for example, diabetes) and can lead to states of hypofibrinolysis, possibly due to alterations in fibrin structure.15,16 If indeed a feature of DVT, C3-induced alteration of fibrin structure may make successful treatment with plasminogen activators or plasmin very difficult as this may lead to reduced exposure of fibrin binding sites. This situation would result in impaired thrombolysis and the need to use higher doses to achieve a desirable clinical outcome. Plasminogen activators have a narrow margin of safety because they systemically stimulate plasminogen activation, leading to increased bleeding risks, with the most serious of these risks being intracranial hemorrhage.37 Using higher doses of plasminogen activators would therefore compromise patient safety.
In conclusion, we here present an effective model for thrombus induction and subsequent treatment via catheter-directed thrombolysis. There was no evidence of activation of systemic complement (C5a, C5b9, C3a) at either 0 or 24 h after thrombolytic treatment. In addition, levels of a known prothrombotic marker in DVT, soluble P-selectin,25 was significantly elevated in the plasmin group at the 24 h time point compared with the other treatment groups. Therefore, treatment with plasmin may lead to a prothrombotic state and may be what led to the increased IVC+thrombus weight in this group. These features may therefore limit the usefulness of this therapeutic agent for the treatment of DVT. In addition, our data indicated local deposition of complement (C3) in the thrombus itself, suggesting that complement activation in DVT may be localized to the site of the thrombus.15,16 No significant alterations in coagulation or hematologic parameters were noted at the time points examined, indicating that treatment effects were localized and transient. Additional research in this area is warranted to better establish the role of different thrombolytic agents in venous disease, specifically DVT, as well as examining additional acute time points (for example, 3 h, 6 h) after thrombolytic therapy to better establish the role of complement activation during thrombolytic therapy.
Acknowledgments
This study was supported by NIH 1P01HL089407–01A5, Animal Core A, and NIH 1K01 HL080962–01A5. We also thank The Unit for Laboratory Animal Medicine for sponsoring the comparative medicine training program for Dr Katherine A Shuster.
References
- 1.Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Brückner UB, Nilsson B, Gebhard F, Lambris JD, Huber-Lang M. 2010. Molecular intercommunication between the complement and coagulation systems. J Immunol 185: 5628–5636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.André P, Hartwell D, Hrachovinová I, Saffaripour S, Wagner DD. 2000. Procoagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci USA 97:13835–13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett WR, Yawn DH, Migliore PJ, Young JB, Pratt CM, Raizner AE, Roberts R, Bolli R. 1987. Activation of the complement system by recombinant tissue plasminogen activator. J Am Coll Cardiol 10:627–632 [DOI] [PubMed] [Google Scholar]
- 4.Bernardi E, Bagatella P, Frulla M, Simioni P, Prandoni P. 2001. Postthrombotic syndrome: incidence, prevention, and management. Semin Vasc Med 1:71–80 [DOI] [PubMed] [Google Scholar]
- 5.Bourget JL, Clark J, Joy N. 1997. Comparing preincisional with postincisional bupivacaine infiltration in the management of postoperative pain. Arch Surg 132:766–769 [DOI] [PubMed] [Google Scholar]
- 6.Castellino FJ, Ploplis VA. 2005. Structure and function of the plasminogen–plasmin system. Thromb Haemost 93:647–654 [DOI] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. [Internet] Data and statistics, DVT/PE–NCBDDD. [Cited 15 March 2012]. Available at: http://www.cdc.gov/ncbddd/dvt/data.html.
- 8.Collen D. 1980. On the regulation and control of fibrinolysis. Edward Kowalski Memorial Lecture. Thromb Haemost 43:77–89 [PubMed] [Google Scholar]
- 9.Del Conde I, Crúz MA, Zhang H, López JA, Afshar-Kharghan V. 2005. Platelet activation leads to activation and propagation of the complement system. J Exp Med 201:871–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaffney PJ. 1993. D-dimer. History of the discovery, characterization, and utility of this and other fibrin fragments. Fibrinolysis 7 Suppl 2:2–8 [Google Scholar]
- 11.Haskard DO, Boyle JJ, Mason JC. 2008. The role of complement in atherosclerosis. Curr Opin Lipidol 19:478–482 [DOI] [PubMed] [Google Scholar]
- 12.Heit JA, Mohr DN, Silverstein MD, Petterson TM, O'Fallon WM, Melton LJ. 2000. Predictors of recurrence after deep vein thrombosis and pulmonary embolism: a population-based cohort study. Arch Intern Med 160:761–768 [DOI] [PubMed] [Google Scholar]
- 13.Henke PK, Wakefield T. 2009. Thrombus resolution and vein wall injury: dependence on chemokines and leukocytes. Thromb Res 123: S72–S78 [DOI] [PubMed] [Google Scholar]
- 14.Hertle E, van Greevenbroek MM, Stehouwer CD. 2012. Complement C3: an emerging risk factor in cardiometabolic disease. Diabetologia 55:881–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hess K, Alzahrani SH, Mathai M, Schroeder V, Carter AM, Howell G, Koko T, Strachan MWJ, Price JF, Smith KA, Grant PJ, Ajjan RA. 2012. A novel mechanism for hypofibrinolysis in diabetes: the role of complement C3. Diabetologia 55: 1103–1113 [DOI] [PubMed] [Google Scholar]
- 16.Howes JM, Richardson VR, Smith KA, Schroeder V, Somani R, Shore A, Hess K, Ajjan R, Pease RJ, Keen JN, Standeven KF, Carter AM. 2012. Complement C3 is a novel plasma clot component with antifibrinolytic properties. Diab Vasc Dis Res 9:216–225 [DOI] [PubMed] [Google Scholar]
- 17.Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, Niho Y. 1997. C5a induces tissue factor activity on endothelial cells. Thromb Haemost 77:394–398 [PubMed] [Google Scholar]
- 18.Institute for Laboratory Animal Research 2011. Guide for the care and use of laboratory animals, 8th ed. Washington (DC): The National Academies Press [Google Scholar]
- 19.Marder VJ. 2008. Preclinical studies of plasmin: superior benefit-to-risk ratio of plasmin compared to tissue plasminogen activator. Thromb Res 122: S9–S15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marder VJ. 2009. Thrombolytic therapy for deep vein thrombosis: potential application of plasmin. Thromb Res 123: S56–S61 [DOI] [PubMed] [Google Scholar]
- 21.Marder VJ, Landskroner K, Novokhatny V, Zimmerman TP, Kong M, Kanouse JJ, Jesmok G. 2001. Plasmin induces local thrombolysis without causing hemorrhage: a comparison with tissue plasminogen activator in the rabbit. Thromb Haemost 86:739–745 [PubMed] [Google Scholar]
- 22.Marder VJ, Novokhatny V. 2010. Direct fibrinolytic agents: biochemical attributes, preclinical foundation, and clinical potential. J Thromb Haemost 8:433–444 [DOI] [PubMed] [Google Scholar]
- 23.Meier TR, Myers DD, Jr, Wrobleski SK, Zajkowski PJ, Hawley AE, Bedard PW, Ballard NE, Londy FJ, Kaila N, Vlasuk GP, Schaub RG, Wakefield TW. 2008. Prophylactic P-selectin inhibition with PSI421 promotes resolution of venous thrombosis without anticoagulation. Thromb Haemost 99:343–351 [DOI] [PubMed] [Google Scholar]
- 24.Mewissen MW, Seabrook GR, Meissner MH, Cynamon J, Labropoulos N, Haughton SH. 1999. Catheter-directed thrombolysis for lower extremity deep venous thrombosis: report of a National Multicenter Registry. Radiology 211:39–49 [DOI] [PubMed] [Google Scholar]
- 25.Myers D, Jr, Farris D, Hawley A, Wrobleski S, Chapman A, Stoolman L, Knibbs R, Strieter R, Wakefield T. 2002. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J Surg Res 108:212–221 [DOI] [PubMed] [Google Scholar]
- 26.Myers DD, Hawley AE, Farris DM, Wrobleski SK, Thanaporn P, Schaub RG, Wagner DD, Kumar A, Wakefield TW. 2003. P-selectin and leukocyte microparticles are associated with venous thrombogenesis..J Vasc Surg 38:1075–1089 [DOI] [PubMed] [Google Scholar]
- 27.Myers DD, Wakefield TW. 2005. Inflammation-dependent thrombosis. Front Biosci 10: 2750–2757 [DOI] [PubMed] [Google Scholar]
- 28.Myers DD, Jr, Henke PK, Wrobleski SK, Hawley AE, Farris DM, Chapman AM, Knipp BS, Thanaporn P, Schaub RG, Greenfield LJ, Wakefield TW. 2002. P-selectin inhibition enhances thrombus resolution and decreases vein wall fibrosis in a rat model. J Vasc Surg 36:928–938 [DOI] [PubMed] [Google Scholar]
- 29.Ramacciotti E, Blackburn S, Hawley AE, Vandy F, Ballard-Lipka N, Stabler C, Baker N, Guire KE, Rectenwald JE, Henke PK, Myers DD, Wakefield TW. 2011. Evaluation of soluble P-selectin for the diagnosis of deep venous thrombosis. Clin Appl Thromb Hemost 17:425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramacciotti E, Myers DD, Jr, Wrobleski SK, Deatrick KB, Londy FJ, Rectenwald JE, Henke PK, Schaub RG, Wakefield TW. 2010. P-selectin– PSGL1 inhibitors versus enoxaparin in the resolution of venous thrombosis: a meta-analysis. Thromb Res 125: e138–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sams VG, Lawson CM, Coan P, Bemis D, Newkirk K, Karlstad M, Norwood J, Barlow P, Goldman MH, Daley BJ. 2012. Effect of local anesthetic on microorganisms in a murine model of surgical site infection. J Trauma Acute Care Surg 73: 441–445 [DOI] [PubMed] [Google Scholar]
- 32.Schaiff WT, Eisenberg PR. 1997. Direct induction of complement activation by pharmacologic activation of plasminogen. Coron Artery Dis 8:9–18 [DOI] [PubMed] [Google Scholar]
- 33.Silverstein MD, Heit JA, Mohr DN, Petterson TM, O'Fallon WM, Melton LJ. 1998. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med 158:585–593 [DOI] [PubMed] [Google Scholar]
- 34.Sims PJ, Faioni EM, Wiedmer T, Shattil SJ. 1988. Complement proteins C5b–9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem 263:18205–18212 [PubMed] [Google Scholar]
- 35.Speidl WS, Kastl SP, Huber K, Wojta J. 2011. Complement in atherosclerosis: friend or foe? J Thromb Haemost 9:428–440 [DOI] [PubMed] [Google Scholar]
- 36.Spencer FA, Emery C, Lessard D, Anderson F, Emani S, Aragam J, Becker RC, Goldberg RJ. 2006. The Worcester Venous Thromboembolism Study: a population-based study of the clinical epidemiology of venous thromboembolism. J Gen Intern Med 21:722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart D, Kong M, Novokhatny V, Jesmok G, Marder VJ. 2003. Distinct dose-dependent effects of plasmin and TPA on coagulation and hemorrhage. Blood 101:3002–3007 [DOI] [PubMed] [Google Scholar]
- 38.Syrovets T, Lunov O, Simmet T. 2012. Plasmin as a proinflammatory cell activator. J Leukoc Biol 92:509–519 [DOI] [PubMed] [Google Scholar]
- 39.Syrovets T, Simmet T. 2004. Novel aspects and new roles for the serine protease plasmin. Cell Mol Life Sci 61:873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan CH, Kun KY, Onsiong MK, Chan MK, Chiu WKY, Tai CM. 2002. Postincisional local anaesthetic infiltration of the rectus muscle decreases early pain and morphine consumption after abdominal hysterectomy. Acute Pain 4:49–52 [Google Scholar]
- 41.Wiedmer T, Esmon CT, Sims PJ. 1986. Complement proteins C5b–9 stimulate procoagulant activity through platelet prothrombinase. Blood 68:875–880 [PubMed] [Google Scholar]
- 42.Wu G, Hu W, Shahsafaei A, Song W, Dobarro M, Sukhova GK, Bronson RR, Shi G-P, Rother RP, Halperin JA, Qin X. 2009. Complement regulator CD59 protects against atherosclerosis by restricting the formation of complement membrane attack complex. Circ Res 104:550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]