

Abstract
Studies suggest that Hsf4 expression correlates with its role in cell growth and differentiation. However, the role of Hsf4 in tumorigenesis in vivo remains unexplored. In this article, we provide evidence that absence of the Hsf4 gene suppresses evolution of spontaneous tumors arising in p53- or Arf-deficient mice. Furthermore, deletion of hsf4 alters the tumor spectrum by significantly inhibiting development of lymphomas that are normally observed in the majority of mice lacking p53 or Arf tumor suppressor genes. Using mouse embryo fibroblasts deficient in the hsf4 gene, we have found that these cells exhibit reduced proliferation that is associated with induction of senescence and senescence-associated β-galactosidase (SA-β-gal). Cellular senescence in hsf4-deficient cells is associated with the increased expression of the cyclin-dependent kinase inhibitors, p21 and p27 proteins. Consistent with the cellular senescence observed in vitro, specific normal tissues of hsf4−/− mice and tumors that arose in mice deficient in both hsf4 and p53 genes exhibit increased SA-β-gal activity and elevated levels of p27 compared with wild-type mice. These results suggest that hsf4 deletion-induced senescence is also present in vivo. Our results therefore indicate that Hsf4 is involved in modulation of cellular senescence, which can be exploited during cancer therapy.
Introduction
Heat shock factor 4 (Hsf4) is a member of the heat shock transcription factor family that contains conserved DNA binding and trimerization domains (1). Studies show that Hsf4 is transcriptionally activated during lens epithelial cell differentiation (2, 3). Hsf4 deletion affects the expression of small Hsps in lens epithelial cells (2, 3). Hsf4 mutations have been detected in humans as well as in other species and are associated with Marner, lamellar, and other forms of cataracts (4, 5). We previously reported that Hsf4b interacts with extracellular signal–regulated kinase (ERK)1/2 and is a direct target of ERK/MAPK (mitogen-activated protein kinase; ref. 6). In addition, using a yeast 2-hybrid screen, we identified a dual-specificity tyrosine phosphatase, Dusp26 that interacts with Hsf4 (6). Indeed, in mammalian cells, Hsf4 simultaneously binds to both ERK1/2 and Dusp26, suggesting that Hsf4 may potentially act as a scaffold regulating ERK1/2 signaling. Phosphorylation of Hsf4b, a transcriptional activator isoform, by ERK1/2 leads to an increase in its ability to bind DNA and stimulate transcription (6). A potential role for Hsf4 in regulation of cellular proliferation and differentiation has previously been elucidated (2, 3, 6).
Cellular senescence is characterized by irreversible growth inhibition in the G1 phase of the cell cycle in normal cells and has been expanded to include a premature triggering event through activation of oncogenes, tumor suppressor genes, oxidative stress, and DNA damage (7–11). Cellular senescence occurs both in vitro and in vivo and plays an essential role in suppressing tumor growth (8, 12, 13). Cellular senescence occurs through multiple mechanisms (9–11): replicative senescence occurs through erosion of telomeres in human cells undergoing DNA replication and enhanced expression of cyclin-dependent kinase inhibitors (CDKI; refs. 9, 14). Multiple stimuli can cause replicative senescence leading to p16/INK4a induction and stimulation of DNA damage and activation of the p53 pathway (9). Another mode of cellular senescence, called oncogene-induced senescence (OIS), occurs via expression of oncogenic c-Ha-RasV12 in human fibroblasts (11). OIS also induces p53 through activation of a DNA damage pathway and induction of p19Arf, which also leads to increased expression of p53 (11). A third mode of cellular senescence induction is through the loss of Pten. This mode of cellular senescence is not characterized by a DNA damage response but occurs via p53-dependent translation (9, 11). The Skp2 deletion in mice has recently been shown to also induce cellular senescence through an Arf-p53–independent pathway resulting from induction of Atf4, p21, and p27 (15). The precise mechanism underlying Atf4, p21, and p27 induction of the cellular senescence program is unclear. The CDKI p27 suppresses tumorigenesis in several different human tumors through inhibition of cyclin A- and cyclin E–containing CDK2 complexes (16). Deletion of Hsp70 in mice also induces cellular senescence and upregulation of p21 in a model of mammary tumorigenesis (17). Although induction of cellular senescence was originally discovered in cells in culture, senescence also occurs in vivo (9–11, 15). Discovery of methods to improve detection and quantification of cellular senescence in human tumors and additional modifiers that are involved in promoting cellular senescence are crucial in advancing the use of therapeutics in treatment of cancer and increasing the senescence response in human tumors.
In this study, we show that loss of the hsf4 gene significantly inhibits spontaneous tumorigenesis in p53- or Arf-deficient cancer models. Hsf4 deletion induces cellular senescence that is associated with an increase in the expression of the CDKIs p21 and p27.
Materials and Methods
Mice
All procedures involving mice were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee at GHSU. Generation of hsf4−/− mice (C57BL/6 genetic background) has been previously reported (2). The Trp53 mutant mice (C57BL/6 genetic background) were obtained from Jackson laboratory (Bar Harbor, ME). Hsf4 and p53 heterozygous mice were intercrossed at the F2 generation to generate mice with different genotypes (p53−/−, hsf4−/−p53−/−, p53+/−, and hsf4−/−p53+/−). The Arf-deficient mouse (B6:129; Arf−/−) line was obtained from NIH Mouse Repository (NCI-Frederick; ref. 18). The Arf+/− mice were crossed with hsf4 −/− mice and F1 progenies were intercrossed to generate a cohort of hsf4Arf-deficient mice for further observation.
For tumor-free survival determination, mice were observed 2 times per week until tumor growth was observed or mice exhibited health problems. Tumors were extracted, fixed, frozen in OCT, or in liquid nitrogen for further examination or were analyzed by flow cytometry.
Cell culture
Primary mouse embryonic fibroblasts (MEF) were prepared from 13.5-day-old (E13.5) embryos (2). Primary MEFs were stably transduced with E1A or c-Ha-RasV12 using retroviral vectors (19). Briefly, retroviral vectors containing E1A or c-Ha-RasV12 were transfected into Phoenix cells to generate retroviral particles for infection into MEFs. Positively transformed cells were selected using puromycin (2 μg/mL) or hygromycin (100 μg/mL) and cultured in Dulbecco’s Minimal Essential Medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS; ref. 19). For clonogenic assays, the appropriate number of cells were plated in tissue culture dishes and cultured for 10 days in DMEM supplemented with 10% FCS. Cells were fixed and stained with buffer containing 6% glutaraldehyde and 0.5% crystal violet. Colonies that contained 50 cells or more were counted. For the soft agar colony formation assay, 2 × 104 cells were cultured in DMEM containing 0.7% base agar and 0.35% top agar. Cultured cells were incubated at 37°C for 14 days and colonies were counted. To detect SA-β-gal activity, MEFs were fixed for 3 minutes at 25°C with 3% paraformaldehyde (3% PFA) in PBS and were incubated overnight at 37°C in SA-β-gal staining solution containing 1 mg/mL 5-bromo-4-chloro-3-indolyl- β-D-galactopyranoside (X-gal), 5 mmol/L potassium ferrocyanide, 5 mmol/L potassium ferricyanide, 150 mmol/L NaCl, 2 mmol/L MgCl2, 40 mmol/L citric acid, titrated with NaH2PO4 to pH 6.0. At the end of the incubation period, the SA-β-gal–positive cells were quantified (20). For each group, the percentage of positive cells was determined following counting at least 500 cells. To detect SA-β-gal–positive cells in vivo, 7-μm frozen tissue sections were fixed in 0.5% gluteraldehyde for 15 minutes at 25°C. Tissue sections were rinsed once with PBS and stained with SA-β-Gal in the dark for 16 hours at 25°C. After staining, the sections were rinsed with PBS and stained with eosin for 10 seconds.
Quantitative PCR
Total RNA was isolated from cells using TRIzol (Invitrogen) and reverse transcribed using Improm-II Reverse Transcription System (Promega, WI, USA). Quantitative reverse transcription PCR (qRT-PCR) reaction was conducted using iQ SYBR Green Supermix (Bio-Rad) on a Eppendorf Realplex qPCR system. Relative quantitative plots were constructed for quantity of RNA input and for each gene of interest. qRT-PCR was conducted using gene-specific primers. Primer pairs for p21 were P1-ttgcactctggtgtctgagc and P2-tctgcgcttggagtgataga, and primer pairs for p27 were P1-gtggaccaaatgcctgactc and P2-tctgttctgttggccctttt.
Histologic analyses
To prepare paraffin-embedded tissue sections, tumor tissue was extracted and fixed in 4% PFA. For other procedures, tissues were frozen in OCT or in liquid nitrogen. Hematoxylin and eosin (H&E) staining was carried out using paraffinized tissue sections (7 μm) following the standard procedures (21). Immunohistochemical staining was carried out as described previously (21). Briefly, tissue sections were blocked with 1% bovine serum albumin (BSA) for 30 minutes, incubated with primary antibody overnight at 4°C, rinsed, and incubated with biotin-conjugated secondary antibody for 1 hour at 25°C. Tissue sections were rinsed and incubated with Extravidin-Alkaline Phosphatase (Sigma) for 30 minutes at 25°C. Positively stained cells were visualized using Fast Red (Roche). Sections were counter-stained with hematoxylin. For immunofluorescence, following antigen retrieval, paraffinized tissue sections were blocked in 3% BSA for 60 minutes at 25°C, incubated with primary antibody overnight at 4°C (or 3 hours at 25°C), and then with fluorescence-conjugated secondary antibody for 1 hour at 25°C. Nuclei were stained with 4′, 6-diamidino-2-phenylindole (DAPI) reagent. Antibodies used were as follows: Ki-67 (Thermo Scientific), Desmin (Santa Cruz), Myogenin (Santa Cruz), and p27 (BD Transduction Laboratory). Terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) staining was done with ApoptoTag Red in situ Apoptosis Detection (Chemicon/Millipore). Nuclei were counterstained with DAPI reagent. Photographs were taken using a Zeiss Axio Imager. M1 microscope with a Zeiss Axio Cam MRc, or Zeiss Axio Cam HRc camera using a Axiovision 4.8 software.
Immunoblot analysis
Protein lysate was prepared from tumors and normal tissues using radioimmunoprecipitation assay buffer containing protease inhibitor cocktail (Roche Diagnostics; ref. 21). Thirty micrograms of proteins were fractionated on a SDS-polyacrylamide gel, transferred to Immobilon-P membrane (Millipore), and probed with primary antibody. Membranes were incubated with appropriate horseradish peroxidase–conjugated secondary antibody. Immunoreactive bands were detected with ECL (Amersham). Antibodies used were as follows: α-Tubulin, Atf4, p53, p21, β-actin, GFP, and Ras (Santa Cruz); pRb and Rb (Cell Signaling); E1A (Active Motif).
Statistical analysis
All in vitro experiments were repeated at least 3 times. Tumor-free survival was analyzed using Kaplan–Meier survival analysis. The data are presented as mean ± SD. Statistical significance between experimental groups was assessed using an unpaired 2-sample Student t test or log-rank (Mantel–Cox) test.
Results
Hsf4 potentiates Arf-p53–mediated tumorigenesis
Hsf4 is expressed in many tissues (lymphocytes, muscle, brain, and eye/lens) and is required for cellular proliferation and differentiation (ref. 2, and Y. Hu and N.F. Mivechi; unpublished data). Although the function of Hsf1 family member in tumorigenesis has been examined in a number of model systems, the role of Hsf4 in tumorigenesis remains elusive. To understand the function of Hsf4 in tumorigenesis, we used a p53-deficient mouse model that spontaneously develops tumors within 30 weeks. We observed a cohort of p53−/−, p53+/−, hsf4−/−p53+/−, and hsf4−/−p53−/− mice 2 times per week for signs of tumor development for up to 109 weeks. Consistent with our previous data and that of others (22–24), p53−/− mice developed spontaneous tumors within 30 weeks, and 72% of the tumors that developed in the mice were thymomas or lymphomas in origin following histologic analyses (24). Interestingly, hsf4−/−p53−/− mice exhibited significant suppression of tumor development compared with p53−/− mice (P< 0.001; Fig. 1A). At 30 weeks of age, approximately 48% (11/23) of hsf4−/−p53−/− mice were free of tumors. However, by 59 weeks of age all hsf4−/−p53−/− mice (23/23) succumbed to their tumors. This data suggested that hsf4 deficiency significantly suppresses p53-mediated tumorigenesis. Comparable results were obtained when hsf4−/−p53+/− mice were observed for a period of tumor-free survival compared with p53+/− mice (Fig. 1B). Tumors were identified in 100% of p53+/− mice (22/22) within 90 weeks. However, at the termination of the experiment (109 weeks), only 43% (6/14) of hsf4−/−p53+/− mice had been diagnosed with tumors (Fig. 1B). No tumors were detected in wild-type (n = 30) or hsf4−/− mice (n = 30) during the 2-year period of observation (data not shown). These results showed that Hsf4 is positively involved in tumorigenesis that is induced following disruption of the p53 gene.
Figure 1.
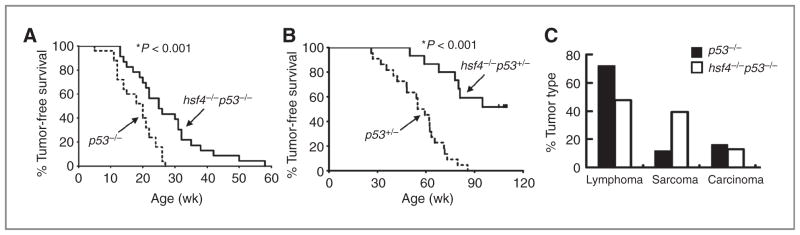
Loss of hsf4 gene suppresses tumorigenesis and alters tumor spectrum in p53-deficient mice. A and B, a cohort of, p53−/− (n = 25), hsf4−/−p53−/− (n = 23), p53+/− (n = 22), hsf4−/−p53+/− (n = 14) mice was observed for development of tumors for a period of 109 weeks. The tumor-free survival time of each genotype of mice was analyzed using Kaplan–Meier survival analysis. P < 0.001 using log-rank (Mantel–Cox) test. C, tumor spectrum for p53−/− and hsf4−/−p53−/− are indicated. The tumors were analyzed and characterized following H&E staining, immunostaining with specific markers, or analyzed by FACS. The tumors were grouped into 3 categories according to their pathologic characteristics: lymphoma/thymoma, sarcoma, and carcinoma (hepatocellular carcinoma, lung carcinoma, and digestive tract carcinoma).
We characterized the tumors that developed in the absence of p53 gene, using histopathologic, immunohistochemical, and fluorescence-activated cell sorting (FACS) analyses (22). As presented in Fig. 1C, the tumors that developed in hsf4−/−p53−/− mice exhibited pathologic diversity. Various tumor types including thymomas/lymphomas, soft tissue sarcomas, and with lesser frequency, carcinomas (liver, lung) were identified. These tumor types were also identified in p53−/− mice, indicating that hsf4 deficiency did not affect the diversity of tumors that normally develop in p53−/− mice.
Although various types of tumors were detected, the tumor spectrum observed in hsf4−/−p53−/− mice was altered compared with the tumors that were observed in p53−/− mice. As has previously been reported, predominant tumors that developed in p53−/− mice were lymphomas and thymomas (72%), and sarcomas and other tumors accounted for 12% and 16%, respectively (23–25). Interestingly, loss of hsf4 in p53−/− mice decreased the incidence of lymphomas or thymomas to 48% and increased the number of sarcomas to 40% (Fig. 1C). These results indicated that hsf4 deficiency impacts p53-mediated tumorigenesis differently, depending on the tissue and cell type.
To exclude the possibility that delayed tumorigenesis observed in hsf4−/−p53−/− was not the consequence of alterations in the tumor spectrum, we also compared tumor-free survival of mice that only developed thymomas, and lymphomas, or sarcomas, and carcinomas. Data indicated that the development of both tumor types in hsf4−/−p53−/− mice were significantly delayed compared with p53−/− mice (data not shown). This showed that suppression of tumorigenesis in hsf4−/−p53−/− mice is not due to alterations in tumor spectrum. These results further confirmed the wide spectrum of antitumor effects of hsf4 deficiency.
Hsf4 is highly expressed in skeletal muscle and is potentially involved in myoblast differentiation (Hu and Mivechi; unpublished data). Histopathologic characterization of representative tumors that were detected in p53−/− and hsf4−/−p53−/− mice are presented in Fig. 2A. Interestingly, 5 of 10 soft tissue sarcomas were positive for expression of Desmin and Myogenin, indicating that these tumors originated from skeletal muscle and showed overabundance of this tumor type among sarcomas in hsf4−/−p53−/− mice compared with p53−/− mice (Fig. 2B). To further examine the positive impact exerted by Hsf4 during tumorigenesis, we investigated whether expression of hsf4-GFP was altered in tumor tissues obtained from p53−/− or hsf4−/−p53−/− mice. In hsf4−/− mice, the expression of GFP reporter is driven by hsf4 promoter elements (2). Therefore, the level of GFP in normal muscle tissue and muscle-derived sarcomas [rhabdomyosarcoma (RMH)] were examined in wild-type or hsf4−/− tumors in the presence or absence of the p53 gene. As presented in Fig. 2C (left panel), the expression of GFP in tumors derived from hsf4−/−p53−/− mice was significantly increased in 4 of 5 tumors tested compared with GFP expression in normal muscle tissue derived from hsf4−/− mice. These results showed that hsf4 promoter-driven GFP transcription is increased in tumor tissues where the p53 gene is absent. To determine whether p53 exerts any effect on Hsf4 promoter activity, expression of GFP in adult muscle of wild-type and hsf4−/− mice, in the presence or absence of p53, was examined (Fig. 2C, right panel). Data indicated that presence or absence of p53 do not alter hsf4-GFP expression. Immunoblot analyses indicated that Hsf4a/b protein expression was also not altered in MEFs that were deficient in p53 compared with wild-type cells (data not shown). Taken together, these data showed that Hsf4 modifies p53-mediated tumorigenesis in vivo.
Figure 2.

hsf4−/−p53−/− sarcomas exhibit lower proliferative potential. A, histopathologic analyses (H&E staining) of representative tumors from the p53−/− (a, d) and hsf4−/−p53−/− (b, c, e, f) mice. (a) lymphoma, (b) HCC, (c) RMH, (d) sarcoma, (e) sarcoma, (f) lung carcinoma. Bar = 100 μm. B, characterization of RMH tumors from hsf4−/−p53−/− mice using immunohistochemical staining; (a) H&E, (b) desmin, and (c) myogenin. Bar = 100 μm. C, immunoblot analyses showing the expression of hsf4-GFP in normal muscle tissue and RMH from hsf4−/−p53−/− mice (left). Cell lysates from p53−/− tumors were used as negative control. α-Tubulin served as loading control. Right panel shows GFP expression in the normal adult muscle in the indicated genotypes. D, Ki67 immunostaining of sarcomas derived in p53−/− (n = 4) versus hsf4−/−p53−/− mice (n = 9). Ki67-positive cells were quantitated per mm2 of tissue section (right). Bars represent mean ± SD. P values were calculated using 2-tailed Student t test. Arrows show Ki67-positive cells. Bar = 10 μm. E and F, TUNEL-positive cells in representative thymoma and sarcoma derived in p53−/− or hsf4−/−p53−/− mice (n = 5–7 per group). TUNEL-positive cells were quantitated per mm2 of tissue sections (right). Bars represent mean ± SD. P values were calculated using 2-tailed Student t test. Arrows show some of the TUNEL-positive cells. Bar = 100 μm.
Tumors derived in hsf4−/−p53−/− versus p53−/− mice were characterized further in terms of proliferative capacity using Ki67 immunostaining (Fig. 2D) and cell death using TUNEL assay (Fig. 2E and F). Cellular proliferation determined by Ki67 immunostaining was significantly decreased in hsf4−/−p53−/− sarcomas compared with sarcomas of the same type that developed in p53−/− mice (Fig. 2D). We also examined whether there were differences in apoptosis that occurred in thymomas and sarcomas that could potentially explain the delayed tumorigenesis in the absence of hsf4 gene compared with wild-type. Data presented in Fig. 2E and F indicate that although there are variations in the number of apoptotic cells detected between different thymomas that were deficient in hsf4 and p53 genes, in general, there was a significantly greater number of apoptotic cells in tumors of these mice compared with those lacking p53 only. The number of apoptotic cells was also higher in sarcomas that developed in hsf4−/−p53−/− mice compared with those in p53−/− mice; however, the data did not reach a level of significance.
The INK4a/Arf locus encodes p16INK4a and p14Arf (p19Arf in mice). This locus is deleted in many human tumors (26). In addition, mice lacking one or both p16Ink4a or p19Arf spontaneously develop tumors (27, 28). Arf normally binds to mdm2 leading to stabilization of p53, and its deletion leads to lower p53 expression and cellular proliferation. Loss of Arf induces multiple types of tumors in mice (27, 28). The impact of hsf4 ablation on spontaneously developed tumors in Arf−/− mice was determined. We observed a cohort of Arf−/− and hsf4−/− Arf−/− mice and found that tumor development in hsf4−/− Arf−/− mice was significantly delayed when compared with Arf−/− mice (P = 0.04). Approximately 21% (4/19) of hsf4−/− Arf−/− mice did not develop tumors at 59 weeks of age at the termination of the experiment. Similar to p53−/− mice, the loss of hsf4 in Arf−/− mice resulted in alterations in the tumor spectrum that favored a significant reduction in the number of lymphomas/thymomas that were observed (22% in hsf4−/− Arf−/− mice vs. 85% in Arf−/− mice). However, the incidence of sarcomas (33% in hsf4−/− Arf−/− mice vs. 85% in Arf−/− mice) and other tumor types, including cancers of the colon, liver, and bone, was increased (22%) in hsf4−/− Arf−/− mice versus Arf−/− (7%) mice (Fig. 3B). Alterations in the tumor spectrum in the absence of Arf suggest that the ability and/or function of Hsf4 in tumorigenesis vary in different tissues or cell types. These results indicated that Hsf4 may carry out a critical role in promoting development of lymphomas/thymomas compared with other tumor types. Taken together, these results further confirmed the inhibitory function of hsf4 deficiency on Arf-p53–mediated tumorigenesis.
Figure 3.

Loss of hsf4 gene suppresses tumorigenesis and alters tumor spectrum in Arf-deficient mice. A, a cohort of Arf−/− (n = 15) and hsf4−/− Arf−/− (n = 19) mice was monitored for development of tumors for 59 weeks. The tumor-free survival was estimated using Kaplan–Meier survival analysis. The tumor types were determined by histologic examination and FACS analyses.*, P =0.04 was determined using log-rank (Mantel–Cox) test. B, percent tumor spectrum observed in Arf−/− and hsf4−/− Arf−/− mice. The tumors were grouped into 3 categories according to the pathologic characteristics: lymphoma/thymoma, sarcoma, and carcinoma (hepatocellular carcinoma, uterine, and colon). No tumors were found in 4 of 19 hsf4−/− Arf−/− mice during observation period of 59 weeks. C, representative histopathologic analyses using H&E staining of tumors from Arf−/− mice (a, lymphoma; c, HCC) and hsf4−/− Arf−/− (b, RMH; d, HCC) are presented. Bar = 100 μm.
Hsf4p53-deficient MEFs exhibit reduced ability to proliferate or to undergo full transformation
Delayed tumorigenesis that was observed in hsf4−/−p53−/− mice, and the fact that tumors that developed in hsf4−/−p53−/− mice proliferated more slowly compared with p53−/− mice, and because ectopic expression of c-Ha-RasV12 in murine fibroblasts is known to cooperate with loss of p53 to immortalize cells (15, 19) prompted us to investigate the proliferation, transformation potential, and cellular senescence response of p53−/− compared with hsf4−/−p53−/− MEFs. Thus, we prepared MEFs from wild-type (hsf4+/+), hsf4−/−, p53−/−, and hsf4−/−p53−/− embryos. We found that primary MEFs deficient in hsf4 (passage 3) exhibit a slower growth rate compared with wild-type MEFs but did not reach a significant level (Fig. 4A and B). Interestingly, MEFs deficient in both hsf4 and p53 genes exhibited senescence and apoptosis between passages 2 to 7 compared with MEFs deficient in p53 alone (data not shown). Therefore, to analyze these cells further and stably transform them using oncogenes, as has been described below, we used cells deficient in p53, or hsf4 and p53 (with or without oncogenes), using passage 10. Nevertheless, even at this passage the slower proliferation rate observed in hsf4−/−p53−/− MEFs was significant compared with p53−/− MEFs (Fig. 4A and B). To determine whether the slower proliferation rate can be overcome following over-expression of c-Ha-RasV12, we examined the proliferation rate of hsf4−/−p53−/− cells expressing c-Ha-RasV12 compared with p53−/− cells expressing Ha-RasV12. The data indicated that cells deleted in the hsf4 gene still proliferate more slowly than the cells expressing hsf4 gene (Fig. 4A and B). This indicated that the proliferative defect observed in hsf4−/− cells cannot be overcome by p53 deficiency and expression of Ha-RasV12. Coexpression of E1A and Ha-RasV12 in cells overcomes cellular senescence induced by Ha-RasV12, which, as noted above, is a p53-dependent cellular senescence pathway (19). The data presented in Fig. 4C and D indicate that coexpression of E1A and Ha-RasV12 could not entirely rescue the slow proliferation ability of hsf4−/− cells. These data indicated that deletion of the hsf4 gene leads to retardation of cellular proliferation, even in the presence of an oncogene and loss of p53 tumor suppressor gene, or when 2 cooperating oncogenes are expressed in these cells. Immunoblot analyses presented in Fig. 4B–D confirm expression of Ha-RasV12 and E1A in the appropriate MEF cell lines.
Figure 4.

Loss of hsf4 gene leads to inhibition of MEF proliferation in the presence of oncogenic Ha-Ras or loss of p53. A–D, representative growth curves of MEFs for the indicated genotypes infected with empty vector or with Ha-RasV12 or E1A. A total of 2 × 105 cells were cultured, and cell number was determined for a period of 6 days. Data represents mean ± SD. P values were determined using 2-tailed Student t test. P < 0.001 for all groups; except for hsf4+/+ and hsf4−/− cells expressing E1A where P < 0.01. Phase contrast photographs showing representative culture dish for each group are presented in panels B and D. Immunoblot analyses show the expression of E1A and Ras in the specified cell lines. Presence or absence of these proteins has been shown as positive or negative control. β-Actin represents loading control.
Coexpression of E1A plus Ha-RasV12, or as noted above, p53 loss plus ectopic expression of Ha-RasV12 in MEFs leads to cellular transformation (19). To examine the cellular transformation potential of cells without the hsf4 gene, we carried out colony formation assays in tissue culture and anchorage-independent growth in soft agar (Fig. 5A and B). The data indicated that cells deficient in hsf4, but transformed with E1A plus Ha-RasV12 or deficient in p53−/−, exhibited significantly less plating efficiency (Fig. 5A) as well as a lesser capacity to form colonies in soft agar (Fig. 5B). The data showed that, indeed, cells harboring loss of both hsf4 and p53 genes, but expressing Ha-RasV12, exhibited significant reduction in their ability to form colonies on culture dish or in soft agar (Fig. 5). Interestingly, hsf4−/− cells harboring both E1A and Ha-RasV12 showed approximately comparable levels (although reduced) to hsf4+/+ MEFs carrying these oncogenes in terms of transformation ability as detected by colony formation in soft agar (Fig. 5B). These data indicated that deletion of the hsf4 gene significantly reduces the transformation ability of MEFs deficient in p53 gene even when they express Ha-RasV12, as measured by anchorage-independent growth.
Figure 5.

Loss of hsf4 in MEF reduces anchorage-independent cell growth. A, colony formation assay: 102 MEF cells from the indicated genotypes were plated and cultured for 10 days. Colonies were stained by crystal violet, and the total number of colonies containing 50 cells or more were quantitated. Left panel shows representative culture plate for the indicated cell lines. Right panel shows average plating efficiency (number of colonies counted/number of cells plated) calculated from 3 independent experiments. Data represent mean ± SD. P values were determined using 2-tailed Student t test.*, P < 0.05, ***, P <0.001. B, soft agar assay: 2 × 104 cells of the indicated genotypes were plated and cultured on a plate containing 0.7% base agar and 0.35% top agar at 37°C for 14 days. Total number of colonies was quantified. All experiments were repeated at least 3 times. Left panel shows representative plates with colonies formed on soft agar. Right panel shows average colony numbers quantified per plate averaged from 3 independent experiments. Data represents mean ± SD. P values were determined using 2-tailed Student t test; **, P < 0.01; ***, P < 0.001.
Hsf4-deficient cells exhibit enhanced cellular senescence that is associated with increased expression of p21 and p27 and this, in part, is independent of cellular p53 status
The reduced ability of hsf4−/−p53−/− MEFs to proliferate and form colonies on soft agar, even in the presence of Ha-RasV12, prompted us to determine whether hsf4-deficient cells undergo cellular senescence. Thus, we examined SA-β-gal activity in passage 3 primary MEFs. The data showed a significant increase in SA-β-gal in primary MEFs deficient in the hsf4 gene (Fig. 6A). The hsf4−/−p53−/− MEFs also exhibited significant increase in SA-β-gal activity compared with hsf4+/+p53−/− cells (Fig. 6B). Hsf4-deficient MEFs carrying no p53 gene and expressing Ha-RasV12 also showed cellular senescence, but not significantly more than those cells expressing the hsf4 gene (data not shown). Quantification of the data is presented in the panels below the figures indicating that the senescence in hsf4−/−p53−/− MEFs is independent of p53 status.
Figure 6.

Loss of hsf4 increases cellular senescence that is associated with upregulation of p21 and p27. A and B, wild-type and hsf4−/− primary (A), p53−/−, or hsf4−/−p53−/− without Hsf4b (B), MEFs were stained for SA-β-gal activity. Arrows indicate positively stained cells. Panel B also shows SA-β-gal activity in hsf4−/−p53−/− MEFs that were transduced stably with Hsf4b cDNA showing the reversal of cellular senescence. Percent number of SA-β-gal–positive cells from 3 independent experiments are presented. Bar graphs represent mean ± SD. P values were determined using 2-tailed Student t test; **, P < 0.01. Magnification 400×. C and D, immunoblot analyses of MEFs isolated from the indicated genotypes probed for expression of senescence-specific cellular markers. β-Actin is presented as loading control. E, qPCR showing relative the mRNA levels of p21 and p27 in MEFs for the indicated genotypes. Bar graphs represent mean ± SD. Level of GAPDH mRNA was used as control.
Expression of a number of proteins has been shown to affect cellular senescence in a p53-dependent and p53-independent manner (15, 19, 29). Immunoblotting experiments presented in Fig. 6C indicate that expression of p53, p21, and p27 was higher in primary MEFs deficient in the hsf4 gene, which may explain increased cellular senescence observed in these cells. Increased expression of p21 and p27 (4- and 3-fold, respectively) proteins still persisted in cells deficient in both the hsf4 and p53 genes compared with cells that contained the hsf4 gene (Fig. 6C). Although expression of p21 was higher (3-fold) in hsf4−/−p53−/− cells expressing HaRasV12, expression of p27 was comparable between these cells and cells expressing Hsf4 (data not presented). Expression of Atf4 that has been previously correlated with increased cellular senescence in a p53-independent manner did not show significant changes (15). As noted above, hsf4−/− primary MEFs exhibited increased p53 levels, partly increasing the p21 expression level. However, the p21 expression levels persisted in hsf4−/−p53−/− cells without, or with, ectopic expression of Ha-RasV12 (data not presented), suggesting that increased expression of p21 in the absence of the hsf4 gene may be one reason for the cellular senescence observed in these cells. The p-Rb level was elevated in both hsf4+/+ primary MEFs and in hsf4+/+p53−/− cells, and its levels decreased in the absence of the hsf4 gene, indicating the presence of cell-cycle block in the absence of hsf4 gene (30). These data indicated that absence of the hsf4 gene leads to increased cellular senescence and this, at least in part, is dependent on p21 expression.
To determine whether Hsf4 deficiency is the cause of the increased cellular senescence, and the higher expression levels of p21 and p27 in MEFs, we ectopically expressed Hsf4b (transcriptional activator isoform) in hsf4−/−p53−/− cells. Data presented in Fig. 6B indicate a significant reduction in the number of SA-β-gal–positive cells. In addition, immunoblotting experiments presented in Fig. 6D indicate that expression of both p21 and p27 returns to the levels observed in p53−/− MEFs when Hsf4b is overexpressed in hsf4−/−p53−/− MEFs. We also asked whether there was an increase in the levels of p21 and p27 mRNAs in hsf4−/− MEFs in the presence, or absence of p53 gene, which could explain the increase in the protein levels for these genes. We found that the p21 and p27 mRNA expression levels were comparable in the presence, or absence of p53 gene between wild-type and hsf4−/− MEFs (Fig. 6E). However, although mRNA expression level of p21 was reduced in the absence of p53, there was no reduction in the levels of p27 mRNA in the absence of p53 gene in wild-type and hsf4−/− MEFs. These data could suggest an increase in p21 and p27 protein stability in the absence of hsf4 gene (Fig. 6E).
Hsf4 deficiency induce senescence in vivo
The fact that Hsf4 deficiency in the primary as well as in p53-deficient MEFs induces cellular senescence in vitro prompted us to examine whether tumor suppression could be due to activation of cellular senescence in vivo. We therefore examined a number of tissues from the 4-month-old wild-type and hsf4−/− mice for the presence of SA-β-gal activity. The data indicated that we could detect increased SA-β-gal activity in tissues such as thymus, liver, and kidney of hsf4−/− mice compared with wild-type mice (Fig. 7A). A number of other tissues, such as spleen, was also positive for SA-β-gal activity in hsf4−/− mice whereas other tissues, such as skin and small intestine, were negative for SA-β-gal in both hsf4−/− and wild-type mice (data not presented).
Figure 7.

Hsf4 deficiency mediates senescence in normal and tumor tissue in vivo. A, representative normal tissue sections stained for SA-β-gal activity (blue staining). Bar = 10 μm. B, representative tumor tissue sections stained for SA-β-gal activity (blue staining). Bar = 10 μm. C, immunofluorescence staining of p27 and p21 in representative histologic sections of thymomas (n = 4–6) and sarcomas (n = 5–7) that developed in p53 and hsf4p53-deficient mice. Arrows show representative cells with nuclear staining for p27 or p21. Bar = 5 μm.
Cellular senescence that is also induced in vivo can lead to tumor suppression (14, 15). We then examined whether SA-β-gal activity can be detected in sections of thymomas and sarcomas derived from p53−/− or hsf4−/−p53−/− mice. The data presented in Fig. 7B indicate that indeed tumors arising in hsf4−/−p53−/− mice exhibit SA-β-gal activity in contrast to those same tumor types that developed in p53−/− mice, which were devoid of SA-β-gal activity. The presence of cellular senescence in sarcomas and thymomas derived from hsf4−/−p53−/− mice indeed were associated with increased expression of nuclear p27 compared with those tumors derived in p53−/− mice (Fig. 7C, arrowheads). The p21 expression in sarcomas and thymomas derived from hsf4−/−p53−/− mice were more complex and seemed to be associated with the nuclei in some cells and cytoplasm in others (Fig. 7C, arrowheads show nuclear staining). The p53−/− tumors that were tested did not express p21 or p27 (Fig. 7C).
These data indicated that specific tissues of hsf4−/− mice exhibit some degree of cellular senescence in contrast to wild-type mice in which this was not observed. In addition, increased cellular senescence could also be detected in thymomas and sarcomas that developed in hsf4−/−p53−/− mice. These data indicated that induction of cellular senescence may contribute to suppression of tumorigenesis observed in hsf4-deficient mice.
Discussion
Hsf4 is widely expressed in many tissues including the hematopoietic compartment (31). Hsf4 expresses in 2 isoforms that have activator (Hsf4b) and repressor (Hsf4a) properties; however, the significance of these splice variants in terms of cellular physiology is not entirely understood. Hsf4 regulates the expression of Hsps and has been shown to regulate differentiation and proliferation of lens fiber cells and lens epithelial cells in mice (2, 3). Hsf4 is also expressed in C2C12 myoblasts and regulates their differentiation (Hu and Mivechi; unpublished observation). Our studies show that Hsf4 is expressed in 89% of CD8+ and 87% of CD4+ T cell population and seems to express at low levels in B220+ B cells (31).
Hsf1, another Hsf family member, has been shown to significantly promote tumorigenesis (22, 32, 33). However, the function of Hsf4 in the tumorigenesis process remains unexplored. To reveal the role of Hsf4 in Arf-p53–mediated tumorigenesis, we observed cohorts of hsf4p53 and hsf4Arf doubly deficient mice and found significant inhibition of tumorigenesis in mice deficient for the hsf4 gene compared with mice that express wild-type levels of Hsf4. As noted above, Hsf4 is highly expressed in T cells and its deletion significantly delayed development of lymphomas and thymomas in both Arf- and p53-deficient models. Arf-deficient mice exhibited less delay in tumorigenesis compared with p53-deficient mice in absence of hsf4 gene. This could be due to the fact that these mice were in mix genetic background compared with hsf4p53-deficient mice that were in C57BL/6. The tumorigenesis phenotype observed in hsf4p53-deficient mice compared with p53-deficient mice is comparable with our previously published data, suggesting that p53 deficiency significantly reduced development of lymphomas in the absence of the hsf1 gene as well (22). The likely cause of reduction in thymomas and lymphomas in the absence of the hsf4 (or hsf1) gene when p53 is lost may be due to the induction of apoptosis (Fig. 2) and senescence (Fig. 7) in lymphoid cells that undergo transformation. This is because both Hsf1 and Hsf4 regulate expression of Hsps that not only protect cells against apoptosis but also their absence induces cellular senescence (2, 3, 17, 22, 34, 35). Analyses of various cellular compartments of lymphoid tissues of normal mice deficient in the hsf4 gene did not reveal any overt defects in the number of lymphoid cells in the absence of the hsf4 gene (data not shown); however, we were able to detect some evidence of SA-β-gal activity in the thymus and spleen, suggesting the presence of cells undergoing senescence. Hsf4 is also expressed in myoblasts and in skeletal muscle, and Hsf4 expression is enhanced in muscle progenitors C2C12 cells during their differentiation (data not shown). The role of Hsf4a (transcriptional repressor) and Hsf4b (transcriptional activator) isoforms in myoblast differentiation is currently under investigation in our laboratory (Hu and Mivechi; unpublished data).
Induction of cellular senescence that occurs in vivo can potentially lead to suppression of tumor cell proliferation. Induction of Arf/p53-dependent cellular senescence following overexpression of mutant Ras or by inactivation of pten is the major pathway that limits tumor progression (36). However, p53-independent cellular senescence has also been discovered, potentially increasing the number of pathways that can be targeted in tumor cells to induce senescence (36). Interestingly, MEFs derived from hsf4−/− mice exhibit a lower level of cellular proliferation and transformation ability, as well as enhanced cellular senescence that is, in part, independent of p53. Notably, expression of 2 oncogenes (E1A and Ha-RasV12) in hsf4-deficient cells could partly overcome the reduction in transformation and cellular senescence that is observed in the absence of the hsf4 gene. However, cellular proliferation was nevertheless significantly slower in hsf4-deficient cells expressing E1A and Ha-RasV12 than those cells that carried the hsf4 gene.
Our explanation for the increase in cellular senescence in the absence of the hsf4 gene is that hsf4−/− cells express high levels of the CDKIs p21 and p27 in primary MEFs as well as following deletion of the p53 gene. Examples of p21 expression in cells that exhibit senescence have recently been reported (9, 15, 17, 30). In the study by Guo and colleagues, authors show that expression of TAp63 isoform in cells induces cellular senescence through Arf/p53-independent pathway, but through an increase in the expression of p21 and p-Rb. TAp63-associated cellular senescence overrides the Ras-induced cellular transformation observed in p53−/− cells inhibiting cellular transformation (30). Our data show that p21 is highly expressed in hsf4−/− primary MEFs compared with wild-type MEFs, and this correlates with the induction of p53 in these cells. However, hsf4−/−p53−/− cells also express high levels of p21, suggesting that hsf4-induced senescence in part occurs independent of p53 but dependent on p21. The elevated p21 expression was still persistent when Ha-RasV12 was expressed in hsf4−/−p53−/− cells, and this correlated with reduced cellular proliferation and reduced ability of these cells to form colonies in soft agar compared with cells that carried the hsf4 gene. These data indicate that hsf4p53 deficiency plus overexpression of Ha-RasV12 is perhaps comparable with TAp63 expression in cells in which in spite of a reduction in cellular senescence, the transformation ability of MEFs is still compromised (30).
The p27 expression level is tightly controlled by several signaling pathways. Inhibition of the Ha-Ras/MAPK pathways has been shown to increase the levels of p27, resulting in inhibition of cellular proliferation (16). An increase in nuclear p27 level has also been shown to induce cellular senescence (15, 16, 37). Overexpression of p27 inhibits prostatic intraepithelial neoplasia to invasive prostate cancer in vivo (37). Both p21 and p27 proteins are degraded by Skp2 E3 ligase, and skp2−/− cells in the presence of tumor suppressors exhibit increased cellular senescence (15). Reduction in the levels of molecular chaperones (Hsp25 and αB-crystallin) reduces protein degradation via ubiquitin proteasome system (38–40), and elevated levels of p21 and p27 in the absence of hsf4 gene could be the result of inefficient degradation of these CDKIs as well. Previous studies indicate that Hsf4 regulates the expression of Hsp25 and αB-crystallin (2, 3).
In conclusion, we show that loss of the hsf4 gene suppresses Arf/p53-mediated tumorigenesis. Hsf4 inhibition induces cellular senescence, but inhibits cellular proliferation and transformation that is in part dependent on the expression of p21 and p27, and is independent of p53. This indicates that Hsf4 most likely impacts cellular senescence both before and after tumorigenesis has been initiated. Thus, similar to other Hsf family members, Hsf4 may be a good target for tumor therapy.
Acknowledgments
Grant Support
This work was supported by NIH grants CA132640 and CA062130 and in part by VA Award 1I01BX000161 to N.F. Mivechi and NIH CA121951 grant to D. Moskophidis.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: X. Jin, D. Moskophidis, N.F. Mivechi.
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): X. Jin, B. Eroglu, W. Cho.
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, and computational analysis): X. Jin, W. Cho, N.F. Mivechi.
Writing, review, and/or revision of the manuscript: X. Jin, N.F. Mivechi.
Development of methodology: B. Eroglu, Y. Yamaguchi.
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): W. Cho.
Study supervision: D. Moskophidis, N.F. Mivechi.
References
- 1.Nakai A, Tanabe M, Kawazoe Y, Inazawa J, Morimoto RI, Nagata K. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol Cell Biol. 1997;17:469–81. doi: 10.1128/mcb.17.1.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Min JN, Zhang Y, Moskophidis D, Mivechi NF. Unique contribution of heat shock transcription factor 4 in ocular lens development and fiber cell differentiation. Genesis. 2004;40:205–17. doi: 10.1002/gene.20087. [DOI] [PubMed] [Google Scholar]
- 3.Fujimoto M, Izu H, Seki K, Fukuda K, Nishida T, Yamada S, et al. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J. 2004;23:4297–306. doi: 10.1038/sj.emboj.7600435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bu L, Jin Y, Shi Y, Chu R, Ban A, Eiberg H, et al. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet. 2002;31:276–8. doi: 10.1038/ng921. [DOI] [PubMed] [Google Scholar]
- 5.Enoki Y, Mukoda Y, Furutani C, Sakurai H. DNA-binding and transcriptional activities of human HSF4 containing mutations that associate with congenital and age-related cataracts. Biochim Biophys Acta. 2010;1802:749–53. doi: 10.1016/j.bbadis.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Hu Y, Mivechi NF. Association and regulation of heat shock transcription factor 4b with both extracellular signal-regulated kinase mitogen-activated protein kinase and dual-specificity tyrosine phosphatase DUSP26. Mol Cell Biol. 2006;26:3282–94. doi: 10.1128/MCB.26.8.3282-3294.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 8.Ohtani N, Mann DJ, Hara E. Cellular senescence: its role in tumor suppression and aging. Cancer Sci. 2009;100:792–7. doi: 10.1111/j.1349-7006.2009.01123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Callado M, Serrano M. Senescence in tumors: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–7. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Callado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–33. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Lanigan F, Geraghty JG, Bracken AP. Transcriptional regulation of cellular senescence. Oncogene. 2011;30:2901–11. doi: 10.1038/onc.2011.34. [DOI] [PubMed] [Google Scholar]
- 12.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 13.Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20:150–5. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Cosme-Blanco W, Shen MF, Lazar AJ, Pathak S, Lozano G, Multani AS, et al. Telomere disfuction suppresses spontaneous tumorigenesis in vivo by initiating p53-dependent cellular senescence. EMBO Report. 2007;8:497–503. doi: 10.1038/sj.embor.7400937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–9. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res. 2011;17:12–8. doi: 10.1158/1078-0432.CCR-10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng L, Hunt C, Yaglom JA, Gabai VL, Sherman MY. Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene. 2011;30:2836–45. doi: 10.1038/onc.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 19.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 20.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eroglu B, Moskophidis D, Mivechi NF. Loss of Hsp110 leads to age-dependent tau hyperphosphorylation and early accumulation of insoluble amyloid beta. Mol Cell Biol. 2010;30:4626–43. doi: 10.1128/MCB.01493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Min JN, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF. Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene. 2007;26:5086–97. doi: 10.1038/sj.onc.1210317. [DOI] [PubMed] [Google Scholar]
- 23.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 24.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 25.Harvey M, McArthur MJ, Montgomery CA, Jr, Bradley A, Donehower LA. Genetic background alters the spectrum of tumors that develop in p53-deficient mice. FASEB J. 1993;7:938–43. doi: 10.1096/fasebj.7.10.8344491. [DOI] [PubMed] [Google Scholar]
- 26.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 27.Sharpless NE, Ramsey MR, Balasubramanian P, Castrillon DH, DePinho RA. The differential impact of p16(INK4a) or p19(ARF) deficiency on cell growth and tumorigenesis. Oncogene. 2004;23:379–85. doi: 10.1038/sj.onc.1207074. [DOI] [PubMed] [Google Scholar]
- 28.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–22. [PubMed] [Google Scholar]
- 29.Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogen signaling. Genes Dev. 1998;12:3008–19. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo X, Keyes WM, Papazoglu C, Zuber J, Li W, Lowe SW, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol. 2009;11:1451–7. doi: 10.1038/ncb1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin X, Eroglu B, Moskophidis D, Mivechi NF. Targeted deletion of hsf1, 2, and 4 genes in mice. Methods Mol Biol. 2011;787:1–20. doi: 10.1007/978-1-61779-295-3_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin X, Moskophidis D, Mivechi NF. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011;14:91–103. doi: 10.1016/j.cmet.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–18. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;2:476–83. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- 35.Xanthoudakis S, Nicholson DW. Heat-shock proteins as death determinants. Nat Cell Biol. 2000;2:E163–5. doi: 10.1038/35023643. [DOI] [PubMed] [Google Scholar]
- 36.Chan CH, Gao Y, Moten A, Lin HK. Novel ARF/p53-independent senescence pathways in cancer repression. J Mol Med (Berl) 2011;89:857–67. doi: 10.1007/s00109-011-0766-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14:146–55. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin X, Moskophidis D, Hu Y, Phillips A, Mivechi NF. Heat shock factor 1 deficiency via its downstream target gene alphaB-crystallin (Hspb5) impairs p53 degradation. J Cell Biochem. 2009;107:504–15. doi: 10.1002/jcb.22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Homma S, Jin X, Wang G, Tu N, Min J, Yanasak N, et al. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J Neurosci. 2007;27:7974–86. doi: 10.1523/JNEUROSCI.0006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin DI, Barbash O, Kumarm KG, Weber JD, Harper JW, Klein-Szanto AJ, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF (FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355–66. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]