

Abstract
Translocations occur through the aberrant joining of large stretches of non-contiguous chromosomal regions. The substrates for these illegitimate rearrangements can arise as a result of damage incurred during normal cellular processes, such as transcription and replication, or through the action of genotoxic agents. In lymphocytes many translocations bear signs of having originated from abnormalities introduced during programmed recombination. Although recombination is tightly controlled at different levels, mistakes can occur leading to cytogenetic anomalies that include deletions, insertions, amplifications and translocations, which are an underlying cause of leukemias and lymphomas. In this review we focus on recent studies that provide insight into the origins of translocations that arise during the two lymphocyte specific programmed recombination events: V(D)J and class switch recombination (CSR).
Introduction
Lymphocytes undertake two distinct programmed recombination events to generate diversity within their antigen receptor loci. The first occurs during B and T cell development, targeting the variable (V), diversity (D) and joining (J) gene segments of the immunoglobulin (Ig) or T cell receptor (Tcr) loci to create a vast repertoire of receptors that form the foundations of the adaptive immune response. Rearrangement involves recombinase (RAG1/2) mediated cutting and rejoining of different combinations of V, D and J gene segments that leads to the genesis of distinct loci. The products of functionally rearranged immunoglobulin (Igh, Igk and Igl) and T cell receptor (Tcra, Tcrb, Tcrd and Tcrg) loci are expressed respectively as single specificity B and T cell receptors, that recognize and combat foreign antigen and protect against disease [1–6] (Figure 1).
Figure 1. Programed genomic rearrangements in the immune system.
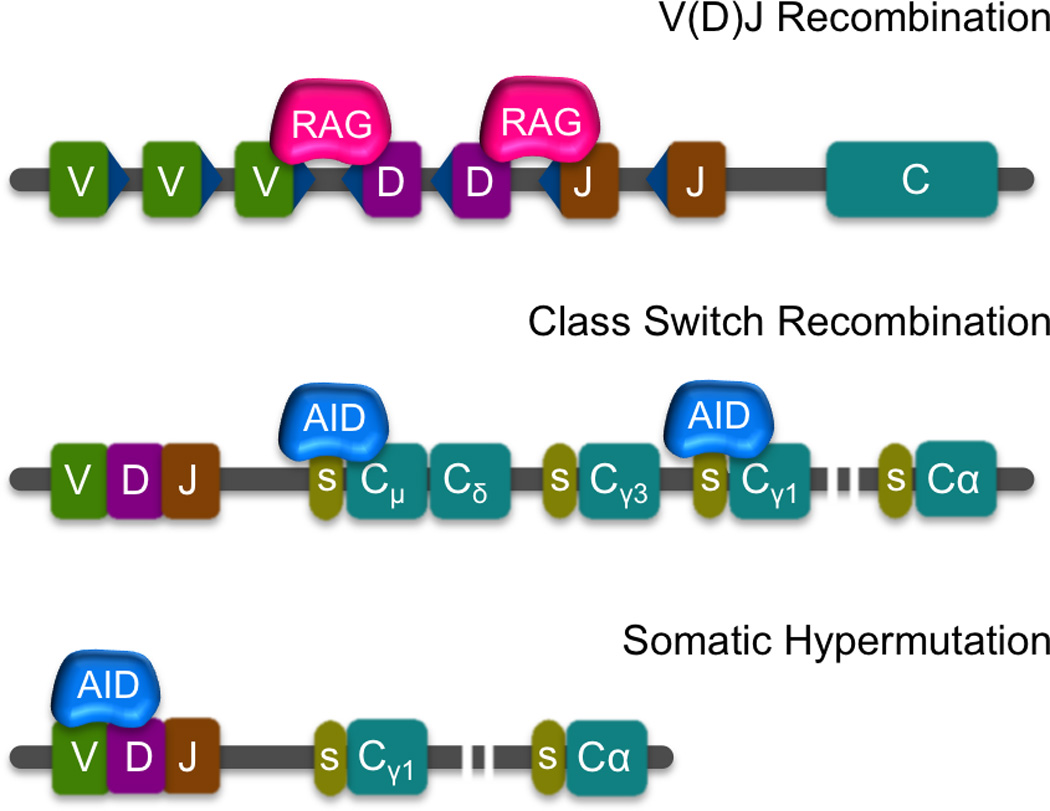
V(D)J recombination is catalyzed by RAG which targets recombination signal sequences (RSS – blue triangles) flanking V,D and J segments in developing B and T cells. The different combinatorial outcomes generate a wide variety of antigen recognition sequences. Class Switch Recombination (CSR) and Somatic Hypermutation (SHM) occur in mature B cells and are both initiated by AID. CSR exchanges the constant region of Igh from Cµ regions such as Cγ1. This occurs after AID induces DSBs on the switch regions (green ovals) located upstream of each constant region, promoting recombination and deletion of the intervening sequences. SHM results in mutations in the variable portion of the immunoglobulin loci that can increase affinity towards antigen.
In B cells a second recombination event, class switch recombination (CSR), occurs in mature germinal center B cells after contact with antigen. CSR targets the C terminal end of the V(D)J rearranged Igh locus, replacing the Igµ constant region with another constant region gene (Igγ1, Igα or Igε, etc) (Figure 1). This process alters the effector function of the antibody molecule so that the immune system is able to mount a tailored response to whichever antigen is present [7–9]. In addition, the introduction of mutations that target the variable regions of all Ig loci, can lead to high affinity antibody molecules as a result of somatic hypermutation (SHM) [10–12] (Figure 1). Although both CSR and SHM are mediated by the activation-induced cytidine deaminase enzyme, AID [13–16] (Figure 1), DSBs are normally only introduced during switching. AID acts on single-stranded DNA in switch regions of the immunoglobulin heavy chain (Igh) converting deoxycytidine to deoxyuridine residues. Double strand breaks (DSBs) are subsequently generated by the combined action of the uracil-N-glycosylase, UNG, which is part of the base excision repair (BER) pathway, and mismatch repair (MMR) proteins [17,18]. Despite differences, both CSR and SHM have the potential to contribute to genomic instability [19–21]. Indeed, the risks associated with these processes are heightened because binding of AID is not tightly regulated and therefore hits many other off-target genes (such as Myc) that can partner with its principal target, Igh, in oncogenic translocations [22–26].
AID-generated substrates for recurrent translocations are distributed throughout the genome
Although widely implicated in many cancers, the salient features associated with genomic regions involved in aberrant translocations have not been clearly defined. To examine this, both the Alt and Nussenzweig labs performed genome-wide analyses of translocations resulting from I-Sce1 induced breaks in cells isolated from mice engineered to contain one or more copies of the I-Sce1 meganuclease target sequence in the Igh or Myc loci [27,28]. Both labs examined translocations in short-term cultured splenic B cells, stimulated under conditions that promote Sµ to Sγ1 joining and switching to IgG1. In both cases translocation partners with the I-Sce1 site were identified by deep-sequencing following a PCR amplification step that relied on primers hybridizing to the I-Sce1 side of the translocation (Figure 2). The Alt lab examined translocations in wild-type and AID deficient (Aicda−/−) B cells, while the Nussenzweig lab used Aicda−/− cells with retrovirally overexpressed levels of AID for most of their analysis. When compared to wild-type levels, it is clear that overexpression of AID led to the discovery of increased levels of AID-mediated translocations [27], which is consistent with the finding that increased expression of AID leads to increased damage [29–31]. Despite differences in their systems and the saturation level of breaks, the Alt and Nussenzweig labs reach very similar conclusions. Although rearrangements are distributed throughout the genome, the majority of translocations, whether AID dependent or independent, occur with loci on the same chromosome within a short distance (up to 350 kb) of the I-Sce1 site and the frequency of these decline inversely with increasing separation from the ISce1 induced break, up to a distance of 50Mb. Furthermore, translocations occur near active transcription start sites (TSSs) and their distribution is highly correlated with gene density as expected from previous studies [32–37]. Importantly, the majority of hotspots – rearrangements that occur at significantly high frequency – were found by both labs to be AID dependent, and included many previously identified targets of AID [38]. These data indicate that AID produces substrates for translocations, that impact on the translocation landscape of the cells, and, not surprisingly, the Nussenzweig lab found an overlap of AID-mediated translocation partners in cells harboring I-Sce1 sites in Igh or in Myc [27].
Figure 2. Techniques used for identification of chromosomal translocations and characterization of nuclear organization.
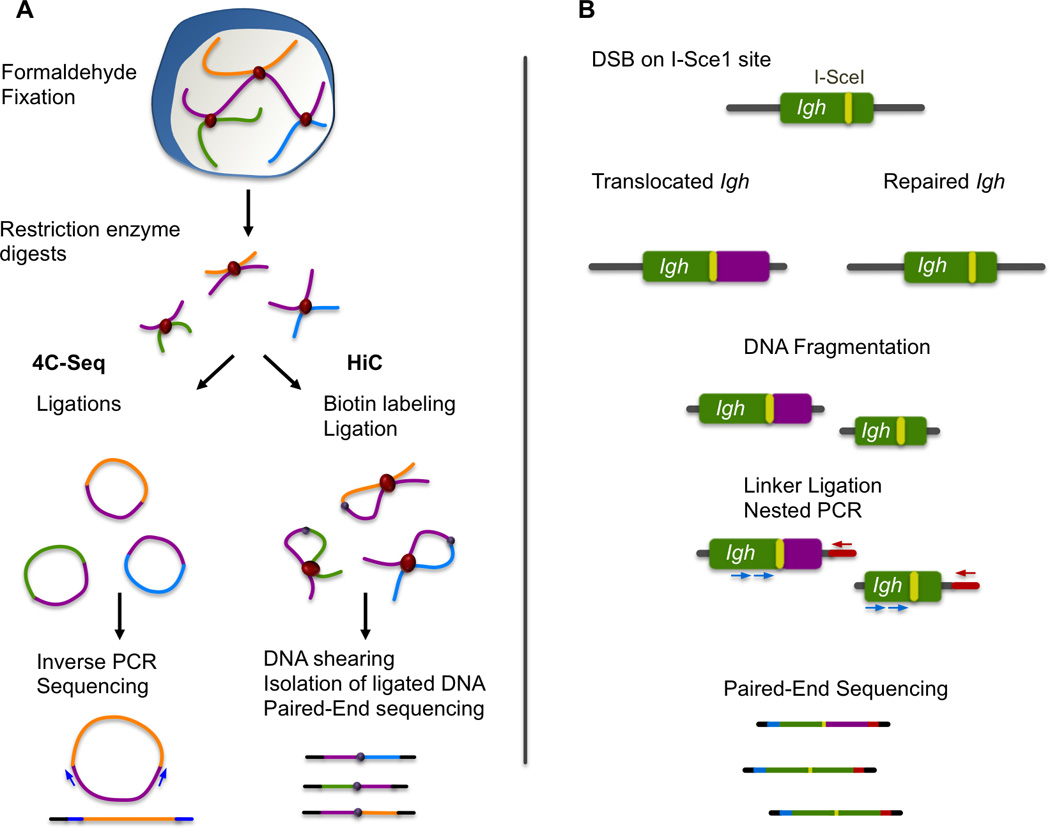
(A) 4C-seq and HiC identify chromosomal interactions through formaldehyde fixation and subsequent digestion and ligation of chromatin segments found in close proximity in the cell nucleus. 4C-seq is used to find all the interaction partners of a certain locus through inverse PCR and massively parallel sequencing. An additional digestion and ligation step (not represented here for simplification purposes) is required in this method. In HiC, all fragments that represent chromosomal interactions are isolated through biotin/streptavidin purification and then identified through genomewide sequencing. (B) Simplified scheme of one of the techniques used for genome-wide identification of Igh translocation partners. An engineered I-SceI site on Igh is used to induce breaks and translocations. These are identified through ligation-mediated PCR that takes advantage of the fact that the I-SceI site provides the precise location of one of the translocation partners.
Both the Alt and Nussenzweig labs found a significant level of AID dependent rearrangement between the I-Sce1 site located in Sµ (Nussenzweig) or Sγ1 (Alt) with Sγ1 or Sµ, respectively, as well as Sε. This is expected because under the stimulation conditions used for these experiments, breaks should be introduced into all three switch regions. Both labs found that cells harboring I-Sce1 sites in Myc had a good representation of translocations with the same switch regions as found for I-Sce1 sites in Igh, even though this locus is located on a different chromosome. These data indicate that hotspots have a dominant role in determining the spectrum of translocations.
The impact of nuclear organization on translocation frequency
Although break frequency is an important factor in determining the translocation landscape, it is also clear that nuclear proximity contributes. Indeed, out of the 234 AID mediated translocation hotspots associated with the IghI-SceI site found by the Nussenzweig lab [27], 40% were on the same chromosome as the I-SceI site (chromosome 12) while only 11% were identified on chromosome 15. In contrast, when analyzing AID mediated translocation hotspots associated with the MycI-SceI allele (located on chromosome 15) they found that the number of translocations to chromosome 12 dropped to 10%, while translocations within chromosome 15 increased to 33%. Since the location of AID generated breaks is not dependent on the location of the I-SceI site, these changes can only be explained by differences in the frequency of interactions in cis versus trans [39] which gives rise to a higher number of translocation hotspots in whatever chromosome contains the I-SceI site. This finding strongly implies a link between nuclear organization and AID mediated translocation hotspot frequency.
To investigate the relationship between AID mediated translocation frequency and proximity to Igh the Casellas lab performed chromosome conformation capture with deep sequencing (4C-seq) [40,41]. In addition, our lab performed a similar analysis using the same Igh translocation capture sequence data set (TC-seq generated by Klein et al.,) [27] and data from our own 4C-seq experiments (Figure 2) [42]. Surprisingly, both our labs arrive at different conclusions. While we found that close nuclear proximity to Igh predisposes to AID mediated translocations, the Casellas lab concluded that these two factors are not linked and that the frequency of translocations is dependent on the frequency of damage alone. This, despite their finding that the chromosome containing the I-SceI site has the highest number of AID mediated translocations hotspots [27]. Consistent with these data (and in agreement with the data from other chromatin conformation capture studies [39]) we find that interactions on the same chromosome dominate the 4C profile and the strength of these signals declines with increasing distance from the Igh locus over a distance of 60Mb. In this sense, the TC-seq profile mirrors our 4C-seq interaction profile.
Different approaches for 4C-seq analyses can impact on the results
We speculate that the different conclusions between the Skok and Casellas labs can be explained by differences in our 4C-seq analyses and the interpretation of the results obtained with this technique. The Casellas lab segments the genome into 200Kb non-overlapping intervals and assigns the same 4C-seq signal to each gene with a TSS that lies within a particular window according to the reads mapped therein (Figure 3A). In contrast, we took two different approaches for analysis of 4C-seq data (Figure 3B). The first involved segmentation of the genome into 200Kb windows centered around every annotated TSS, as this is where AID enrichment is found. Thus, we assigned each gene its own individual 4C-seq signal. However, biases are introduced in any chromosome conformation capture experiment that uses a fixed window size approach for analysis because the mapping of reads depends on the location of restriction enzyme sites used in preparation of the 4C template, and their number can vary widely from one window to another. To bypass these limitations we examined enrichment of 4C-seq signal in domains using increasing length scales of five to thirty HindIII sites (Figure 3C) [43,44]. Since the fixed window size and the domainogram approach gave rise to a distinct sets of results, we performed DNA FISH analyses (the gold standard for measuring gene interactions) to determine which was the more robust. Our results clearly indicate that the FISH data were most closely aligned with the domainogram analyses, underscoring the strengths of this approach. Using this methodology we performed a comparative analysis of TC-seq [27] and 4C-seq and found that genomic domains, which contact Igh at significant frequency incorporate the vast majority of known AID hotspot target genes and translocation partners of Igh.
Figure 3. Methods to analyze 4C-seq.
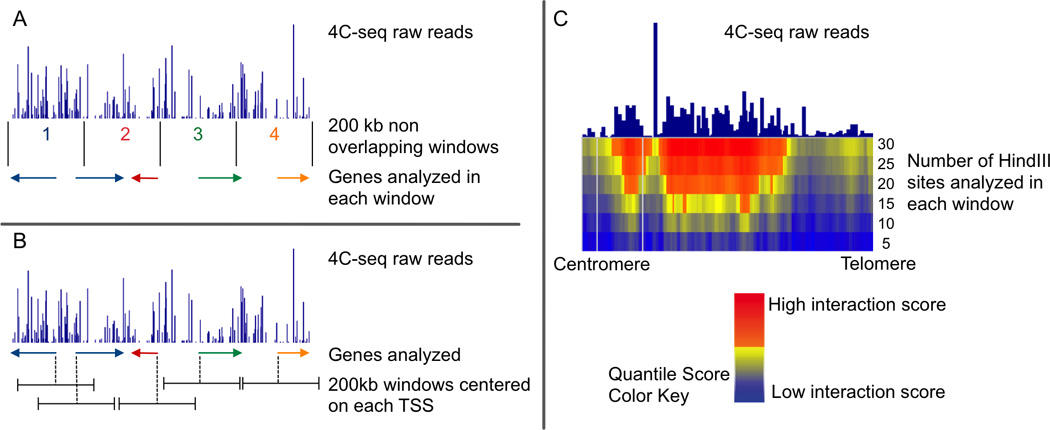
A and B represent different approaches to gene centric 4C-seq analyses. (A) The method used by the Casellas lab divides the genome into non overlapping 200 kb windows. The total number of 4C-seq reads that map to each window are quantified and normalized relative to the total number of genome-wide reads. Genes are assigned within a window depending on their most upstream RefSeq genomic location (NCBI Reference Sequence). All genes that start within the same 200 kb window will have the same 4C score. (B) In contrast, the method we used for gene-centric 4C analysis assigns each gene to its own distinct 200 kb window, which is centered on the location of its transcription start site (TSS). To identify which genes have a statistically significant interaction with the bait, signals associated with each gene are compared with the overall 4C-seq signal using an empirical resampling approach. (C) We also used a domainogram approach to analyze 4C-seq. For the construction of domainograms, no anchors (such as gene location or windows based on genomic positions) are used. For each mapped read, a Quantile Score is calculated using windows of increasing numbers of restriction enzyme sites. The enzyme HindIII is used to define window size as the reads in a 4C-seq experiment can only be mapped to these sites. This method calculates the significance of an interaction by comparing local enrichment of the 4C signal.
High frequency DSBs and spatial proximity influence translocations in developing B cells
Our conclusions are in agreement with those of the Dekker and Alt labs who examined the impact of nuclear organization and translocations in a V(D)J recombining G1-arrested mouse pro-B cell line using TC-seq in conjunction with Hi-C (Figure 2) [45]. In contrast to mature cell lines undergoing AID-mediated CSR, there is almost no detectable genome instability in these cells, likely because compared to AID, RAG targeting is less promiscuous and depends on selective binding to conserved recombination signal sequences (RSSs) within individual antigen receptor loci. To compensate for the lack of damage, the Alt lab generated translocation libraries in a repair compromised (Ataxia-Telangiectasia mutated (ATM) kinase) deficient setting, using cell lines harboring I-Sce1 sites in three different chromosomes. With this system they found that up to 25% of translocations occurred with RAG targeted antigen receptor loci independent of their genomic location. In agreement with the findings from splenic B cells, these data indicate that high frequency DSBs dominate the translocation landscape.
To obtain a broader distribution of DSBs and to normalize for the skewed high frequency of breaks in antigen receptor loci, cells were subjected to γ-irradiation. This reduced the relative frequency of rearrangements with antigen receptor loci and increased breaks elsewhere in the genome, thereby leveling the playing field. Consistent with previous findings [27,28,42], the Alt and Dekker labs found that rearrangements occurred predominantly on the same chromosome as the I-Sce1 site. Furthermore, using allele specific analysis they were able to confirm that rearrangements were only enriched on the cis chromosome as opposed to the trans homologue. Moreover, the intra-chromosomal interaction profile was strikingly similar to the translocation profile, in line with our results obtained in mature B cells [42]. In addition, when the genome-wide distribution of breaks was normalized by γ-irradiation of cells, inter-chromosomal translocation and interaction frequencies were highly correlated. Importantly, for I-Sce1 sites located on different chromosomes the translocation profile differed in a manner that was linked to inter-chromosomal interaction frequency. These data indicate that chromosome folding and nuclear proximity have an important influence on translocation frequency.
Conclusions
The data from these papers provide important insight into the origins of recurrent translocations in lymphocytes. It is clear that in both developing and mature B cells, loci with a high frequency of DSBs will dominate the rearrangement spectrum. Indeed this is the case for hotspots associated with both AID and RAG [27,28,45] where translocation frequency is determined by the break frequency of targeted loci. This is demonstrated in ATM kinase impaired developing B cells where the frequency of rearrangement between I-Sce1 sites and the various antigen receptor loci is dependent on the frequency of breaks, rather than proximity to the I-Sce1 site [45]. Similarly, for AID rearrangements: although we find that proximity is important for targeting and rearrangement, the frequency of translocations will always be more highly correlated with the frequency of DSBs (which is influenced by several gene intrinsic factors such as the level of Polymerase stalling and presence of AID recognition sites) than to positioning relative to Igh. Thus, the analyses performed by the Casellas lab showing that TC-seq and 4C-seq signal on hotspots are not highly correlated is entirely expected [41]. Nonetheless, this finding does not undermine the fact that nuclear/genomic proximity plays an important role in predisposing to AID mediated translocations as shown by our analyses (Figure 4) [42].
Figure 4. AID targets are located in chromosomal domains that are found in close proximity to Igh.
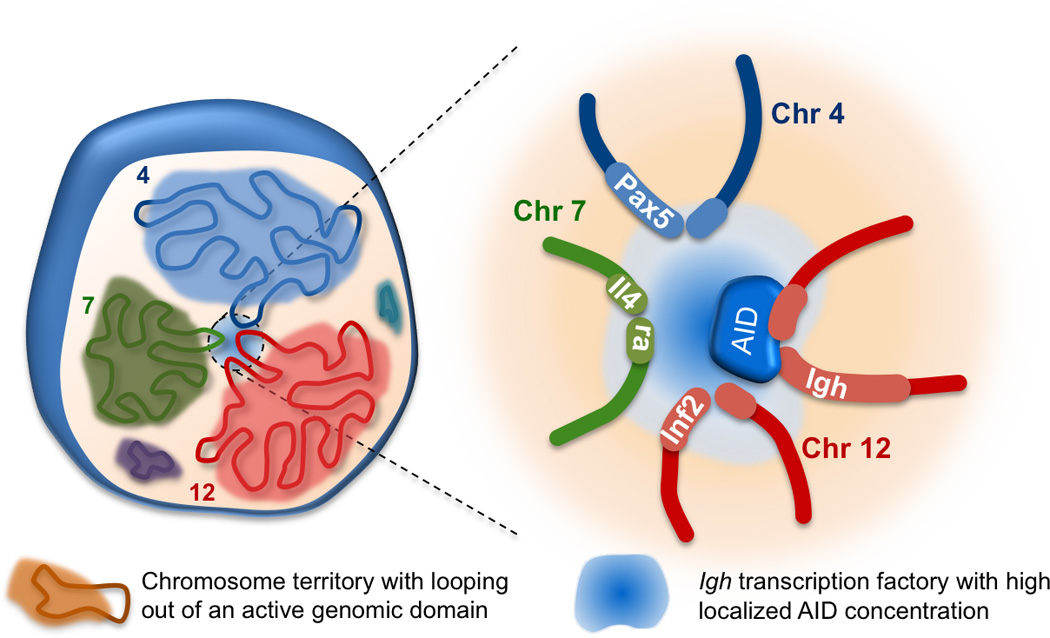
Chromosomes occupy discrete locations in the nucleus known as chromosome territories. Inactive genes are mostly found within these territories while active genes can loop out and engage in intra- and interchromosomal interactions in transcription or recombination factories [46]. Igh on chromosome 12 associates with many other actively transcribed genes in B cells undergoing CSR. Indeed, the vast majority of AID off-target hotspot genes such as Pax5, Il4ra and Inf8 are all located in chromosomal domains that have a significant interaction with Igh. We propose that this predisposes them to AID targeting and potential translocations with Igh as the local concentration of AID in these areas is higher in the transcription factories frequented by Igh.
Finally, it should be noted that these genome-wide studies rely on artificially induced breaks at ISce1 sites, therefore, although they provide general information about the origins of translocations, they cannot inform us about rearrangements that occur as a result of genetic mutations in an in vivo setting. This is the next step then: to examine the influence of specific factors in generating translocations in different lineages and stages of development.
Acknowledgements
We would like to thank the Skok lab, specifically Julie Chaumeil, for insightful discussions and critical reading of this manuscript. This work is supported by the National Institute of Health: R01GM086852, R56AI1099111. J.A.S is an LLS scholar and P.R. is supported by an NCC fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
* Special Interest ** Outstanding Interest
- 1.Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal v(d)j recombination. Cell. 2002;109(Suppl):S45–S55. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- 2.Johnson K, Hashimshony T, Sawai CM, Pongubala JM, Skok JA, Aifantis I, Singh H. Regulation of immunoglobulin light-chain recombination by the transcription factor irf-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28(3):335–345. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 3.Hewitt SL, Chaumeil J, Skok JA. Chromosome dynamics and the regulation of v(d)j recombination. Immunological reviews. 2010;237(1):43–54. doi: 10.1111/j.1600-065X.2010.00931.x. [DOI] [PubMed] [Google Scholar]
- 4.Feeney AJ. Epigenetic regulation of antigen receptor gene rearrangement. Current opinion in immunology. 2011;23(2):171–177. doi: 10.1016/j.coi.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schatz DG, Ji Y. Recombination centres and the orchestration of v(d)j recombination. Nat Rev Immunol. 2011;11(4):251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- 6.Schatz DG, Swanson PC. V(d)j recombination: Mechanisms of initiation. Annu Rev Genet. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- 7.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin class-switch DNA recombination: Induction, targeting and beyond. Nat Rev Immunol. 2012;12(7):517–531. doi: 10.1038/nri3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato L, Stanlie A, Begum NA, Kobayashi M, Aida M, Honjo T. An evolutionary view of the mechanism for immune and genome diversity. Journal of immunology. 2012;188(8):3559–3566. doi: 10.4049/jimmunol.1102397. [DOI] [PubMed] [Google Scholar]
- 10.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, Scharff MD. The biochemistry of somatic hypermutation. Annu Rev Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 11.Neuberger MS. Antibody diversification by somatic mutation: From burnet onwards. Immunol Cell Biol. 2008;86(2):124–132. doi: 10.1038/sj.icb.7100160. [DOI] [PubMed] [Google Scholar]
- 12.Liu M, Schatz DG. Balancing aid and DNA repair during somatic hypermutation. Trends Immunol. 2009;30(4):173–181. doi: 10.1016/j.it.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (aid), a novel member of the rna-editing deaminase family in germinal center b cells. The Journal of biological chemistry. 1999;274(26):18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 14.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (aid), a potential rna editing enzyme. Cell. 2000;102(5):553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 15.Petersen-Mahrt SK, Harris RS, Neuberger MS. Aid mutates e. Coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418(6893):99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 16.Delker RK, Fugmann SD, Papavasiliou FN. A coming-of-age story: Activation-induced cytidine deaminase turns 10. Nature immunology. 2009;10(11):1147–1153. doi: 10.1038/ni.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011;32(5):194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gazumyan A, Bothmer A, Klein IA, Nussenzweig MC, McBride KM. Activation-induced cytidine deaminase in antibody diversification and chromosome translocation. Adv Cancer Res. 2012;113:167–190. doi: 10.1016/B978-0-12-394280-7.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141(1):27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gostissa M, Alt FW, Chiarle R. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol. 2011;29:319–350. doi: 10.1146/annurev-immunol-031210-101329. [DOI] [PubMed] [Google Scholar]
- 21.Robbiani DF, Nussenzweig MC. Chromosome translocation, b cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol. 2013;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 22.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, Nussenzweig MC. Aid is required for c-myc/igh chromosome translocations in vivo. Cell. 2004;118(4):431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, Eisenreich TR, Chen J, Dickins RA, Lowe SW, Nussenzweig A, et al. Role of genomic instability and p53 in aid-induced c-myc-igh translocations. Nature. 2006;440(7080):105–109. doi: 10.1038/nature04495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-ig loci in activated b cells. Molecular cell. 2011;41(2):232–242. doi: 10.1016/j.molcel.2011.01.007. * This study presents the first genome-wide unbiased identification of AID off-targets during CSR using ChIP-Chip with an anti-Nbs1 antibody, which is part of the MRN complex which has been shown to be recruited to the switch regions of Igh.
- 25.Kato L, Begum NA, Burroughs AM, Doi T, Kawai J, Daub CO, Kawaguchi T, Matsuda F, Hayashizaki Y, Honjo T. Nonimmunoglobulin target loci of activation-induced cytidine deaminase (aid) share unique features with immunoglobulin genes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(7):2479–2484. doi: 10.1073/pnas.1120791109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Y, Soong TD, Wang L, Melnick AM, Elemento O. Genome-wide detection of genes targeted by non-ig somatic hypermutation in lymphoma. PLoS One. 2012;7(7):e40332. doi: 10.1371/journal.pone.0040332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, Casellas R, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in b lymphocytes. Cell. 2011;147(1):95–106. doi: 10.1016/j.cell.2011.07.048. ** References 28 and 29 used engineered I-SceI sites in Igh and Myc to identify translocation partners of these loci in B cells activated to undergo CSR. Both studies identify the genomic locations of hotspots, with a localized enrichment of translocations with Igh and Myc. They confirmed previous studies that indicate that AID hits many loci across the genome but preferentially targets actively transcribed genes with a high-incidence of polymerase stalling.
- 28. Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, Neuberg D, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in b cells. Cell. 2011;147(1):107–119. doi: 10.1016/j.cell.2011.07.049. ** References 28 and 29 used engineered I-SceI sites in Igh and Myc to identify translocation partners of these loci in B cells activated to undergo CSR. Both studies identify the genomic locations of hotspots, with a localized enrichment of translocations with Igh and Myc. They confirmed previous studies that indicate that AID hits many loci across the genome but preferentially targets actively transcribed genes with a high-incidence of polymerase stalling.
- 29.Martin A, Scharff MD. Somatic hypermutation of the aid transgene in b and non-b cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(19):12304–12308. doi: 10.1073/pnas.192442899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takizawa M, Tolarova H, Li Z, Dubois W, Lim S, Callen E, Franco S, Mosaico M, Feigenbaum L, Alt FW, Nussenzweig A, et al. Aid expression levels determine the extent of cmyc oncogenic translocations and the incidence of b cell tumor development. The Journal of experimental medicine. 2008;205(9):1949–1957. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, Honjo T. Constitutive expression of aid leads to tumorigenesis. The Journal of experimental medicine. 2003;197(9):1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lebecque SG, Gearhart PJ. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5' boundary is near the promoter, and 3' boundary is approximately 1 kb from v(d)j gene. The Journal of experimental medicine. 1990;172(6):1717–1727. doi: 10.1084/jem.172.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances aid-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nature immunology. 2003;4(5):452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 34.Pham P, Bransteitter R, Petruska J, Goodman MF. Processive aid-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424(6944):103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 35.Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the aid antibody diversification enzyme. Nature. 2003;422(6933):726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 36.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, Nieland TJ, et al. Activation-induced cytidine deaminase targets DNA at sites of rna polymerase ii stalling by interaction with spt5. Cell. 2010;143(1):122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase aid and its cofactor rpa in b lymphocytes. Nat Immunol. 2011;12(1):62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the b cell genome during somatic hypermutation. Nature. 2008;451(7180):841–845. doi: 10.1038/nature06547. ** Here, the authors preformed an extensive sequencing of the B cell genome upon induction of Somatic Hypermutation in germinal centers. This was the first large-scale identification of non-Ig AID target genes. In addition, this study shows that while some of the AID off-target genes accumulate mutations others are repaired by a high-fidelity mechanism.
- 39.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van de Werken HJ, Landan G, Holwerda SJ, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Oz Y, Bouwman BA, Verstegen MJ, et al. Robust 4c-seq data analysis to screen for regulatory DNA interactions. Nat Methods. 2012;9(10):969–972. doi: 10.1038/nmeth.2173. [DOI] [PubMed] [Google Scholar]
- 41. Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, Mathe E, et al. DNA damage defines sites of recurrent chromosomal translocations in b lymphocytes. Nature. 2012;484(7392):69–74. doi: 10.1038/nature10909. * This manuscript identifies sites of DNA damage caused by AID using ChIP-seq with an anti-RPA antibody in 53BP1−/−. Translocation rates of AID-mediated hotspots directly correlate with RPA enrichment.
- 42. Rocha PP, Micsinai M, Kim JR, Hewitt SL, Souza PP, Trimarchi T, Strino F, Parisi F, Kluger Y, Skok JA. Close proximity to igh is a contributing factor to aid-mediated translocations. Molecular cell. 2012;47(6):873–885. doi: 10.1016/j.molcel.2012.06.036. * This study provides a high-resolution map of all chromosomal interactions that the Igh locus undergoes during CSR. It demonstrates that the vast majority of AID-mediated translocation hotspots are found within large chromosomal domains that interact frequently with Igh and that genes within these domains are at risk of being targeted by AID and translocating with Igh.
- 43.Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, Tixier V, Mas A, Cavalli G. Polycomb-dependent regulatory contacts between distant hox loci in drosophila. Cell. 2011;144(2):214–226. doi: 10.1016/j.cell.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 44.de Wit E, Braunschweig U, Greil F, Bussemaker HJ, van Steensel B. Global chromatin domain organization of the drosophila genome. PLoS Genet. 2008;4(3):e1000045. doi: 10.1371/journal.pgen.1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, Becker MS, Alt FW, Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148(5):908–921. doi: 10.1016/j.cell.2012.02.002. ** This report describes a high-resolution Hi-C spatial organization map of the G1-arrested mouse pro-B cell genome and compares it to the genome-wide distribution of translocations in the same cells. Upon ionizing radiation translocations were shown to be directly related to pre-existing spatial proximity.
- 46.Chaumeil J, Micsinai M, Panagiotis N, Deriano L, Wang J, Ji Y, Nora E, Rodesch J, Aifantis I, Kluger Y, Schatz DG, et al. Higher-order looping and nuclear organization of antigen receptor loci facilitate targeted rag cleavage and regulated rearrangement in recombination centers. Cell Reports. 2013 doi: 10.1016/j.celrep.2013.01.024. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]