


Abstract
QA-RecombineIt provides a web interface to assess the quality of protein 3D structure models and to improve the accuracy of models by merging fragments of multiple input models. QA-RecombineIt has been developed for protein modelers who are working on difficult problems, have a set of different homology models and/or de novo models (from methods such as I-TASSER or ROSETTA) and would like to obtain one consensus model that incorporates the best parts into one structure that is internally coherent. An advanced mode is also available, in which one can modify the operation of the fragment recombination algorithm by manually identifying individual fragments or entire models to recombine. Our method produces up to 100 models that are expected to be on the average more accurate than the starting models. Therefore, our server may be useful for crystallographic protein structure determination, where protein models are used for Molecular Replacement to solve the phase problem. To address the latter possibility, a special feature was added to the QA-RecombineIt server. The QA-RecombineIt server can be freely accessed at http://iimcb.genesilico.pl/qarecombineit/.
INTRODUCTION
The availability of experimentally determined high resolution protein structures has significantly improved the understanding of biological mechanisms and has greatly facilitated rational drug design (1–4). Although the rate at which protein structures are being determined by experimental methods (mainly X-ray crystallography and nuclear magnetic resonance spectroscopy) has increased dramatically during the past years, this progress has not been efficient enough to match the amount of data on predicted protein sequences generated by genome sequencing efforts. At the time of writing, there are ∼83 000 protein structures in the PDB (5,6), but ∼23 million protein sequences in the non-redundant databases (non-redundant GenBank CDS translations, PDB, SwissProt, PIR and PRF) (7). As a complement to experimental methods, many computational methods have been developed to address the sequence-structure gap. However, to generate a 3D structural model of the target protein, typical users of structure modeling methods (e.g. biologists willing to predict a structure of their favorite protein) use only a few fully automated servers, usually the ones that have performed well in the most recent Critical Assessment of Techniques for Protein Structure Prediction (CASP) experiment (8). The recommended procedure is then to choose the model ranked the highest according to a model quality assessment program (MQAP), a method that predicts the accuracy of the model with respect to the true structure (without knowing the true structure) (9). Even though the model with the best overall score is selected, it does not mean that its local conformation over the entire length of the target sequence is always the closest to the native structure when compared with the conformations of the remaining models. Thus, even if more accurately modeled regions exist in the remaining models, they cannot be easily used to improve the main model.
To address this challenging problem, we developed the QA-RecombineIt web server. Our method operates in two stages (Figure 1). In the first stage (QA-mode), our server predicts the global quality of input models and provides estimates of local quality as the deviation between C-α atoms in the models and corresponding atoms in the unknown native structure. Together with the input models, these predictions subsequently become the input for the second stage (RecombineIt-mode), in which fragments predicted to be better than others are judiciously combined to generate hybrid (consensus) models. Finally, hybrid models are scored by the MQAPs implemented in the QA-mode and then presented to the user.
Figure 1.

Flowchart describing the main functionalities of the QA-RecombineIt server. Protein sequence in FASTA format and 3D structure models of the target protein must be provided to execute the server. QA-RecombineIt implements two modules, including QA-mode for assessment of protein models and RecombineIt-mode for merging the best quality fragments derived from the input models. By default, these modules operate in a fully automatic way. However, more advanced users can modify the operation of the fragment recombination algorithm (RecombineIt-mode) by selecting the method according to which best fragments and/or models will be picked (box A), and/or manually identifying the models (box B) and/or fragments(s) (box C) on the base of which the hybrid model(s) will be created.
Although other freely available servers exist that either predict protein structure from the amino acids sequence (10–14) or assess the quality of protein models (15–19), QA-RecombineIt is unique in providing an integrated underlying methodology for both scoring and improving models. QA-RecombineIt can be run for a set of models generated by a variety of protein structure methods, including fold recognition/de novo modeling, template-based modeling and loop refinement, among others. Essentially, our server can be used at the last step of any protein structure prediction procedure.
MATERIALS AND METHODS
Model quality assessment
QA-RecombineIt relies on five different MQAP procedures. The MQAPs can be divided into two categories: (i) single-model MQAPs, i.e. methods that assess the quality of a single model and (ii) clustering MQAPs, i.e. methods that operate by structural comparisons between a number of alternative models generated for the target sequence. It was shown that in cases where many models based on varied prediction methods are available, clustering approaches significantly outperform single-model MQAPs (18,20–22). However, in cases where only one or few alternative models are available, the gap between the clustering MQAPs and single-model MQAPs is marginal, with single-model MQAPs outperforming clustering MQAPs in some cases (18). Table 1 gives a brief overview of the MQAPs integrated by the QA-RecombineIt server. In the following sections, these methods are presented in detail.
Table 1.
MQAPs implemented in QA-RecombineIt
| Name | MetaMQAP | ProQ2 | DFIRE | GOAP | MQAPmulti |
| Type | LG/S | LG/S | G/S | G/S | LG/C |
S, single-model MQAP; C, clustering MQAP; L, MQAP that predicts local accuracy of a model; G, MQAP that predicts the global accuracy of a model.
MetaMQAP
MetaMQAP is a meta-predictor that combines output from a number of original methods used for predicting global and/or local accuracy of protein structural models, including VERIFY3D (23), PROSA (24), BALA-SNAPP (25), ANOLEA (26), PROVE (27), TUNE (28), REFINER (29) and PROQRES (30). In addition, MetaMQAP analyzes the following features of individual residues: secondary structure, solvent accessibility and depth within the structure. Together with linear regression, these parameters are used to assess the deviations of C-α atoms for a given model. Then, based on these predictions, the global accuracy of the model is calculated and expressed according to the Global Distance Test (GDT_TS) (31). This indicator corresponds to the average value of fractions of C-α atoms in the model that are placed within the distances of 1, 2, 4 or 8 Å from corresponding C-α atoms in the experimentally determined structure.
ProQ2
ProQ2 evaluates the following features (16): atom–atom and residue–residue contacts, surface accessibility, predicted secondary structure, predicted surface exposure and evolutionary information calculated over a sequence window. Based on these features, a support vector machine algorithm predicts the value of S-score (32) as the measure of accuracy for each residue in a protein model. This score is defined as
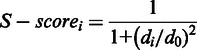 |
where di is the distance for residue i between the experimentally determined structure and the model, and d0 is a distance threshold. As the threshold is set to 3 Å, the S-score is sensitive to errors ranging from 0 to 3 Å. Once the local quality prediction is performed, the global quality prediction for a model is achieved by summing the local predictions and then dividing the sum by the length of the target sequence.
Statistical potentials: DFIRE and GOAP
In general, statistical or knowledge-based potential is a pseudo-energy function derived from known protein structures. The majority of statistical potentials use the inverse Boltzmann law to convert the ratio of observed and expected frequencies of interactions into a scoring function that assesses the likelihood that a given interaction is ‘good’ or ‘bad’ (33,34).
DFIRE is a distance-dependent, structure-derived, all-atom statistical potential based on a distance-scaled, finite, ideal-gas reference state of uniformly distributed non-interacting ideal gas points. The DFIRE potential was found to be efficient in the refinement of protein structures, in particular segments that contain secondary structure elements or loops (35,36). A detailed description of the potential was presented by Zhou and Zhou. (37).
GOAP was developed for atomic-resolution modeling and refinement. Zhou and Skolnick improved the description of pairwise atomic interactions, similar to that of DFIRE, by introducing the orientation-dependence of all individual heavy atoms. To achieve this, a plane-like object is introduced for each atom by using two of its bonded neighboring atoms and itself. Then, a potential value is calculated for each pair of interacting atoms. To do so, the method analyzes the distance between a given pair of atoms and the mutual orientation of the planes assigned to the atoms. The GOAP potential has been described in detail by Zhou and Skolnick (38).
MQAPmulti
MQAPmulti is the only method implemented in QA-RecombineIt that clusters the protein models being scored. This method takes both protein models and the amino acids sequence of the protein under analysis as input. MQAPmulti executes three separate modules to calculate intermediate scores and then combines them into one final score. First, MQAPmulti predicts protein structure features from the target sequence using third-party methods; these features include secondary sequence (39–41), solvent accessibility (42,43) and contact maps (44). The predictions obtained are compared with values of the corresponding features calculated directly from the 3D structural models under evaluation. These (dis)agreement terms, together with in-house implementation of the DFIRE statistical potential and the number of unsatisfied hydrogen bond donors/acceptors, are used to estimate a global accuracy of each of the input models. To do so, a linear regression is applied. It should be emphasized that at this stage, every model is scored independently of the others; thus, the predicted global quality is single-model based. In the remaining part of this manuscript, we will call this score ‘True-MQAPmulti score’. Second, models are clustered according to their structural similarity. Two measures of similarity between two models are applied: GDT (31) (see earlier in the text) and Q-score (45,46), which measures the structural similarity between two models by comparing their internal residue distances. Third, MQAPmulti combines the True-MQAPmulti scores of all models with pairwise similarity of these models to enhance the model quality assessment when only a few models are considered. It is based on the assumption that values of single-model scoring function, on average, decrease as models become more similar to the native structure. It was postulated and then proven (47) that the model closest to the native structure should provide the highest correlation coefficient of a score (provided by such a single-model MQAP) versus distance, when used as the reference in pairwise comparisons with the remaining models. For a set of models, MQAPmulti calculates the correlation coefficient for each model and uses the obtained correlation coefficient as a score of model correctness. MQAPmulti uses two sets of such Pearson’s correlation coefficients: between the score provided by the True-MQAPmulti score and either GDT_TS or Q-score. Finally, the predictions provided by the aforementioned three modules (eight different scores in total) are used, together with a support vector machine algorithm (48,49), to predict the GDT_TS score of the models concerned. The method was trained on CASP7 and CASP8 single-domain protein targets.
Tertiary structure prediction using fragment recombination
RecombineIt is an automatic fragment recombination algorithm that was inspired by both the Frankenstein’s monster approach to comparative modeling—a manual methodology developed previously in our laboratory (50,51)—and many years of experience of our laboratory members in protein structure prediction, in particular in building models for challenging cases that include combination of template-based and template-free modeling [e.g. (52)]. The RecombineIt algorithm comprises four steps. First (stage I), five models are picked according to the highest global score provided by MQAPmulti (or MetaMQAP score, if <50 models are submitted by a user); these models will be used later on as the ‘leading’ models. Simultaneously (stage II), based on the PSIPRED (39) secondary structure prediction, the target sequence is divided into partially overlapping blocks (10 overlapping residues), initially containing one secondary structure element and half of each loop region connected to this element. For long sequences, if needed, these initial blocks are merged (with neighboring blocks in the sequence) to reduce the total number of blocks to ≤15. Then, based on these block boundaries, the input models are cut into fragments. Next (stage III), all possible combinations of the fragments are ranked (without explicitly generating 3D models for each combination). To rank a given combination of fragments, the sum of local MQAPmulti scores (or MetaMQAP scores if <50 models are submitted) is calculated for all residues. In addition, the combination is penalized if its fragments are as follows: (i) derived from models that have different folds from each other (i.e. the TM-score between a pair of models is below 0.3); (ii) derived from models with folds that differ from the folds of the ‘leading’ models (TM-score < 0.3); and (iii) if the similarity of overlapping regions of two merged together fragments is measured to have TM-score smaller than 0.9 (53). If the number of possible combinations of fragments is higher than 108, an in-house developed genetic algorithm is used to find the best combinations of the fragments. Finally (stage IV), 3D models are built for each of the 100 top-scored combinations of fragments by using MODELLER (54) (version 9v3) in a multi-template mode. In this last step, each fragment is considered to be a single template. As the fragments of a given combination only partially overlap in their sequences, it would be impossible for the MODELLER program to model the 3D structure without additional distance restraints between the fragments. Thus, for each fragment, a set of distances between its C-α atoms and the remaining C-α atoms from the model, from which this fragment was derived, is measured. Then, the data are provided for the MODELLER program. Notably, for a given combination of fragments that are derived from many models, there are always two variants of a pair for given residues X and Y. One variant corresponds to the situation in which X is contained in a fragment derived from model Z′, and Y is contained in the rest of the same model. However, for a different model (Z′′), the situation is reversed: Y is located in a fragment, whereas X is found within the rest of the model Z′′. Thus, for cases where the conformations of models Z′ and Z′′ are not identical to one another, the distances between C-α atoms of residues X and Y in model Z′ may not be equal to those observed in model Z′′. To avoid overwriting such conflicting distance constraints assigned to the same pair of residues, only the distances between C-α atoms of odd-numbered residues within a fragment and C-α atoms of even-numbered residues within the remaining part of the model are taken into account.
By default, the QA-RecombineIt server uses MQAPmulti in stages I and III (or MetaMQAP if <50 models are submitted), and MetaMQAP is used by default at stage IV. However, a user can select another set of MQAPs (those described in the previous subsection) to be executed and/or used by RecombineIt.
DESCRIPTION OF THE QA-RecombineIT WEBSERVER
The QA-RecombineIt web server operates through two stages. In the first stage, called the QA-mode, starting from protein sequence and computational models of the protein, our server predicts both global and local accuracy of these models. In the second stage, called RecombineIt-mode, the server runs an algorithm that performs a ‘recombination’ of the best ranked parts of the input models into new hybrid structures that are likely to be better than the input models themselves. Such an approach, when used manually by human predictors, has proven to generate, on average, more accurate models than corresponding input models (50,51). Finally, QA-RecombineIt, in the last step, predicts the model quality for both the original input models as well as the hybrid models resulting from recombination and then selects the highest ranked model(s).
INPUT AND OUTPUT
QA-mode
The only data required as input are a protein sequence of the target protein either as a one-letter sequence or in the FASTA format and 3D structure models for that protein sequence. The models can be produced by a variety of protein structure predictors. Although a model does not need to have all of its residues modeled, the only restriction is that the number of a residue has to be in agreement with its number in the target sequence. On sequence submission to the server, a unique URL for the output of model quality assessment (QA-mode) is generated. The user may bookmark this URL. If a user provides an email address, she or he will be sent a reminder with the link to an output page once the job has been completed (Figure 2A). The results page is split into five panels. The ‘Summary’ panel does not only summarize the output but also allows users to select an MQAP to rank models or to visualize their local correctness. The ‘Tools’ panel allows users to either download the selected models annotated according to the local model quality (B-factor values modified according to predictions provided by MetaMQAP or ProQ2 or MQAPmulti) or to use the TM-score program (53) to superimpose the selected models and then visualize this superposition by JMOL (55) (Supplementary Figure S1). The ‘RecombineIt submission’ panel allows users to execute the RecombineIt algorithm by pressing the ‘Submit recombination of models’ button. Advanced users may want to activate an option, by which they can define ‘leading’ models and/or fragments; this can be accomplished by pressing the ‘advanced mode’ button (Figure 2B). This will force RecombineIt to build models that are as close as possible to one of the selected ‘leading’ models and/or to select fragments that are similar to those selected as ‘leading’ fragments for recombination. Here, it is important to emphasize that manual selection of ‘leading models’ can have a significant impact on the results. Thus, this functionality is only recommended to advanced users with experience in template selection for a given type of targets. The best template does not necessarily have to exhibit the highest sequence similarity with the target. Other important factors that should be considered include the accuracy of the structure (e.g. resolution and R-free of a crystallographic structure or the number of restrains per residue in an nuclear magnetic resonance structure) and the similarity between the physiological conditions (e.g. solvent, pH, ligands, quaternary interactions) of the template and the physiological conditions in which the user wants the target to be modeled. For example, this advanced option can be useful to those wanting to model a ligand-bound protein structure; users can select a model created on the base of a structure with a ligand as the ‘leading’ model. Selecting ‘leading’ fragments helps to have control over the local conformation (e.g. active site) of models built and to avoid the inclusion of templates that may have higher sequence similarity or better resolution, but exhibit ‘wrong’ conformation owing to interactions with other molecules or the absence thereof. The next panel, ‘Sequence 1D information’, shows secondary structure, solvent accessibility and disorder regions predicted for the target protein (see ‘Materials and Methods’ section). Each line of the ‘Model quality assessment’ (Figure 2B) panel contains global quality assessment for a given model. Local model quality (MetaMQAP or ProQ2 or MQAPmulti) is also reported in the form of a heat map (a spectrum of colors from blue to red represents the spectrum of residues predicted to be correct or incorrect). The last panel ‘Detailed information’ shows (dis)agreements between secondary structure and solvent accessibility predicted (from sequence) versus observed (in the model). Also the predictions of global and local accuracies are reported. The latter is presented as interactive charts of residue deviations (in Ångströms) in the function of a residue number. Finally, for each model, the image of its 3D structure is shown and colored according to the predicted local quality.
Figure 2.

QA-RecombineIt outputs. The example of the output of QA-mode (A). (B)—global and local quality of models. Local quality of each model is presented as a heat map. Once the user activates the ‘advance mode’, they can modify the operation of the fragment recombination algorithm by manually identifying the fragments and/or entire models on the base of which the hybrid model(s) will be created. The last panel (C) summarizes the fragment composition of the hybrid models generated. Word balloons indicate and explain the most important features of the results page by which the user can interact with the page; LBM, left mouse button.
RecombineIt-mode
Once a user clicks ‘Submit recombination of models’ button, the QA-mode output becomes an input to the RecombineIt-mode. As in the QA-mode, a unique URL for the output of RecombineIt-mode is generated, which the user may bookmark. The output page of the RecombineIt-mode is similar to that of the QA-mode, except for three differences. First, in contrast to the QA-mode, the RecombineIt-mode has an additional panel named ‘Model fragments’ that summarizes the fragment composition of the generated hybrid models (Figure 2C). At the top of this panel, an interactive model cloud is presented, where the font sizes correspond to the importance of each model in the recombination process, i.e. font size represents the number of times that a fragment from a given model has been applied to generate a hybrid model. Below the model cloud, a linear combination of fragments is presented for each hybrid model. Second, additional fields, named ‘Fragment definition’, are added to the output of the RecombineIt-mode to show the boundaries of the fragments used by RecombineIt. The last difference between the aforementioned two outputs is the lack of a ‘RecombineIt options and submission’ panel.
Molecular replacement mode
In protein crystallography, the determination of a 3D structure of a protein of interest entails gathering the intensity and phase information for a crystal diffraction pattern [review (56)]. Unfortunately, in a typical macromolecular X-ray diffraction experiment, only the intensities of reflections are measured, and information about phases is lost. The Molecular Replacement (MR) technique is one solution to this problem. MR approximates the phase information using a 3D structure of a related protein or a theoretical model of the protein under investigation (called a ‘search model’) (57). Recently, we have shown that using comparative models only marginally increases (by 4.5%) the MR success ratio in comparison with the structures of templates. However, as we have demonstrated, the situation changes dramatically once the comparative models are used together with their local accuracy (58). One of the ways to improve the utility of theoretical models of protein structure in MR is to recalculate the B-factor of each atom on the base of its predicted accuracy and then use such a modified search model with MR programs that take into account B-factor values as the indicator of uncertainties of atomic positions. By doing that, we have shown that predicted local accuracy of a model increases the MR success ratio by 45% compared with corresponding templates (58). Inspired by these findings, we have added functionality to QA-RecombineIt. By using the “Tools” panel, users can download models with B-factor values modified according to the MQAPmulti local score. Such models can be used as search models by MR programs that take into account atoms’ B-factor values, e.g. AMoRe (59) or MOLREP (60).
VALIDATION
Both components of QA-RecombineIt were tested in the CASP9 experiment, a community-wide blind assessment of computational methods for protein structure prediction. MQAPmulti was ranked the 4th best MQAP in predicting local model quality (http://predictioncenter.org/casp9/doc/presentations/CASP9_QA.pdf) and among the 10 best performing MQAPs for global model quality assessment (http://predictioncenter.org/casp9/doc/presentations/CASP9_QA.pdf). For local quality assessment (expressed in Ångströms), the Pearson’s correlation between the correct and predicted accuracy was 0.58. For global model quality (GDT_TS), the correlation was 0.96.
In the case of tertiary structure prediction in the CASP9 experiment, all targets were assigned two expiration dates: first, for automatic server predictors that can meet a 3-day deadline, and, second, for all other predictors, including slower automated methods and human groups. Models submitted for the first deadline were made available for predictors targeting the second deadline. During the CASP9 experiment, RecombineIt operated in a fully automatic mode and used as input only the models of automated server predictors submitted for the first deadline (see Supplementary Figure S2 for more details). Our method was run in its basic mode, i.e. neither ‘leading’ models nor fragments were selected manually. For the targets with at least one template to predict protein structure, including 52 template-based modeling (TBM) targets and 3 TBM/free modeling (FM) targets, the ‘Zhang’ group performed the best among all predictors (server and regular ones), achieving the sum of GDT_TS Z-scores = 51.89. The RaptorX server was ranked as the best among servers and 16th among all predictors (sum of GDT_TS Z-scores = 44.77). RecombineIt was ranked 4th with the sum of GDT_TS Z-scores = 49.69. Hence, RecombineIt run in its ‘basic’ mode produced predictions that were on the average better than all fully automated servers that generated models, which served as the RecombineIt input. Our method also outperformed most of the human expert groups, whose performance surpassed that of all of the servers and whose models were not used by our method.
Although RecombineIt performs well for template-based models, its predictive power is limited in cases of template-free modeling (Supplementary Table S1). For the FM targets, where no template existed to predict the target protein structures, the top-scoring ‘Eloffson’ group achieved the sum of GDT_TS Z-scores = 27.14, whereas the highest ranked server, QUARK, scored 23.08. In this category, our method achieved the GDT_TS Z-score sum of 21.31, which was worse that the score of the QUARK method, whose models were used as an input. Thus, RecombineIt can be safely recommended as a ‘consensus predictor’ for cases of difficult TBM, where the protein to be modeled may exhibit a known fold that is likely to be present in at least some of the input models. However, in the case of structures with completely new folds, and if good models are absent in the starting data set, our procedure cannot guarantee any improvement over the best starting model, even though it is likely to propose a solution that is better than the average.
CONCLUSIONS
The QA-RecombineIt server provides a unified interface for quality assessment and recombination of protein structure models. The algorithms underlying the QA-RecombineIt server were independently tested in the recent CASP9 competition and were found to be competitive in several categories, including model quality assessment and protein structure prediction of TBM targets and TBM/FM targets. As our server can be run with an input consisting of models generated by a variety of protein structure methods (both template-based and template-free), QA-RecombineIt can be used as the final step in any protein structure prediction procedure.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–2.
ACKNOWLEDGEMENTS
The authors thank Jan Kosinski for discussions. They are grateful to Katarzyna Cieslik, Kristian Rother, Lukasz Kozlowski and Nicolle Etchart for editing and proofreading the manuscript.
FUNDING
The 7th Framework Program of the European Commission [EC FP7, REGPOT grant HEALTH-PROT, 229676]. Polish Ministry of Science [N301 10632/3600 to M.P.]. A.B and J.M.B. and the implementation of the QA-RecombineIt software as a server were additionally supported by the Innovative Economy–European Regional Development Fund of the European Union [EU, grant POIG.02.03.00-00-003/09]. Funding for open access charge: [EU grant POIG.02.03.00-00-003/09].
Conflict of interest statement. None declared.
REFERENCES
- 1.Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray CW, Blundell TL. Structural biology in fragment-based drug design. Curr. Opin. Struct. Biol. 2010;20:497–507. doi: 10.1016/j.sbi.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Van Montfort R, Workman P. Structure-based design of molecular cancer therapeutics. Trends Biotechnol. 2009;27:315. doi: 10.1016/j.tibtech.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Scapin G. Structural biology and drug discovery. Curr. Pharm. Des. 2006;12:2087–2097. doi: 10.2174/138161206777585201. [DOI] [PubMed] [Google Scholar]
- 5.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein FC, Koetzle TF, Williams GJ, Meyer-EE J, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The protein data bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 7.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007;35:D5–D12. doi: 10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. Proteins. 1995;23:ii–iv. doi: 10.1002/prot.340230303. [DOI] [PubMed] [Google Scholar]
- 9.Kryshtafovych A, Fidelis K. Protein structure prediction and model quality assessment. Drug Discov. Today. 2009;14:386–393. doi: 10.1016/j.drudis.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z, Eickholt J, Cheng J. MULTICOM: a multi-level combination approach to protein structure prediction and its assessments in CASP8. Bioinformatics. 2010;26:882–888. doi: 10.1093/bioinformatics/btq058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng J, Xu J. RaptorX: exploiting structure information for protein alignment by statistical inference. Proteins. 2011;79:161–171. doi: 10.1002/prot.23175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benkert P, Kunzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37:W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ray A, Lindahl E, Wallner B. Improved model quality assessment using ProQ2. BMC Bioinformatics. 2012;13:224. doi: 10.1186/1471-2105-13-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGuffin LJ. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics. 2008;24:586–587. doi: 10.1093/bioinformatics/btn014. [DOI] [PubMed] [Google Scholar]
- 18.Pawlowski M, Gajda MJ, Matlak R, Bujnicki JM. MetaMQAP: a meta-server for the quality assessment of protein models. BMC Bioinformatics. 2008;9:403. doi: 10.1186/1471-2105-9-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, Eickholt J, Cheng J. APOLLO: a quality assessment service for single and multiple protein models. Bioinformatics. 2011;27:1715–1716. doi: 10.1093/bioinformatics/btr268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGuffin LJ. Benchmarking consensus model quality assessment for protein fold recognition. BMC Bioinformatics. 2007;8:345. doi: 10.1186/1471-2105-8-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cozzetto D, Kryshtafovych A, Ceriani M, Tramontano A. Assessment of predictions in the model quality assessment category. Proteins. 2007;69(Suppl. 8):175–183. doi: 10.1002/prot.21669. [DOI] [PubMed] [Google Scholar]
- 22.Kryshtafovych A, Fidelis K, Tramontano A. Evaluation of model quality predictions in CASP9. Proteins. 2011;79(Suppl. 10):91–106. doi: 10.1002/prot.23180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 24.Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 25.Krishnamoorthy B, Tropsha A. Development of a four-body statistical pseudo-potential to discriminate native from non-native protein conformations. Bioinformatics. 2003;19:1540–1548. doi: 10.1093/bioinformatics/btg186. [DOI] [PubMed] [Google Scholar]
- 26.Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J. Mol. Biol. 1998;277:1141–1152. doi: 10.1006/jmbi.1998.1665. [DOI] [PubMed] [Google Scholar]
- 27.Pontius J, Richelle J, Wodak SJ. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996;264:121–136. doi: 10.1006/jmbi.1996.0628. [DOI] [PubMed] [Google Scholar]
- 28.Lin K, May AC, Taylor WR. Threading using neural nEtwork (TUNE): the measure of protein sequence-structure compatibility. Bioinformatics. 2002;18:1350–1357. doi: 10.1093/bioinformatics/18.10.1350. [DOI] [PubMed] [Google Scholar]
- 29.Boniecki M, Rotkiewicz P, Skolnick J, Kolinski A. Protein fragment reconstruction using various modeling techniques. J. Comput. Aided Mol. Des. 2003;17:725–738. doi: 10.1023/b:jcam.0000017486.83645.a0. [DOI] [PubMed] [Google Scholar]
- 30.Wallner B, Elofsson A. Identification of correct regions in protein models using structural, alignment, and consensus information. Protein Sci. 2006;15:900–913. doi: 10.1110/ps.051799606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levitt M, Gerstein M. A unified statistical framework for sequence comparison and structure comparison. Proc. Natl Acad. Sci. USA. 1998;95:5913–5920. doi: 10.1073/pnas.95.11.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sippl MJ. Calculation of conformational ensembles from potentials of mena force: an approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 1990;213:859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- 34.Miyazawa S, Jernigan RL. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules. 1985;18:534–552. [Google Scholar]
- 35.Zhang C, Liu S, Zhou Y. Accurate and efficient loop selections by the DFIRE-based all-atom statistical potential. Protein Sci. 2004;13:391–399. doi: 10.1110/ps.03411904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J, Xie L, Honig B. Structural refinement of protein segments containing secondary structure elements: local sampling, knowledge-based potentials, and clustering. Proteins. 2006;65:463–479. doi: 10.1002/prot.21085. [DOI] [PubMed] [Google Scholar]
- 37.Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou H, Skolnick J. GOAP: a generalized orientation-dependent, all-atom statistical potential for protein structure prediction. Biophys. J. 2011;101:2043–2052. doi: 10.1016/j.bpj.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 40.Pollastri G, Przybylski D, Rost B, Baldi P. Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins. 2002;47:228–235. doi: 10.1002/prot.10082. [DOI] [PubMed] [Google Scholar]
- 41.Dor O, Zhou Y. Real-SPINE: an integrated system of neural networks for real-value prediction of protein structural properties. Proteins. 2007;68:76–81. doi: 10.1002/prot.21408. [DOI] [PubMed] [Google Scholar]
- 42.Faraggi E, Xue B, Zhou Y. Improving the prediction accuracy of residue solvent accessibility and real-value backbone torsion angles of proteins by guided-learning through a two-layer neural network. Proteins. 2009;74:847–856. doi: 10.1002/prot.22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng J, Randall AZ, Sweredoski MJ, Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005;33:W72–W76. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tegge AN, Wang Z, Eickholt J, Cheng J. NNcon: improved protein contact map prediction using 2D-recursive neural networks. Nucleic Acids Res. 2009;37:W515–W518. doi: 10.1093/nar/gkp305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eastwood M, Hardin C, Luthey-Schulten Z, Wolynes P. Evaluating protein structure-prediction schemes using energy landscape theory. IBM J. Res. Dev. 2001;45:475–497. [Google Scholar]
- 46.Goldstein RA, Luthey-Schulten ZA, Wolynes PG. Optimal protein-folding codes from spin-glass theory. Proc. Natl Acad. Sci. USA. 1992;89:4918–4922. doi: 10.1073/pnas.89.11.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stumpff-Kane AW, Feig M. A correlation-based method for the enhancement of scoring functions on funnel-shaped energy landscapes. Proteins. 2006;63:155–164. doi: 10.1002/prot.20853. [DOI] [PubMed] [Google Scholar]
- 48.Cortes C, Vapnik V. Support-vector networks. Mach. Learn. 1995;20:273–297. [Google Scholar]
- 49.Chang CC, Lin CJ. LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2001;2:27. [Google Scholar]
- 50.Kosinski J, Cymerman IA, Feder M, Kurowski MA, Sasin JM, Bujnicki JM. A “FRankenstein's monster” approach to comparative modeling: merging the finest fragments of Fold-Recognition models and iterative model refinement aided by 3D structure evaluation. Proteins. 2003;53(Suppl. 6):369–379. doi: 10.1002/prot.10545. [DOI] [PubMed] [Google Scholar]
- 51.Kosinski J, Gajda MJ, Cymerman IA, Kurowski MA, Pawlowski M, Boniecki M, Obarska A, Papaj G, Sroczynska-Obuchowicz P, Tkaczuk KL. FRankenstein becomes a cyborg: the automatic recombination and realignment of fold recognition models in CASP6. Proteins. 2005;61:106–113. doi: 10.1002/prot.20726. [DOI] [PubMed] [Google Scholar]
- 52.Kolinski A, Bujnicki JM. Generalized protein structure prediction based on combination of fold-recognition with de novo folding and evaluation of models. Proteins. 2005;61(Suppl. 7):84–90. doi: 10.1002/prot.20723. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 54.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 55.Herráez A. Biomolecules in the computer: Jmol to the rescue. Biochem. Mol. Biol. Educ. 2006;34:255–261. doi: 10.1002/bmb.2006.494034042644. [DOI] [PubMed] [Google Scholar]
- 56.Wlodawer A, Minor W, Dauter Z, Jaskolski M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures. FEBS J. 2008;275:1–21. doi: 10.1111/j.1742-4658.2007.06178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rossmann MG, Blow DM. The detection of sub-units within the crystallographic asymmetric unit. Acta Crystallogr. 1962;15:24–31. [Google Scholar]
- 58.Pawlowski M, Bujnicki JM. The utility of comparative models and the local model quality for protein crystal structure determination by molecular replacement. BMC Bioinformatics. 2012;13:289. doi: 10.1186/1471-2105-13-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr. A. 1994;50:157–163. [Google Scholar]
- 60.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997;30:1022–1025. [Google Scholar]