


Abstract
Identifying which mutation(s) within a given genotype is responsible for an observable phenotype is important in many aspects of molecular biology. Here, we present SigniSite, an online application for subgroup-free residue-level genotype–phenotype correlation. In contrast to similar methods, SigniSite does not require any pre-definition of subgroups or binary classification. Input is a set of protein sequences where each sequence has an associated real number, quantifying a given phenotype. SigniSite will then identify which amino acid residues are significantly associated with the data set phenotype. As output, SigniSite displays a sequence logo, depicting the strength of the phenotype association of each residue and a heat-map identifying ‘hot’ or ‘cold’ regions. SigniSite was benchmarked against SPEER, a state-of-the-art method for the prediction of specificity determining positions (SDP) using a set of human immunodeficiency virus protease-inhibitor genotype–phenotype data and corresponding resistance mutation scores from the Stanford University HIV Drug Resistance Database, and a data set of protein families with experimentally annotated SDPs. For both data sets, SigniSite was found to outperform SPEER. SigniSite is available at: http://www.cbs.dtu.dk/services/SigniSite/.
INTRODUCTION
Whether conducting research in vaccine design or trying to elucidate the intimate details of a given receptor::ligand interaction, genotype–phenotype correlation is a powerful tool to enhance the understanding of the minute subtleties, often characterizing research within the field of molecular biology.
The traditional approach for wet-laboratory analysis of genotype–phenotype correlations involves site-directed mutagenesis and subsequent quantification of mutation-impact on the phenotype, e.g. binding-affinity or catalytic efficiency. This approach of mutating all amino acid residues in a given protein is a time consuming and tedious task. Random mutagenesis has the advantage of introducing a large number of random mutations throughout the protein. One example of application of random mutagenesis is to increase the signal from near-infrared fluorescent proteins (1). In such a panel of sequenced variants with multiple mutations, it is a complex task to systematically pinpoint the exact amino acid residue(s), i.e. the genotype, associated with a given phenotype (e.g. fluorescence). Another area of application is genotype–phenotype association studies in proteins, which show inherent natural variability, as is the case for instance for proteins involved in the pathogenesis of malaria (2).
Here, we present SigniSite, an online application for subgroup-free residue-level genotype–phenotype correlation in protein multiple sequence alignments (MSAs). A number of methods have been developed for the identification of functional sites in protein sequences (3–10), most requiring a definition of functional subgroups before analysis. However, if the phenotype associated with the sequences is not categorical (e.g. substrate-specificity) but continuous (e.g. catalytic efficiency), a pre-division of sequences subgroups is none trivial. In contrast, SigniSite does not require any subgroup division or binary classification. Instead, SigniSite directly analyses the raw sequences and associated continuous values. The main novelty of SigniSite is that unlike conventional methods for the prediction of specificity determining positions (SDP), it not only predicts the positions in the MSA determining a given protein function but also makes a statistical evaluation of which types of amino acid residue substitutions (genotype) are associated with the observable phenotype at the SDP.
The web server implementation of the SigniSite method described here is an automatized online application with an easy-to-interpret graphical output. The application is easy to use for the non-expert end-user and aims at aiding researchers in the analysis of sequence data, where the phenotype is quantified by a real number. A list of abbreviations is available in the Supplementary Data.
THE WEB SERVER
User interface
The SigniSite server is intended to provide the non-expert user with a simple interface. At default settings, an amino acid residue is considered significantly associated with the MSA phenotype, if the P-value for the specific residue is smaller than or equal to 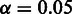 after Bonferroni Single-Step Correction for Multiple Testing (CMT) (11). On the submission page, sequences can be submitted to the server either as paste-in or via the file upload field. On submission, SigniSite will check whether the submitted sequences are aligned. If not, an MSA will be created using MAFFT (12). SigniSite will exclude any characters other than the one-letter representation of the 20 standard proteogenic amino acids from the analysis.
after Bonferroni Single-Step Correction for Multiple Testing (CMT) (11). On the submission page, sequences can be submitted to the server either as paste-in or via the file upload field. On submission, SigniSite will check whether the submitted sequences are aligned. If not, an MSA will be created using MAFFT (12). SigniSite will exclude any characters other than the one-letter representation of the 20 standard proteogenic amino acids from the analysis.
Input
As input SigniSite takes an MSA in FASTA-format (minimum two sequences). Each sequence must have an associated real number, stated white-space-separated as the last element in its FASTA header. At least two different values must exist in the MSA. The MSA is assumed pre-sorted, if the end-placed value is absent. A section with options for customizing the analysis is available. The following parameters are user-adjustable: (i) the level of significance ‘α’, 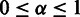 (default is 0.05). (ii) The method for CMT: ‘Bonferroni Single-Step’ (default), ‘Holm Step-Down’ (11) or ‘no correction’. (iii) The sorting of the sequences: ‘Decreasing’, highest sequence-associated value is considered the strongest, e.g. fluorescent protein signals, and vice versa for ‘Increasing’, e.g. binding affinity. Furthermore, the user can choose a reference sequence to assign sequence-specific positional output numbering. This is useful, when the MSA contains insertions. Finally, the user can modify the logo output by choosing to include either ‘Significant positions’ (default, displays all residues at positions where at least one amino acid residue has been identified as significantly associated with the data set phenotype), ‘Significant Residues’ (as for significant positions, but only including significant residues) or ‘Full Logo’ (all residues at all positions). At the results page, a button below the generated logo allows the user to fully customize the logo using Seq2Logo (13).
(default is 0.05). (ii) The method for CMT: ‘Bonferroni Single-Step’ (default), ‘Holm Step-Down’ (11) or ‘no correction’. (iii) The sorting of the sequences: ‘Decreasing’, highest sequence-associated value is considered the strongest, e.g. fluorescent protein signals, and vice versa for ‘Increasing’, e.g. binding affinity. Furthermore, the user can choose a reference sequence to assign sequence-specific positional output numbering. This is useful, when the MSA contains insertions. Finally, the user can modify the logo output by choosing to include either ‘Significant positions’ (default, displays all residues at positions where at least one amino acid residue has been identified as significantly associated with the data set phenotype), ‘Significant Residues’ (as for significant positions, but only including significant residues) or ‘Full Logo’ (all residues at all positions). At the results page, a button below the generated logo allows the user to fully customize the logo using Seq2Logo (13).
Output
The SigniSite output is intended to provide the end-user with an easily interpretable graphical representation of the statistical evaluations performed by SigniSite. An example of a sequence logo (13) generated by SigniSite is shown in Figure 1. The logo gives an overview of residue associations. See Figure 1 legend for further details. SigniSite will also generate a heatmap (Figure 2). The heatmap is intended to give a graphic overview of ‘hot’ and ‘cold’ regions in the MSA, with respect to the data set phenotype. See Figure 2 legend for details.
Figure 1.

Sequence logo. Example of sequence logo (13) output from SigniSite from the analysis of the ATV ∼Antivirogram multiple sequence alignment (MSA), truncated to p1 – p35 for the purpose of illustration (see ‘Materials and Methods’ section). The analysis was performed with default settings. On the x-axis are the MSA positions p and on the y-axis the Z-scores for each amino acid residue a (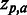 ). The height of each letter representing the residues is proportional to
). The height of each letter representing the residues is proportional to 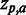 , i.e. the strength of the statistical association between the residue and the data set-phenotype. Residues above the Z = 0 line have a
, i.e. the strength of the statistical association between the residue and the data set-phenotype. Residues above the Z = 0 line have a 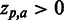 , i.e. enhances the phenotype, whereas residues below the Z = 0 line have a
, i.e. enhances the phenotype, whereas residues below the Z = 0 line have a 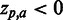 , i.e. inhibits the phenotype, e.g. the presence of a certain residue with favourable chemical properties may enhance binding (
, i.e. inhibits the phenotype, e.g. the presence of a certain residue with favourable chemical properties may enhance binding (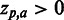 ), whereas a residue with unfavourable properties may inhibit binding (
), whereas a residue with unfavourable properties may inhibit binding (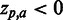 ). Colour-coding: acidic [DE]: red, basic [HKR]: blue, hydrophobic [ACFILMPVW]: black and neutral [GNQSTY]: green (14).
). Colour-coding: acidic [DE]: red, basic [HKR]: blue, hydrophobic [ACFILMPVW]: black and neutral [GNQSTY]: green (14).
Figure 2.

SigniSite heatmap from the analysis of the ATV ∼Antivirogram multiple sequence alignment (MSA), truncated to p1 – p35 for the purpose of illustration (see ‘Materials and Methods’ section). The analysis was performed with default settings. On the x-axis are the 20 proteogenic amino acids a and on the y-axis the positions p in the analysed MSA. The colour coding of the fields is such that fields reflecting 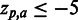 are blue, whereas
are blue, whereas 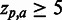 results in a red field. For
results in a red field. For 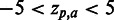 , nuances in between are used. If a residue has a
, nuances in between are used. If a residue has a 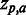 of 0, the cell is coloured grey. Absent residues are coloured black. If only one grey cell is present at a given position, this implies that the position is fully conserved, harbouring only this residue. If more grey cells are present, their associated P-values have become
of 0, the cell is coloured grey. Absent residues are coloured black. If only one grey cell is present at a given position, this implies that the position is fully conserved, harbouring only this residue. If more grey cells are present, their associated P-values have become 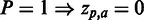 after correction for multiple testing.
after correction for multiple testing.
RESULTS
As an initial performance evaluation, we chose to analyse 18 human immunodeficiency virus type 1 (HIV-1) MSAs compiled from the Stanford University HIV Drug Resistance Database (15,16) (HIVdb) using Spearman’s rank correlation (SCC) to correlate the obtained SigniSite Z-scores (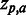 for each residue a at each position p) with the table of resistance mutation scores (RMS) also available from the HIVdb (see ‘Materials and Methods’ section), i.e.
for each residue a at each position p) with the table of resistance mutation scores (RMS) also available from the HIVdb (see ‘Materials and Methods’ section), i.e. 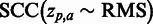 . Results are given in Table 1.
. Results are given in Table 1.
Table 1.
Benchmark results
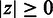
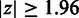
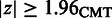


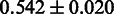
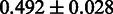


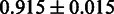



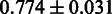

aCalculated against the RMS.
bCalculated against the (RMS + IAS)mut.
Measures are means ± SE. CMT: corrected for multiple testing, SCC: Spearman’s rank correlation, MCC: Matthews Correlation Coefficient, SENS: sensitivity, SPEC: specificity.
As the SCC evaluation is threshold dependent, a threshold-independent performance evaluation was added using the area under the receiver operator characteristics curve (AUC) measure, resulting in 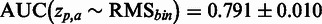 . Certain mutations not included in the RMS were repeatedly identified by SigniSite. As the majority of these mutations were found in the binary resistance annotations from the international antiviral society-USA (IAS) (17), we enriched the RMSbin with the IAS and re-calculated the AUC, obtaining a significant performance increase of
. Certain mutations not included in the RMS were repeatedly identified by SigniSite. As the majority of these mutations were found in the binary resistance annotations from the international antiviral society-USA (IAS) (17), we enriched the RMSbin with the IAS and re-calculated the AUC, obtaining a significant performance increase of 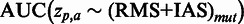
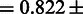 0.011(P = 5.16 · 10−4), two-tailed paired t-test).
0.011(P = 5.16 · 10−4), two-tailed paired t-test).
Furthermore, we evaluated the performance of SigniSite using performance measures: Matthew’s correlation coefficient (MCC), sensitivity (SENS) and specificity (SPEC) against (RMS + IAS)mut. See Table 1 for results.
Having obtained good results for both the threshold-dependent and -independent performance evaluations, we turned to benchmark SigniSite against similar existing methods. In a 2009 benchmark study (18), SPEER (5,19) was identified as the state-of-the-art method for prediction of specificity definition positions (SDP). We, therefore, here compared the performances of SigniSite and SPEER on each of their original benchmarks data sets (see ‘Materials and Methods’ section) against (RMS + IAS)pos. The results are shown in Figure 3. The results show that SigniSite outperforms SPEER on both data sets. The difference in predictive performance was, however, only found to be statistically significant for the HIVdb data set.
Figure 3.

Measures are mean (AUC) ± SE. Columns are: HIV [SPEER/SIGNI], SPEER and SigniSite’s predictions on the HIVdb data set. SDP [SPEER/SIGNI] SPEER and SigniSite’s predictions on the SDP data set. P-values quantifying the significance of the difference in performance were obtained using a two-tailed paired t-test.
DISCUSSION
SigniSite aims at providing a simple-to-use method for subgroup-free residue-level genotype–phenotype correlation in protein MSAs. SigniSite, thus, addresses a long-existing challenge in molecular biology; genotype-phenotype mapping. Genotype–phenotype mapping has a wide range of purposes in molecular biology, e.g. structural regions responsible for immunity (2), identifying protein-variants responsible for the severity of a disease (20) or coupling receptor polymorphisms to surface expression (21) etc.
Site-directed mutagenesis in proteins and subsequent quantification of mutation-impact on a given phenotype is a time consuming and tedious task. High-throughput methods such as e.g. random mutagenesis (1) have, therefore, been developed. However, the challenge of analysing the increasingly larger volumes of data being generated only becomes greater. Additionally, large genotype–phenotype data sets (GPDs) can be compiled from publicly available databases, such as the HIVdb (15,16). SigniSite addresses this exact challenge.
SigniSite was benchmarked on publicly available GPDs and RMS from the Stanford University HIV Drug Resistance Database (HIVdb) (15,16). We observed that for each of the 18 different benchmark data sets, SigniSite consistently identified certain residues, not annotated in the RMS table, as significantly associated with anti-viral drug resistance. We compared these identifications with binary resistance annotations from the International Antiviral Society-USA (IAS) (17) and found that the majority were indeed annotated as resistance impacting. This observation suggests that the RMS data are not exhaustive, and that the obtained correlation should rather be regarded as a lower bound of the true predictive performance.
As the SDP method SPEER (5,19) was found to be the state-of-the-art method in a 2009 benchmark study (18), we chose to compare SigniSite to SPEER. We observed that SigniSite significantly outperformed SPEER on the HIVdb data set (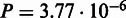 ) and for the SDP data set (as defined in the SPEER paper), SigniSite likewise outperformed SPEER, approaching a significant difference (
) and for the SDP data set (as defined in the SPEER paper), SigniSite likewise outperformed SPEER, approaching a significant difference (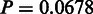 ). Furthermore, SigniSite was much faster, taking only a few minutes to analyse the largest of the MSA (
). Furthermore, SigniSite was much faster, taking only a few minutes to analyse the largest of the MSA (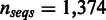 ). SPEER on the other hand requires to be compiled in a slower version, when
). SPEER on the other hand requires to be compiled in a slower version, when 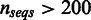 , taking ∼2 h to complete the analysis.
, taking ∼2 h to complete the analysis.
In conclusion, SigniSite provides two important novel features: (i) SigniSite does not require any manual annotation of the data before analysis, e.g. binder/non-binder classification, SigniSite requires only sequences and associated values. (ii) Unlike conventional SDP prediction methods like SPEER, SigniSite will not only identify positions impacting the phenotype but also pinpoint the exact amino acid residue substitution(s) responsible for the impact detected at the identified position. To the best of our knowledge, this level of resolution has so far not been available.
MATERIALS AND METHODS
Benchmark data sets
Summary, see Supplementary Data for details.
HIVdb resistance mutation scores
The table of RMS was downloaded from the HIVdb (15,16), available at http://hivdb.stanford.edu/DR/cgi-bin/rules_scores_hivdb.cgi?class=PI. The table of RMS contains information about positions known to harbour mutations (n = 688) compared with wild-type (WT) and their impact on resistance towards eight different protease inhibitors (PIs). Positive scores range is [3,60] (n = 296) and indicates that the mutation increases the resistance towards a given PI. Negative score range is 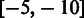 (n = 15) and indicates a decreased resistance. Scores of 0 (n = 377) indicate lack of resistance impact. At each position annotated in the table of RMS, the consensus residue was assigned an RMS of 0.
(n = 15) and indicates a decreased resistance. Scores of 0 (n = 377) indicate lack of resistance impact. At each position annotated in the table of RMS, the consensus residue was assigned an RMS of 0.
IAS resistance annotations
Protease mutations known to impact PI resistance were retrieved from the table ‘mutations in the protease gene associated with resistance to protease inhibitors’, in the International Antiviral Society USA (IAS)’s Update of the Drug Resistance Mutations in HIV-1: March 2013 (17). Also here, the consensus residue at annotated resistance positions was assigned an IAS score of 0.
Table transformations
The following table transformations were performed: 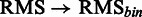 , such that
, such that 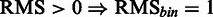 , otherwise
, otherwise 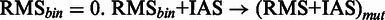 , such that
, such that 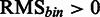 or
or 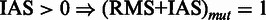 , otherwise
, otherwise 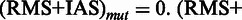
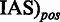 , such that for each position in (RMS + IAS)mut the resulting
, such that for each position in (RMS + IAS)mut the resulting 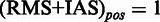 if at least one
if at least one 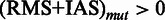 , otherwise
, otherwise 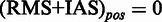 . In all tables, any score
. In all tables, any score 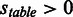 is considered an actual positive and any score
is considered an actual positive and any score 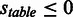 is considered an actual negative (Table 2).
is considered an actual negative (Table 2).
Table 2.
Overview of target table notation
| Notation | Format | Level | Annotating |
|---|---|---|---|
| RMSa | Real num. | Residue | Fold-change in PI resistance |
| IASb | Binary | Residue | PI ass. resistance mutations |
| RMSbinc | Binary | Residue | PI ass. resistance mutations |
| (RMS + IAS)mutd | Binary | Residue | PI ass. resistance mutations |
| (RMS + IAS)pose | Binary | Position | Positions ass. with PI resistance |
aIt is used when calculating SCC, bit is used to look up mutations not annotated in 1, but repeatedly identified by SigniSite, cit is used when calculating AUC, dit is used for the enriched AUC calculation and when calculating the MCC, SENS and SPEC, eit is used as positional targets, when comparing the predictive performances of SigniSite and SPEER.
‘num.’, ‘ass.’, ‘PI’ abbreviates ‘numbers’, ‘association’ and ‘protease inhibitor’. In all tables, any score 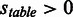 is considered an actual positive and any score
is considered an actual positive and any score 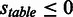 is considered an actual negative.
is considered an actual negative.
MSAs from the HIVdb protease GPDs
GPDs were downloaded from the Stanford University HIV Drug Resistance Database (HIVdb) (15,16) Version 5.0, March, 2012, available at http://HIVdb.stanford.edu/cgi-bin/GenoPhenoDS.cgi. MSAs were compiled from the GPDs. Each MSA contains the sequences of a set of HIV-1 protease variants with measured fold change in resistance (compared with WT) towards the same PI, measured using the same assay. Only PIs present in both the table of RMS and the GPDs were used limiting the analysis to 6 PIs: ATV, IDV, LPV, NFV, SQV and TPV each of which was assayed using the three assays: ‘Antivirogram’ (Virco™), ‘PhenoSense’ (ViroLogic™) and ‘All Others’. A total of 12 714 sequences were constructed and compiled into 18 MSAs. The length of each of the protease variants is 99 amino acid residues.
The SPEER program and SDP benchmark data
SPEER, MSAs and corresponding experimentally annotated specificity determining sites were downloaded from the SPEER repository available at: ftp://ftp.ncbi.nih.gov/pub/SPEER/ (5,19). We downloaded the latest curated version of the data as described by Chakrabarti and Panchenko (18).
The SigniSite method
The method takes a set of (protein) sequences as input. If the sequences are not aligned, Signisite will use MAFFT (12) to make an MSA from the input sequences. Subsequently, the sequences are ranked with respect to a real number associated with each sequence, e.g. the replicative capacity or catalytic efficiency. For each amino acid at each position in the MSA, a non-parametric test is performed to test whether the observed ranks deviate significantly from the expected ranks. CMT of the resulting P-values may be performed using Bonferroni single-step or Holm step-down procedures. The resulting Z-scores per residue are visualized in a logo plot and a heatmap.
Brief description of the method underlying SigniSite
(see Supplementary Data for details). Initially each sequence is assigned a rank by sorting the sequence associated values (either ascending or descending depending on type of value) and then assigning a rank of ‘1’ to the first sequence after sorting, ‘2’ to the second and so forth. Each amino acid residue a observed at position p (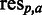 ) in the MSA is then assigned the rank of the sequence to which it belongs. This way each
) in the MSA is then assigned the rank of the sequence to which it belongs. This way each 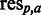 is associated with a specific rank. At each position in the MSA, the mean ranks of each residue type are then calculated and placed in a rank matrix, where each row corresponds to a position in the MSA and each column to one of the 20 standard proteogenic amino acids, sorted according to A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y and V (SigniSite will exclude any characters but these 20).
is associated with a specific rank. At each position in the MSA, the mean ranks of each residue type are then calculated and placed in a rank matrix, where each row corresponds to a position in the MSA and each column to one of the 20 standard proteogenic amino acids, sorted according to A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y and V (SigniSite will exclude any characters but these 20).
Subsequently, SigniSite evaluates for each position and residue type the difference between the mean of the observed and expected ranks. The mean of the expected ranks is the mean of the ranks we would observe if the residue type 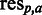 was randomly distributed over the column p in the MSA. This difference between observed and expected ranks is quantified by a Z-score assigned to each residue type at each position, yielding a Z-score-matrix. If a given position is fully conserved, z = 0 is assigned to the conserved residue. If a given residue type is absent at a given position,
was randomly distributed over the column p in the MSA. This difference between observed and expected ranks is quantified by a Z-score assigned to each residue type at each position, yielding a Z-score-matrix. If a given position is fully conserved, z = 0 is assigned to the conserved residue. If a given residue type is absent at a given position, 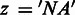 is assigned.
is assigned.
The non-parametric statistics, on which SigniSite is based, are similar to that of Wilcoxon test statistics (22), where the obtained evaluation scores can be approximated by the standard normal distribution, thus allowing Z-score conversion to P-values by standard method. As one test is performed per residue type, per position, SigniSite will by default apply Bonferroni single-step (11) CMT to adjust the reported P-values.
Benchmarking
For each of the 18 MSAs compiled from the HIVdb GPDs (see ‘Materials and Methods’ section), a set of predictions were made (Z-scores) estimating the strength of the association of each residue type a at each position p (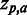 ) to the phenotype of the MSA. The obtained set of
) to the phenotype of the MSA. The obtained set of 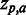 ’s was then correlated with the RMS using Spearman’s rank correlation (SCC) at three significance thresholds: including residues for which: (i)
’s was then correlated with the RMS using Spearman’s rank correlation (SCC) at three significance thresholds: including residues for which: (i) 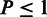 , (ii)
, (ii) 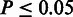 and (iii)
and (iii) 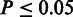 after CMT. The SCC was recorded for each of the 18 MSAs, and the mean and standard error (SE) of the means were calculated.
after CMT. The SCC was recorded for each of the 18 MSAs, and the mean and standard error (SE) of the means were calculated.
For evaluating threshold-independent performance, the AUC measure was applied. The AUC was calculated against two sets of targets: RMSbin and the enriched set of targets (RMS + IAS)mut. The mean AUC and SE were calculated for each set of targets.
Finally, the sensitivity, specificity and MCC were calculated at the same thresholds as the SCC against the enriched set of targets (RMS + IAS)mut. The sensitivity, specificity and MCC were recorded for each of the 18 MSAs, and the means and SEs were calculated.
Comparing SigniSite and SPEER
To compare the performance of SigniSite with that of existing methods, we turned to a 2009 benchmark study by Chakrabarti and Panchenko (18) comparing the predictive performance of five SDP prediction methods, on a set of protein families with experimentally annotated SDPs. As SPEER (5,19) in this benchmark was found to be the best performing method, we here limit our analysis to comparing SigniSite and SPEER by applying both methods to their respective GPDs.
SPEER outputs positional predictions, whereas SigniSite assigns a Z-score for each residue type at each position. To cast the SigniSite Z-scores into one score per positions, the maximum of the absolute Z-scores was chosen.
SigniSite assigns a prediction value to all positions regardless of residue composition, whereas SPEER by default will skip any fully conserved and positions with >20% gaps. To get prediction values for all positions, we assign a value of ‘−100’ to positions not predicted by SPEER (this value is lower than any score predicted by SPEER).
SPEER requires each sequence in an MSA to be subgroup-annotated before analysis. To accommodate this requirement, each HIV MSA was split into two subgroups, by sorting the sequences in the MSA descending on their associated real values and then splitting the sequences into subgroup ‘1’ or ‘2’ on the median of the sorted values.
To perform the rank analysis SigniSite requires that each sequence in the MSA has an associated real number. Of the 20 SDP MSAs, 13 contain only subgroups ‘1’ and ‘2’. We chose to use these 13 MSAs for the benchmark, using ‘1’ or ‘2’ as ‘SigniSite real number values’.
This way the following two comparisons were made: SigniSite versus SPEER on the HIV protease data set and SigniSite versus SPEER in the SDP data set. The AUC measure was used to quantify the performance of each method on each benchmark data set.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary description of the SigniSite Method, Supplementary descriptions of the benchmark data sets, Supplementary section on the impact of chosen seed for random number generation, Supplementary description of the benchmarks strategy, Supplementary Tables of HIV-1 PIs and abbreviations.
FUNDING
National Institutes of Health [HHSN272201200010C]; EU FP7 PepChipOmics: The European Union 7th Framework Program FP7/2007-2013 [222773]; The Center for Genomic Epidemiology (www.genomicepidemiology.org) grant 09-067103/DSF from the Danish Council for Strategic Research; The University of Copenhagen - Program of Excellence. Funding for open access charge: Technical University of Denmark - PhD programme.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Martin Blythe for coming up with the name SigniSite.
REFERENCES
- 1.Shcherbo D, Shemiakina II, Ryabova AV, Luker KE, Schmidt BT, Souslova EA, Gorodnicheva TV, Strukova L, Shidlovskiy KM, Britanova OV, et al. Near-infrared fluorescent proteins. Nat. Methods. 2010;7:827–829. doi: 10.1038/nmeth.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gnidehou S, Jessen L, Gangnard S, Ermont C, Triqui C, Quiviger M, Guitard J, Lund O, Deloron P, Ndam NT. Insight into antigenic diversity of VAR2CSA-DBL5ϵ Domain from multiple Plasmodium falciparum placental isolates. PLoS One. 2010;5:e13105. doi: 10.1371/journal.pone.0013105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt BW, Feenstra KA, Heringa J. Multi-Harmony: detecting functional specificity from sequence alignment. Nucleic Acids Res. 2010;38:35–40. doi: 10.1093/nar/gkq415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capra JA, Singh M. Characterization and prediction of residues determining protein functional specificity. Bioinformatics. 2008;24:1473–1480. doi: 10.1093/bioinformatics/btn214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakrabarti S, Bryant SH, Panchenko AR. Functional specificity lies within the properties and evolutionary changes of amino acids. J. Mol. Biol. 2007;373:801–810. doi: 10.1016/j.jmb.2007.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalinina OV, Novichkov PS, Mironov AA, Gelfand MS, Rakhmaninova AB. SDPpred: a tool for prediction of amino acid residues that determine differences in functional specificity of homologous proteins. Nucleic Acids Res. 2004;32:W424–W428. doi: 10.1093/nar/gkh391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pei J, Cai W, Kinch LN, Grishin NV. Prediction of functional specificity determinants from protein sequences using log-likelihood ratios. Bioinformatics. 2006;22:164–171. doi: 10.1093/bioinformatics/bti766. [DOI] [PubMed] [Google Scholar]
- 8.Ye K, Feenstra KA, Heringa J, Ijzerman AP, Marchiori E. Multi-RELIEF: a method to recognize specificity determining residues from multiple sequence alignments using a Machine-Learning approach for feature weighting. Bioinformatics. 2008;24:18–25. doi: 10.1093/bioinformatics/btm537. [DOI] [PubMed] [Google Scholar]
- 9.Buslje CM, Teppa E, Domnico TD, Delfino JM, Nielsen M. Networks of high mutual information define the structural proximity of catalytic sites: implications for catalytic residue identification. PLoS Comput. Biol. 2010;6:e1000978. doi: 10.1371/journal.pcbi.1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 11.Dudoit S, Yang YH, Callow MJ, Speed TP. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Stat. Sin. 2002;12:111–139. [Google Scholar]
- 12.Katoh K, Misawa K, Kuma KI, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomsen MCF, Nielsen M. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 2012;40:W281–W287. doi: 10.1093/nar/gks469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lund O, Nielsen M, Lundegaard C, Kesmir C, Brunak S. Immunological Bioinformatics. Cambridge, MA, London, England: The MIT Press; 2005. [Google Scholar]
- 15.Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;30:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shafer RW. Rationale and uses of a public HIV drug-resistance database. J. Infect. Dis. 2006;194:S51–S58. doi: 10.1086/505356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson VA, Calvez V, Gnthard HF, Paredes R, Pillay D, Shafer R, Wensing AM, Richman DD. Update of the drug resistance mutations in HIV-1: March 2013. Top Antivir. Med. 2013;21:6–14. [PMC free article] [PubMed] [Google Scholar]
- 18.Chakrabarti S, Panchenko AR. Ensemble approach to predict specificity determinants: benchmarking and validation. BMC Bioinformatics. 2009;373:801–810. doi: 10.1186/1471-2105-10-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakraborty A, Mandloi S, Lanczycki CJ, Panchenko AR, Chakrabarti S. SPEER-SERVER: a web server for prediction of protein specificity determining sites. Nucleic Acids Res. 2012;40:W242–W248. doi: 10.1093/nar/gks559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dendrou CA, Plagnol V, Fung E, Yang JH, Downes K, Cooper JD, Nutland S, Coleman G, Himsworth M, Hardy M, et al. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat. Genet. 2009;41:1011–1015. doi: 10.1038/ng.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armitage P, Berry G, Matthews JNS. Statistical Methods in Medical Research. Malden, MA, USA: Blackwell Publishing Company; 2002. [Google Scholar]