

Abstract
During the past decade, the field of mass spectrometry imaging (MSI) has greatly evolved, to a point where it has now been fully integrated by most vendors as an optional or dedicated platform that can be purchased with their instruments. However, the technology is not mature and multiple research groups in both academia and industry are still very actively studying the fundamentals of imaging techniques, adapting the technology to new ionization sources, and developing new applications. As a result, there are an important variety of data file formats used to store mass spectrometry imaging data and concurrent to the development of MSi collaborative efforts have been undertaken to introduce common imaging data file formats. However, few free software packages to read and analyze files of these different formats are readily available. We introduce here MSiReader, a free open source application to read and analyze high resolution MSI data from the most common MSi data formats. The application is built on the Matlab platform (Mathworks, Natick, MA) and includes a large selection of data analysis tools and features. People who are unfamiliar with the Matlab language will have little difficult navigating the user friendly interface, and users with Matlab programming experience can adapt and customize MSiReader for their own needs.
INTRODUCTION
Mass Spectrometry imaging (MSi) is a technology that combines the mass spectrometric data collected over a sample surface with spatial information to reconstruct an abundance heat map of a specific m/z value. Nowadays, mass spectrometer manufacturers and third parties have released mass spectrometry sources that include a complete suite of software, providing an interface for the user to input the imaging parameters and an interface to the imaging source of the mass spectrometer instrument. Software suites often include an application to load the instrument specific MSi data, reconstruct an image and offer post processing tools to navigate through the data, enhance images, and help with data interpretation including FlexImaging (Bruker, Billerica, MA), ImageQuest (Thermofisher Scientific, Bremen, Germany) and Tissueview (AB Sciex, Foster City, CA). Other instrument makers have implemented imaging capability on their MALDI instruments and offer converter tools for their proprietary file format so that data can be analyzed using third party software. This is the case for Shimadzu (Kyoto, Japan) and Waters (Manchester, UK) that implemented tools to convert their native formats to formats that can be viewed and processed using Biomap (Novartis, Basel, Switzerland), a free processing and viewing software for mass spectrometry imaging data. Biomap[1], together with DatacubeV0.7, developed by AMOLF, are two free vendor neutral imaging software packages that are very popular with research groups developing their own imaging source and with users who wish to compare data obtained from different instruments.
Other imaging packages have been developed during the past few years that have been shelved, marked as “in-development”, or that were not readily available for download at the time that this article was written. Among those projects are MITICS [2], MIRION [3], SpectViewer (CEA), and a new version of Datacube that is currently being developed [4]. This enumeration excludes all software that is instrument specific such as MSImageView[5] (FlashQuant, ABSciex 4000).
MSiReader is a very good option for users that are investigating their own imaging sources as well as researchers in industry who want to compare mass spectrometry data collected with different instruments and have full control over the data processing. MSiReader currently supports most common open data formats for storage and exchange of mass spectrometry data (e.g. mzXML, imzML, img (Analyze 7.5), ASCII).
RESULTS AND DISCUSSION
MSiReader was first designed as a custom application to process and view imaging data acquired from a IR-MALDESI imaging source, developed in our laboratory [6], coupled to a Fourier transform-ion cyclotron resonance mass spectrometer (FT-ICR MS). As mentioned earlier, at the moment of the submission of this article, two free software packages were readily available to process and view mass spectrometry imaging data and both offer great features yet have unfortunate limitations such as the maximum allowed intensity value and the mass resolution (minimum distance between m/z data points. MSiReader was thus designed to maintain the mass resolution of the original instrument data without limiting the dynamic range of the data on the abundance scale. It was decided to program MSiReader using the Matlab platform since it is a common programming language for data analysis and viewing, offers a rich set of vector and matrix processing tools and it is easy to modify and customize in an open source environment. MSiReader requires the following Matlab toolboxes: Statistics, Bioinformatics and Image Processing.
Some foreseeable limitations of the Matlab language for the analysis of mass spectrometry imaging data include a slower computation speed (compared to java and C++) and maximum dataset size (limited by computer’s RAM). MSiReader V0.00 has been designed and tested with Matlab R2012a and R2012b under the Windows OS WIN7 (64-bit) and 8 Gb of RAM. Under these conditions, files containing 5000 spectra and where the number of m/z data points per scan varied from 20,000 to 90,000 have been routinely processed. Recent testing with 16 Gb of RAM on an Intel i7 PC (3.4GHz, WIN7, 64-bit) show that data sets containing more than 20,000 spectra are easily processed.
However, Matlab is well known as a good platform for the rapid development and testing of prototype algorithms. A comparison of Datacube, Biomap and MSiReader is presented in Table 1.
Table 1.
Comparison of free MS imaging software packages readily available for download.
| Software | Authors | Input file format | Advantages | Limitations |
|---|---|---|---|---|
| Biomap V3.8.0.4† | Rausch, Stoeckli |
|
|
|
| DataCube V0.7† | AMOLF |
|
|
|
| MSiReader V0.0 | Robichaud et al. |
|
|
|
Latest version available at the moment of writing the manuscript
MSiReader was first programmed to be used with mzXML [7] files, which is an open XML (extensible markup language) format developed to store, share and process MS data generated by different instrument manufacturers (mostly liquid chromatography MS data). Free applications to convert MS files from different instrument native formats (e.g. Thermo, Agilent, ABSciex, Waters, Bruker) to mzXML such as MSConvert, developed by the ProteoWizard project [8, 9] are available online [10]. Additional common open data formats for storage and exchange of mass spectrometry data were later added such as the imZML format (both processed and continuous), developed by the EU project Computis [11, 12] specifically for sharing MSi data. There are multiple converters that can be used to go directly from the vendor’s format to imzML such as RawtoImzmlConverter (Thermo), HDI (Waters). The msimaging website [5] keeps an updated version of these conversion applications in their imzML section. Users can also use MSConvert to convert vendor’s format to HUPO-PSI’s mzML[13] and then use the recently developed imzMLConverter[14] to convert the mzML file to an imzML file. Vendors are showing more and more interest in those formats to the point where most have committed to provide mzML support in the next release of their software [13].
Analyze 7.5 was recently added as a data format supported by MSiReader. Analyze 7.5 was originally developed by the Mayo clinic to share MRI imaging data [15] and adapted for mass spectrometry imaging by Stoeckli et al. [1] while developing the Biomap application. The various file conversion pathways from vendor’s format to MSiReader are summarized in Figure 1.
Figure 1.
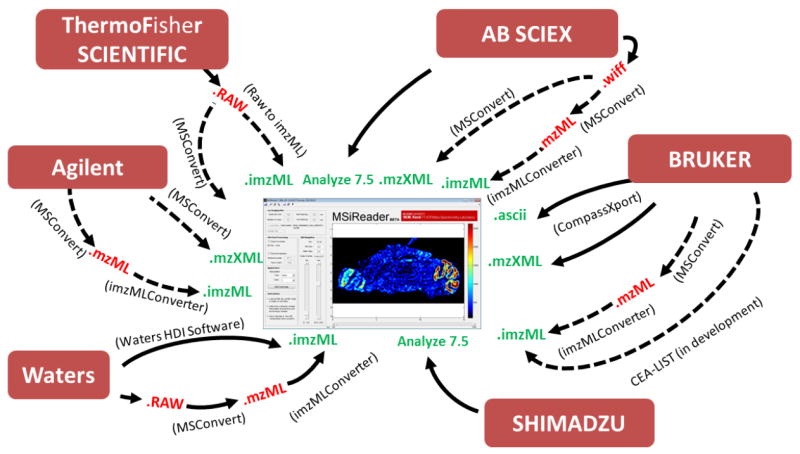
Different file conversion pathways from vendors’ file formats to MSiReader compatible files (green).
Even though Matlab software is necessary to run MSiReader, users do not need to be fluent in the Matlab programming language to use the application since a user-friendly interface was built using the GUI design environment (GUIDE). Knowledge of Matlab language is only necessary if a user wishes to customize functions in the script or add new capabilities.
A screenshot of the interface is shown in Figure 2. MSiReader currently includes a peak detection and feature recognition algorithm, where supervised analysis is used to identify molecules that are more abundant in a user defined region of interest (ROI) on the tissue when compared to a reference ROI also picked by the user. Peaks are detected by comparing their average intensities over the 2 ROI’s as well as their occurrence. Those criteria for detection are well defined in the MSiReader interface and can be easily modified to maximize output (see Figure S1). A mass spectrum showing the superposed averaged signal for both the interrogated and the reference zones is generated and can be used to browse through the list of peaks that are specific to the interrogated zone (see Figure S2). The extracted peak list can also be saved in an Excel workbook where a mass excess plot is automatically generated and compared to the mass excess distribution of lipids [6, 16] from the LipidMaps database [17, 18] (see Figure S3). To calculate the peak centroids, the user is given the option to use the 3-points parabola algorithm[19] or the fully customizable MSpeaks function developed by Matlab. This univariate comparison technique between interrogated and reference zone is particularly useful for peak detection in the context of histology dependent/directed [20] or feature based analysis.
Figure 2.
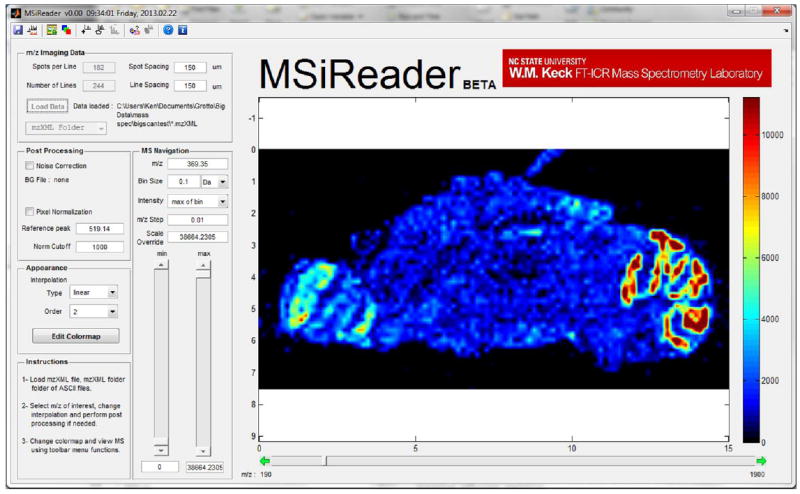
MSiReader user friendly interface. User can choose among several different color scales and adjust intensity using scrollbars. Another scrollbar can be used to navigate through the imaging file or the m/z value can be entered directly as input. All other data analysis features can be accessed in left panel or in toolbar menu.
Among data processing operations that can be routinely performed by MSiReader are spectrum baseline correction, peak normalization (individual scans are normalized to the intensity of a specific m/z value), background signal subtraction (e.g. automatically remove signal from ambient ions or matrix) and extraction of single or averaged mass spectrum over a user defined region of interest (see Figure S4). The heat map appearance can be customized using Matlab’s Colormap editor and several interpolation schemes can be applied at the image level. The intensity scale for the image generation can also be easily adjusted using scrollbars. The user may also choose to display the abundance on the heatmap as the maximum value in the selected bin size or the sum of the data points in that same bin. Units for bin size itself can also be changed from Dalton (Da) to part per million (ppm) at any time. Series of images can be automatically generated from peak list. Finally, a colocalization tool has been integrated in MSiReader so that up to three m/z ion maps can be superposed on the same heatmap image (see Figure S5).
Among the other features that are currently under development and will be included in future updates of the software are the capability to perform internal post-acquisition mass calibration of the data to improve mass accuracy. We are also currently working on processing algorithms to better handle larger imaging files. A user manual is provided with MSiReader explaining in details all its features as well as giving more details about the different algorithms used to process the data. MSiReader is currently released under an open source license (BSD 3)[21].
CONCLUSION
We introduced MSiReader, an open source, powerful interface data built on the Matlab platform to process, analyze and visualize imaging data. MSiReader currently supports most of the common mass spectrometry file sharing formats. Even though it has some limitations regarding imaging file size, MSiReader is an invaluable resource to MSi source developers, researchers, or people in industry who are looking for a free open source interface to interpret or compare MSi data. Users are welcome to download the latest version of MSiReader on http://www.msireader.com and to contact us through the website to give suggestions, report problems with the software or share new features.
Supplementary Material
Acknowledgments
The authors would like to thank Alan M. Race from the University of Birmingham for granting us permission to include his imzML parser function[14] in MSiReader. The authors would like to gratefully acknowledge the financial support received from the National Institutes of Health (R01GM087964), the W. M. Keck Foundation, and North Carolina State University.
References
- 1.Stoeckli M, Staab D, Staufenbiel M, Wiederhold KH, Signor L. Molecular imaging of amyloid beta peptides in mouse brain sections using mass spectrometry. Anal Biochem. 2002;311(1):33–39. doi: 10.1016/s0003-2697(02)00386-x. [DOI] [PubMed] [Google Scholar]
- 2.Jardin-Mathe O, Bonnel D, Franck J, Wisztorski M, Macagno E, Fournier I, Salzet M. MITICS (MALDI Imaging Team Imaging Computing System): a new open source mass spectrometry imaging software. J Proteomics. 2008;71(3):332–345. doi: 10.1016/j.jprot.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Hester A, B W, Leisner A, Maass K, Paschke C, Spengler B. 53rd ASMS Conference on Mass Spectrometry and Allied Topics; 2005. [Google Scholar]
- 4.Smith DF, Kharchenko A, Konijnenburg M, Klinkert I, Pasa-Tolic L, Heeren RM. Advanced Mass Calibration and Visualization for FT-ICR Mass Spectrometry Imaging. J Am Soc Mass Spectr. 2012 doi: 10.1007/s13361-012-0464-1. [DOI] [PubMed] [Google Scholar]
- 5.msimaging. http://www.maldi-msi.org/
- 6.Robichaud G, Barry J, Garrard K, Muddiman D. Infrared Matrix-Assisted Laser Desorption Electrospray Ionization (IR-MALDESI) Imaging Source Coupled to a FT-ICR Mass Spectrometer. J Am Soc Mass Spectr. 2012:1–9. doi: 10.1007/s13361-012-0505-9. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, Aebersold R. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22(11):1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 8.ProteoWizard. http://proteowizard.sourceforge.net/
- 9.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24(21):2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.mzXML converter. http://tools.proteomecenter.org/wiki/index.php?title=Formats:mzXML.
- 11.Römpp A, S T, Hester A, Klinkert I, Heeren R, Stöckli M, et al. Data Mining in Proteomics. Humana Press; New York: 2010. [Google Scholar]
- 12.Schramm T, Hester A, Klinkert I, Both JP, Heeren RM, Brunelle A, Laprevote O, Desbenoit N, Robbe MF, Stoeckli M, Spengler B, Rompp A. imzML - A common data format for the flexible exchange and processing of mass spectrometry imaging data. J Proteomics. 2012;75(16):5106–5110. doi: 10.1016/j.jprot.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 13.Martens L, Chambers M, Sturm M, Kessner D, Levander F, Shofstahl J, Tang WH, Rompp A, Neumann S, Pizarro AD, Montecchi-Palazzi L, Tasman N, Coleman M, Reisinger F, Souda P, Hermjakob H, Binz PA, Deutsch EW. mzML--a community standard for mass spectrometry data. Mol Cell Proteomics. 2011;10(1) doi: 10.1074/mcp.R110.000133. R110 000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Race AM, Styles IB, Bunch J. Inclusive sharing of mass spectrometry imaging data requires a converter for all. J Proteomics. 2012;75(16):5111–5112. doi: 10.1016/j.jprot.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 15.Analyze 7.5. http://eeg.sourceforge.net/
- 16.McDonnell LA, van Remoortere A, de Velde N, van Zeijl RJM, Deelder AM. Imaging Mass Spectrometry Data Reduction: Automated Feature Identification and Extraction. J Am Soc Mass Spectr. 2010;21(12):1969–1978. doi: 10.1016/j.jasms.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH, Jr, Murphy RC, Raetz CR, Russell DW, Subramaniam S. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007;35(Database issue):D527–532. doi: 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LIPID MAPS Structure Database (LDMS) doi: 10.1093/nar/gkl838. http://www.lipidmaps.org/data/structure/index.html. [DOI] [PMC free article] [PubMed]
- 19.Giancaspro C, Comisarow MB. Exact Interpolation of Fourier-Transform Spectra. Appl Spectrosc. 1983;37(2):153–166. [Google Scholar]
- 20.Jones EA, Deininger SO, Hogendoorn PC, Deelder AM, McDonnell LA. Imaging mass spectrometry statistical analysis. J Proteomics. 2012 doi: 10.1016/j.jprot.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 21.Open Source Initiative OSI - The BSD 3-Clause License. http://opensource.org/licenses/BSD-3-Clause.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.