

Abstract
Background. Mycobacterium tuberculosis can grow in the hostile intracellular environment of macrophages by actively evading macrophage-associated antibacterial activities. The stress response factor SigH contributes to this process by modulating β-chemokine and interleukin 6 (Il6) expression. Hence, Il6 is of critical importance for acquired immunity against M. tuberculosis infection. Here, we attempted to better characterize the role of Il6 in the immune response to M. tuberculosis infection.
Methods. A small interfering RNA–based approach was used to silence expression of the Il6 transcript in host macrophages infected with a wild-type strain of M. tuberculosis or an attenuated mutant strain of M. tuberculosis (Mtb:Δ-sigH). The outcome was measured by the analysis of bacterial burden and transcriptome-wide analysis of host gene expression. Transcriptome results were confirmed via quantitative polymerase chain reaction and enzyme-linked immunosorbent assay.
Results. Wild type and Mtb:Δ-sigH infection of host macrophages in which Il6 had been silenced resulted in increased expression of interferon-inducible genes, especially those involved in type I interferon signaling. The expression of Ly-6 genes was significantly higher in cells infected with Mtb:Δ-sigH, compared with those infected with the wild-type strain (P < .05).
Conclusions. M. tuberculosis regulates host Il6 production to inhibit type I interferon signaling and, consequently, disease progression. Mtb:Δ-sigH is associated with delayed activation of macrophages, compared with the wild-type strain, and with delayed inflammatory stimuli as consequence. These findings have important implications for improving understanding of the mechanisms behind M. tuberculosis virulence and pathogenesis and provide an initial road map to further investigate the mechanisms that may account for the deleterious effects of type I interferons in M. tuberculosis infection.
Keywords: M. tuberculosis, Il6, silencing, macrophages, sigma factor
Resistance to Mycobacterium tuberculosis infection requires the host to restrict bacterial replication via a successful T-helper 1 (Th1) response [1]. Hence, proinflammatory cytokines and chemokines induced by M. tuberculosis are crucial for immunity to tuberculosis. Because macrophages have potent antimicrobial functions, they play an important role in the innate immune response to pathogens and, thus, are essential in shaping adaptive immune responses [2, 3]. Nevertheless, M. tuberculosis can evade the functions of macrophages and actively grow within their hostile intracellular environment [4]. As part of this strategy, M. tuberculosis inhibits phagosome maturation and acidification, interferes with responses to interferon γ (Ifng), resists antimicrobial agents that damage the bacterial cell envelope, and counters toxic reactive oxygen and nitrogen intermediates [1, 5]. The evasion of these innate immune defenses allows M. tuberculosis to replicate within the host and escape early immune detection. Therefore, regulation of early immune events by pathogens also interferes with the induction of proinflammatory cytokines and, consequently, with the disease outcome [2, 6, 7].
M. tuberculosis can restrict macrophage activation and proinflammatory responses through the stress response factor SigH [8]. Transcriptional comparison of infected macrophages demonstrated that the Mtb:Δ-sigH mutant strain induced significantly higher levels of interleukin 6 (Il6) than M. tuberculosis, suggesting its critical importance for acquired immunity against tuberculosis. Il6 is a pleiotropic proinflammatory cytokine, and its increased production is a hallmark of many human chronic inflammatory diseases.
Tumor necrosis factor (Tnf) and Il6 are differentially required for protective immune responses in mice infected with M. tuberculosis. However, despite its importance in mediating inflammation, Il6 is not as essential as tumor necrosis factor for antimycobacterial effector mechanisms [9]. Il6 is critical to resistance against tuberculosis after infection with high doses of intravenously delivered M. tuberculosis, but it is dispensable for the control of mycobacterial growth after low-dose aerosol-delivered infection [10–12]. In addition, it has been shown that Il6 is essential for generating protective Th1 immune responses after vaccination with a subunit vaccine against M. tuberculosis [13] but that it has an inhibitory function with respect to Ifng signaling [12]. Hence, we used a small interfering RNA (siRNA)–based approach to further characterize the role of Il6 and the components of the macrophage-signaling machinery that regulate intracellular survival of M. tuberculosis.
MATERIALS AND METHODS
Murine Cell Line and Il-6 siRNA
Macrophage cell lines derived from C57BL6/J wild-type and Toll-like receptor 2 (TLR2)–knockout mice were obtained from BEI Resources (catalog nos. NR-9456, NR-9457, and NR-9567; Manassas, VA). Cell lines were cultured as adherent cells in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum under a humidified 5% CO2 atmosphere, as recommended by BEI Resources. Four distinct ON-TARGETplus SMARTpool siRNA species targeting different sequences of the Il6 transcript were obtained from Dharmacon (catalog no. L-043739-00-0005). Predesigned siRNA obtained from Life Technologies included GAPDH siRNA (catalog no. AM4624) as a positive control and siRNA with a scrambled sequence (catalog no. 12935200) as a negative control.
Infection With M. tuberculosis and Mtb:Δ-sigH Strains
The wild-type M. tuberculosis strain CDC1551 and the mutant strain Mtb:Δ-sigH were cultured as previously described [14, 15]. C57BL/6 macrophages were infected with either M. tuberculosis wild-type or its mutant counterpart at a multiplicity of infection of 1:10 and after 4 hours, cells were washed extensively to remove extracellular bacilli [8, 16].
siRNA Transfection
Transfection was performed by mixing Il6 siRNA and 1 μL of Lipofectamine RNAiMAX (Invitrogen) for 20 minutes at room temperature. The complex was added to a single well of a 24-well plate with 1 × 105 preinfected cells per well. Transfections with positive and negative controls were performed in parallel. All transfections were undertaken in a final volume of 600 μL, with a final siRNA concentration of 100 nM. Transfected cells were harvested 24 or 48 hours after transfection.
Real-Time Polymerase Chain Reaction (PCR)
Real-time PCR was performed with complementary DNA corresponding to 100 ng of each independent RNA sample, using the SYBR green Supermix kit (Applied Biosystems), with specific primers for each target obtained from PrimerBank (available at: http://pga.mgh.harvard.edu/primerbank/) or created by us (Table 1). Data were normalized using 18S ribosomal RNA as an invariant transcript and presented as fold induction over infected macrophages treated with control siRNA using the delta-delta Ct method (ΔΔCt). The average relative levels of expression and the SDs were determined in triplicate.
Table 1.
Primer Sequences and PrimerBank Identification (ID) Numbers
| Target | Primer Sequencea | PrimerBank IDb |
|---|---|---|
| Il6 | Forward: 5′ CCACGGCCTTCCCTACTTC3′; reverse: 5′ TTGGGAGTGGTATCCTCTGTGA3′ | … |
| 18S rRNA | Forward: 5′ TTGACGGAAGGGCACCACCAG 3′; reverse: 5’ GCACCACCACCCACGGAATCG 3′ | … |
| Gapdh | Forward: 5′ CTTTGGCATTGTGGAAGGGCTCAT 3′; reverse: 5′ ACCAGTGGATGCAGGGATGATGTT 3′ | … |
| Rsad2 | … | 237512932c1 |
| Cxcl10 | … | 371940989c1 |
| Cxcl11 | … | 9507070c1 |
| Il15 | … | 363000959c1 |
| Irg1 | … | 950650a1 |
| Ifit1 | … | 145301610c3 |
| Ifit2 | … | 162461505c1 |
| Ifit3 | … | 6754288a1 |
a Oligonucleotide sequences for mouse DNA–specific primers designed by us for quantitative polymerase chain reaction analysis of the specified targets.
b PrimerBank ID nos. used to generate mouse genome–specific primers for the specified targets.
Enzyme-Linked Immunosorbent Assay (ELISA) and 4-Plex Assay
Supernatants collected from infected cells, siRNA-treated cells, and cells on medium alone were assayed by the Mouse Il6 ELISA Kit (Invitrogen) according to the manufacturer's instructions. Supernatants were also used for quantification of secreted IFNg, interleukin 15, CCL2, and CXCL10, using the mouse cytokine 4 milliplex map kit (Millipore).
DNA Microarray Experiments and Analysis
A total of 24 hours after infection and silencing, host transcripts extracted from approximately 3 × 105 cells by means of the RNeasy kit (Qiagen) were used to profile the expression of mouse genome, using 4 × 44 mouse microarrays (Agilent) as described elsewhere [17–19]. Control samples (ie, samples infected with wild type or Mtb:Δ-sigH and treated with negative-control siRNA) were labeled with Cy3, whereas experimental samples (ie, samples infected with wild type or Mtb:Δ-sigH and treated with siRNA to silence Il-6 expression) were labeled with Cy5; methods are described elsewhere [17–19]. Genes whose expression changed by at least 50% (P < .05) were considered to be differentially expressed in a significant manner. For microRNA (miR) analysis, total transcripts extracted from approximately 3 × 105 cells were labeled and hybridized to miRCURY LNA miR Arrays (Exiqon) as described elsewhere [20].
Colony-Forming Unit (CFU) Counts
Intracellular bacteria were obtained by lysing the cells with sterile phosphate-buffered saline (PBS) containing 0.1% saponin (Sigma). The released bacilli were serially diluted in PBS containing 0.01% Tween-80 (Merck) and plated on Middlebrook 7H10/OADC agar in triplicate. CFUs were counted after 21 days of incubation at 37°C.
Statistical Analysis
The statistical significance of findings was determined by analysis of variance and the Mann–Whitney U test, using GraphPad Prism, except for microarray results, for which a t test script in the Spotfire DecisionSite/S+ Array Analyzer was used.
Protocol Approval
All procedures were approved by the Tulane Institutional Biosafety Committee.
RESULTS
Downregulation of Il6 Messenger RNA (mRNA) and Protein by siRNA Delivery in C57BL6/J Macrophages Cells In Vitro
Real-time PCR and ELISA were performed 24 hours after transfection to evaluate the level of Il6 mRNA and protein expression. GAPDH siRNA was used as the silencing reference standard. The difference between GAPDH-transfected samples and the corresponding negative control was used to calculate the percentage of GAPDH mRNA that remained. The siRNA knocked down GAPDH mRNA by >85% in cells infected with wild type or Mtb-ΔsigH (data not shown). Similarly, the Il6 siRNA induced significant reductions of 49% (fold-change [FC], −1.98) and 65% (FC, −2.86; P < .01) in Il6 mRNA levels in macrophages infected with M. tuberculosis and Mtb:Δ-sigH, respectively (Figure 1A). The protein expression levels were also downregulated by 41% (FC, −1.7) and 47% (FC, −1.9; P < .05) in wild type– and Mtb:Δ-sigH–infected cells, respectively (Figure 1B).
Figure 1.
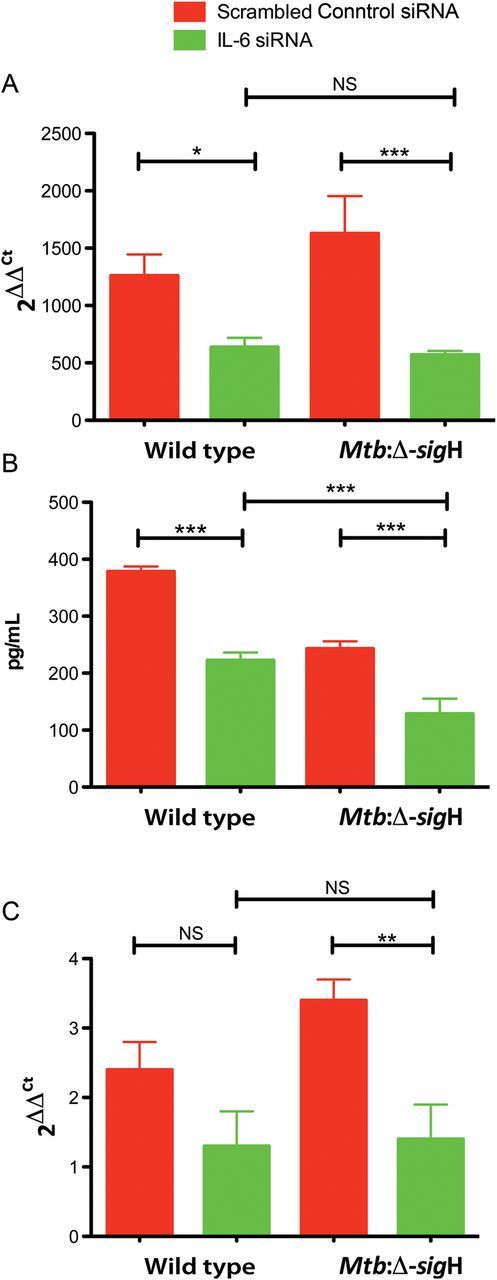
Interleukin 6 (Il6) gene silencing treatment of C57BL6/J macrophages infected with wild-type Mycobacterium tuberculosis or with Mtb:Δ-sigH, an attenuated mutant. A, Wild type–infected cells were incubated in the presence of Il6 small interfering RNA (siRNA) or a scrambled siRNA control for 24 hours. Il6 induction was evaluated by real-time polymerase chain reaction analysis performed as described in Materials and Methods. B, Culture supernatants from cells infected with the wild-type strain were assayed for Il6 by enzyme-linked immunosorbent assay. C, Tlr2−/− cells were incubated in the presence of Il6 siRNA or negative scrambled siRNA control for 24 hours. Data means ± standard errors of 3 independent experiments. *P < .05; **P < .01. Abbreviation: NS, not significant.
M. tuberculosis normally limits the magnitude of Tlr2 activation, thereby preventing robust activation of macrophage proinflammatory responses, including Il-6 expression [21]. Thus, we analyzed the expression and efficacy of Il6 silencing in Tlr2−/− bone marrow macrophages infected with M. tuberculosis. We found that Il6 expression was severely reduced after infection with the wild-type strain, as well as with the mutant strain. The residual levels of Il6 mRNA in wild type–infected cells were higher than but not significantly different from those in Mtb:Δ-sigH–infected cells (Figure 1C). Moreover, the silencing effect was very similar to that in wild-type–infected macrophages. Thus, Il6 mRNA levels were reduced by 47% (FC, −1.88) and 58% (FC, −2.37; P < .01) in Tlr2−/− macrophages infected with wild type and Mtb:Δ-sigH, respectively (Figure 1C). These results show that increased signaling through Tlr2 leads to higher levels of Il-6 by Mtb:Δ-sigH–infected macrophages and suggest that SigH functions by limiting the magnitude of the Tlr2-dependent innate immune response.
Despite similar Il6 mRNA levels, IL6 protein levels were significantly lower in Mtb:Δ-sigH–infected cells, compared with wild type–infected cells (Figure 1B; P < .05). Conversely, Mtb:Δ-sigH induced significantly higher expression of the IL6 gene, compared with wild type (Figure 1A; P < .05). Thus, since miRs regulate protein translation and/or mRNA destabilization, we used a miR array approach to determine whether miRs were involved in Il6 regulation. We analyzed the expression profile of miRs from C57BL6/J macrophages infected with wild type or Mtb:Δ-sigH for 24 hours. Of the miRs that were significantly downregulated, let-7a and miR-142-3p directly target Il6 [22–24]. Here, let-7a and miR-142-3p were downregulated in cells infected with wild type, whereas only miR-142-3p was found to be significantly downregulated in cells infected with Mtb:Δ-sigH (Table 2). Since let-7a and miR-142-3p directly inhibit Il6 [22, 24], we suggest that downregulation of both miRs is the mechanism used by the immune system to induce levels of IL6 protein that are higher during wild type infection, compared with Mtb:Δ-sigH infection.
Table 2.
Fold-Change (FC) in MicroRNA (miR) Levels After Infection With Wild-Type or Mutant Mycobacterium tuberculosis
| miR | Wild Type, FCa | Mutant, FCa |
|---|---|---|
| let-7a | −2.3 | … |
| miR-142-3p | −2.3 | −2.1 |
Cells were infected with wild-type M. tuberculosis and the Mtb:Δ-sigH mutant for 24 hours, and miR levels were assessed in total isolated RNA, as described in Materials and Methods. let-7a and miR-142-3p were examined because they are known to target interleukin 6.
a Fold change (FC) in cells infected with M. tuberculosis (wild-type or mutant) relative to uninfected cells.
Effect of Il6 Knockdown on Bacterial Growth
To determine whether downregulation of Il6 might affect an already established infection with M. tuberculosis, Il6 expression in macrophages was silenced for 24 and 48 hours. Briefly, C57BL6/J macrophages were infected for 4 hours with wild type or Mtb:Δ-sigH at a multiplicity of infection of 1:10, and the expression of Il6 was silenced. Compared with the negative scrambled control, administration of Il6 siRNA for 24 hours had no significant effect on growth of either M. tuberculosis strain. However, after 48 hours of silencing, CFUs of wild type and Mtb:Δ-sigH were significantly increased (P < .01; Figure 2). Hence, reduced Il6 expression resulted in increased susceptibility during experimental M. tuberculosis infection, indicating that Il6 has an effect on the protective immune response.
Figure 2.
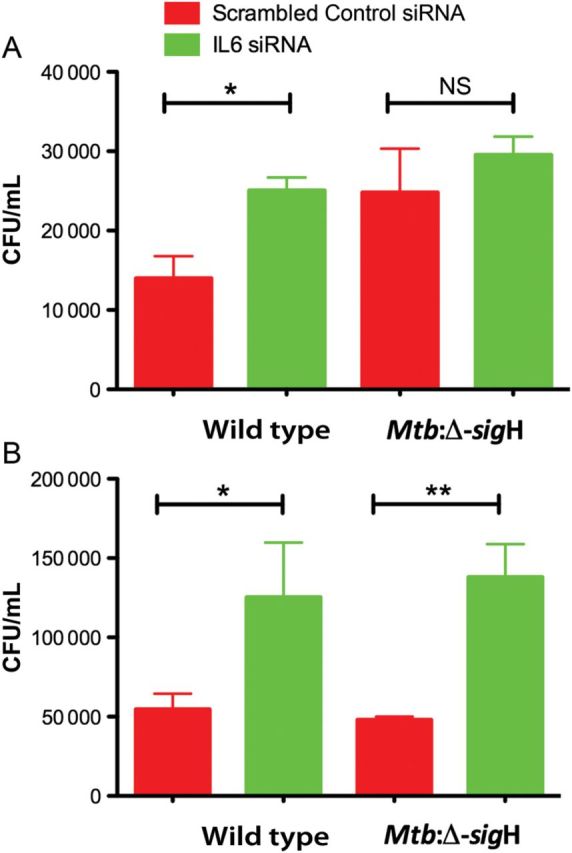
Bacterial burden following treatment with interleukin 6 (Il6) small interfering RNA (siRNA) in C57BL6/J cells infected with wild-type Mycobacterium tuberculosis or Mtb:Δ-sigH, an attenuated mutant. Administration of Il6 siRNA for 24 hours had no significant effect on mycobacterial growth for either M. tuberculosis strain. After 48 hours of silencing, colony-forming units (CFUs) of both silenced M. tuberculosis strains were found to be significantly increased. *P < .05; **P < .01. Data means ± standard errors of 3 biological replicates.
Global Transcriptomic Response to Il6 Silencing
To study the impact of Il6 on host phagocytes, we compared the transcriptome profiles of infected C57Bl6/J macrophages silenced for Il6 to profiles of C57Bl6/J macrophages treated with scrambled siRNA. Genes that showed an alteration in expression by >1.5-fold were considered. Thus, when biological replicates of infected C57bl6/J macrophages silenced for Il6 were compared to the infected nonsilenced controls, wild type and Mtb:Δ-sigH were found to induce expression of 204 genes and 313 genes, respectively, by at least 50%. A total of 141 genes were induced by both strains.
After 24 hours of silencing, interferon-inducible genes were found to be upregulated by both M. tuberculosis strains (Table 3). However, the expression of these genes was not significantly different between the mutant and wild-type strains. In addition, pathway analysis of the expression profiles showed that the majority of the transcripts were associated with type I Ifnab signaling. These results demonstrate that Il6 downregulates interferon responses in murine macrophages infected with either wild type or Mtb:Δ-sigH and plays an important role in the process that leads to disease susceptibility.
Table 3.
Key Immune Function Genes With Elevated Expression After Interleukin 6 Silencing Concomitant With Wild-Type or Mutant Mycobacterium tuberculosis Infection
| Gene | Microarray Expression, FCa |
Quantitative PCR, FCa |
||||
|---|---|---|---|---|---|---|
| Wild Type | Mutant | Wild Type | Pb | Mutant | Pb | |
| Cxcl10 | 2.83 | 3.13 | 2.2 | .05 | 2.7 | .01 |
| Cxcl11 | 2.76 | 2.33 | 1.5 | NS | 1.4 | NS |
| IL15 | 1.92 | 2.14 | 1.89 | NS | 1.73 | NS |
| Ifit1 | 1.72 | 1.60 | 2.14 | .05 | 2.13 | .05 |
| Ifit2 | 2.41 | 2.17 | 1.76 | .01 | 1.65 | .001 |
| Ifit3 | 2.35 | 1.97 | 1.81 | NS | 1.41 | NS |
| Rsad2 | 3.45 | 3.05 | 2.14 | .05 | 2.13 | .05 |
| Irg1 | 2.07 | 2.09 | 1.91 | .001 | 1.72 | .001 |
| Igtp | 1.53 | 1.63 | ND | ND | ND | ND |
P values from real-time PCR.
To confirm the results obtained from microarrays, we performed quantitative PCR for type I Ifn pathway genes. The expression of Cxcl10, Ifit1, Ifit2, Rsad2, and Irg1 was significantly higher in cells infected with either strain when Il6 expression was silenced, compared with when the control scrambled siRNA was used (Table 3). Conversely, even though the expression of Il15, Cxcl11, and Ifit3 was upregulated, these findings were not significant.
The expression of 3 Tlr genes was differentially regulated following infection with either strain. However, the differences between the 2 strains were not significant. Hence, the expression of Tlr2, Tlr3, and Tlr4 was induced by 1.93-, 1.70-, 1.75-fold, respectively, by the silencing of Il6 in wild type–infected cells and by 2.06-, 1.75-, 1.62-fold, respectively, in those infected with the mutant. Tlrs recognize pathogen-associated molecules, which stimulate the efferent limb of the immune system to secrete cytokines and activate macrophages. Exposure of cells to type I Ifn likely involves upregulation of caspases and proapoptotic innate sensors, such as Tlrs and inflammasomes [21, 25]. Thus, our results suggest that the knockdown of Il6 upregulates type 1 IFNs, which in turn induce Tlrs in a positive-feedback manner.
Furthermore, microarray analysis revealed that 4 genes belonging to the lymphocyte antigen 6 complex (Ly6) were induced in response to Il6 silencing. In mice, expression of the Ly6 locus encodes a family of 10–12-kDa proteins that are linked to the cell surface by a glycosylphosphatidylinositol anchor and have cell signaling and cell adhesion properties [26]. Our results showed that, while the expression of all 4 Ly6 genes was higher in the wild-type strain relative to the mutant strain, levels of Ly6c and Ly6i expression, but not of Ly6a and Ly6f expression, were significantly higher (Figure 3A; P < .05). Since type 1 IFN is directly involved in Ly6c monocyte differentiation [27], we suggest that Ly6 genes might play an important role in host defense against M. tuberculosis infection. Moreover, the expression of the other 4 genes (ie, Cish, Csf1r, Ifitm6, and Cxcl2), appeared to be significantly different in cells infected with Mtb:Δ-sigH, compared with wild type, when Il6 expression was silenced (Figure 3B). These genes play crucial roles in immunity against bacterial and viral infections and in governing the extent of disease progression and severity.
Figure 3.
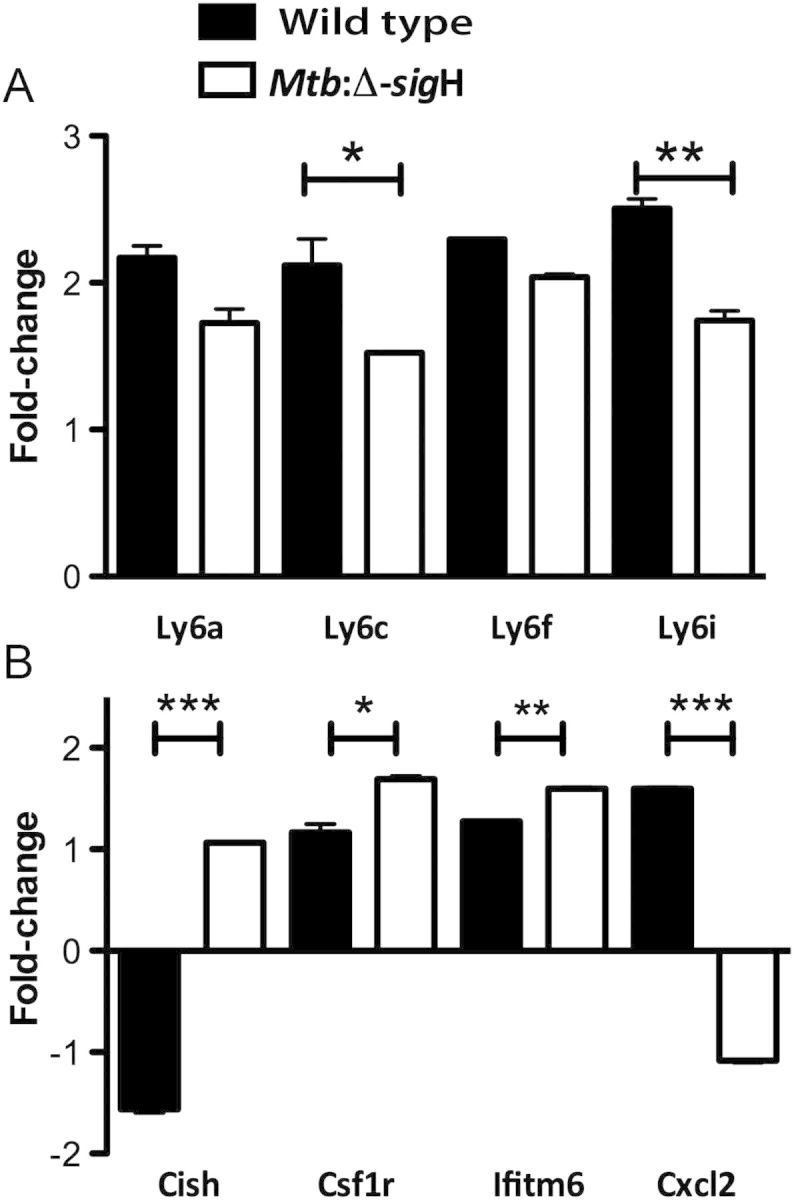
Comparison of host immunity gene expression in C57BL6/J macrophages infected with wild-type Mycobacterium tuberculosis or Mtb:Δ-sigH, an attenuated mutant, and transfected with interleukin 6 (Il6) small interfering RNA (siRNA). A, Expression of Ly6 genes. B, Expression of Cish, Csf1r, Ifitm6, Cxcl2 genes. Expression values are shown for DNA microarray experiments. Values represent responses that were statistically different between the Il-6 siRNA vs scrambled negative control treatments. *P < .05; **P < .01.
Confirmation of Microarray Results at the Protein Level by Cytokine/Chemokine Assay
We performed bead array assays for CXCL10, IFNγ, CCL2, and IL15 in the supernatants obtained from cells that were silenced and infected with either M. tuberculosis strain. IFNγ protein levels were not elevated in the supernatant of macrophages infected with wild type or Mtb:Δ-sigH, although the IFN-inducible chemokine CXCL10 was increased. Hence, similar to the transcript analysis, silenced macrophages infected with the mutant strain induced higher levels of CXCL10 protein (2.2-fold; P < .05), compared with those infected with the wild-type strain (1.5-fold; P > .05; Figure 4A). This result is in accordance with an in vivo study, in which levels of IFNa2 and IFNγ proteins were not elevated but the level of CXCL10 was significantly increased in serum samples from patients with active tuberculosis [28].
Figure 4.
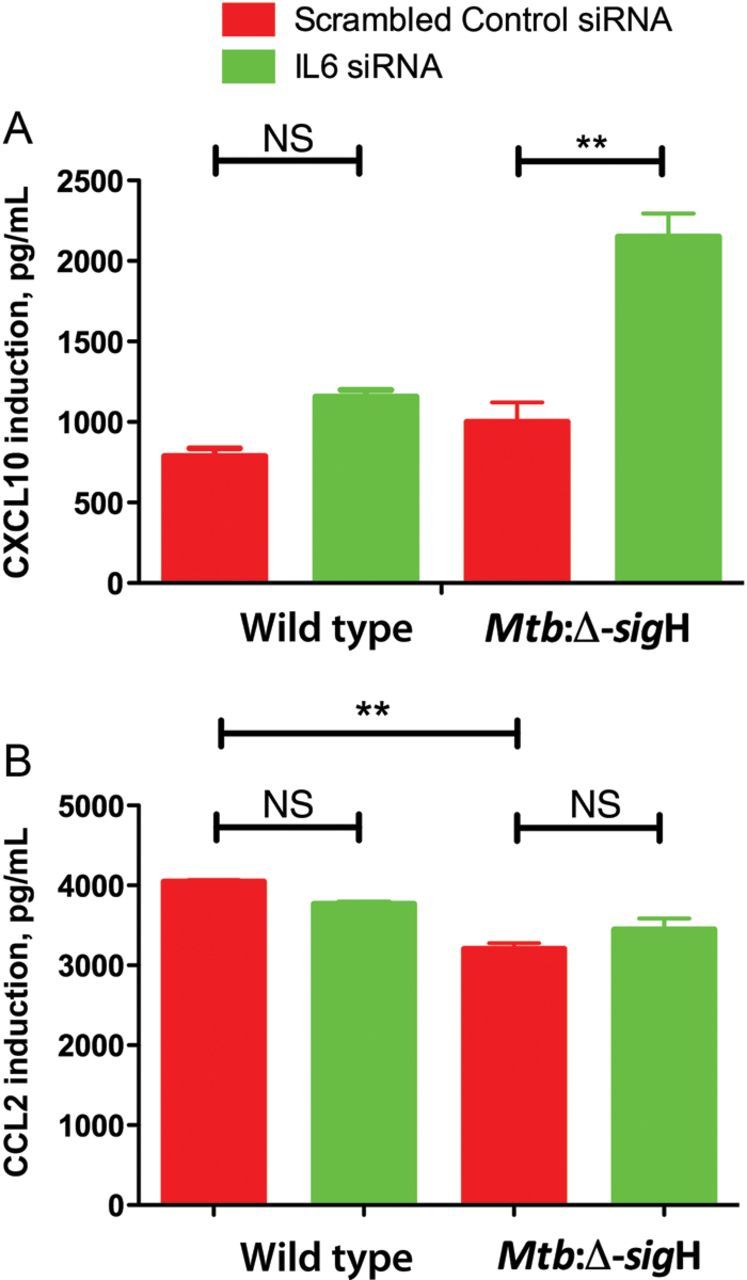
Enhanced induction of Cxcl10 by Il6 siRNA treatment. Macrophages from C57BL6/J mice were infected with wild-type Mycobacterium tuberculosis or Mtb:Δ-sigH, an attenuated mutant, and transfected with Il6 siRNA. Supernatants were collected at 24 hours after silencing and assayed for (A) Cxcl10 and (B) Ccl2 by a cytokine milliplex assay.
Likewise, even though microarray analysis showed upregulation of Il15 expression, levels of IL15 protein could not be detected. This result can be explained by the very short in vivo half-life of soluble Il15, as well as the unique mechanism of Il-15 trans-presentation [29]. Moreover, since Ly6c-high monocytes, also known as inflammatory monocytes (Ly6chigh/Ccr2+/Cx3cr1low), have been shown to be the main producers of Ccl2 in mice [27] and were upregulated in the microarray studies, we looked at CCL2 protein expression. Results showed that silencing of Il-6 had no significant effect on CCL2 protein expression, yet there was a significant difference in the induction between the 2 strains. Thus, the wild-type strain induced higher levels of CCL2 than the mutant strain when Il6 was silenced (P < .05; Figure 4B). This result shows that activation and recruitment of monocytes to the site of infection are delayed in the mutant strain, compared with the wild-type strain. This is consistent with our in vivo observations, in which incorporation of BrdU in bone marrow–derived monocytes was significantly induced in macaques infected with wild-type M. tuberculosis but not in those infected with Mtb:Δ-sigH [18].
DISCUSSION
M. tuberculosis interferes with host signaling pathways activated by Ifng [30–32] for its survival. It exploits Il6 induction as one of the mechanisms to inhibit Ifng [31]. Here, we show that the downregulation of Il6 in murine macrophages infected with virulent or attenuated M. tuberculosis induces transcription of Ifn-related genes (Table 3). This result was validated by significantly increased real-time PCR transcript and protein levels of the Ifn-inducible chemokine Cxcl10 in silenced macrophages (Figure 4A). Although Ifng is known to be protective during immune responses to intracellular pathogens, including mycobacteria, the role of type I Ifnab is less clear [25, 33]. It has been shown that activation of type I Ifn signaling is crucial for defense against viral infections but may be harmful during bacterial infections, including those due to mycobacteria [34–36].
Ifn signaling triggers hematopoietic stem cell proliferation [37] and, thus, mediates monocyte differentiation [27]. A recent report showed that Ifn-I receptor knockout (Ifnar1−/−) mice developed significant defects in the infiltration of Ccl2-producing Ly6chi monocytes in the lung after influenza virus infection [27]. Here, we show that despite the enhanced expression of Ly6-related genes in silenced macrophages, the difference in CCL2 protein levels between silenced and control infected macrophages were not significant (Figure 4B). Nonetheless, macrophages infected with wild type induced higher levels of CCL2 and had less effective silencing of Il-6, compared with macrophages infected with Mtb:Δ-sigH. On the basis of these results and previous in vivo observations [18], we conclude that the wild-type strain induces earlier and more robust macrophage activation, compared with the Mtb:Δ-sigH strain, and, thus, partially impairs silencing effectiveness. This differential activation of macrophages likely gives the wild-type strain, as opposed to the Mtb:Δ-sigH strain, the ability to resist host cellular immunity and progress to active disease or to succumb to the host protective responses.
In this study, silencing of Il6 prompted significant opposite regulatory effects on the transcript levels of Cxcl2 and Cish in cells infected with Mtb:Δ-sigH, relative to levels in cells infected with wild type (Figure 3B). Type I Ifn has been shown to inhibit the production of Cxcl2 during influenza virus infection, thus decreasing neutrophil recruitment and dampening host defense against secondary bacterial infections [25]. Here, the downregulation of Cxcl2 by Mtb:Δ-sigH, as opposed to the upregulation by the wild-type strain, is consistent with the higher protein levels of IFN-inducible chemokine CXCL10 presented by Mtb:Δ-sigH–infected cells, compared with wild type–infected cells (Figure 4A). On the contrary, Cish was found to be upregulated in Mtb:Δ-sigH–infected cells and downregulated in wild type–infected cells. Cish single-nucleotide polymorphisms correlate with increased susceptibility to tuberculosis [38], and it is essential for the maturation of dendritic cells and the generation of an effective cytotoxic T-lymphocyte response [39]. Hence, it appears that M. tuberculosis may use antigens expressed by the SigH regulon during the infection process via yet-to-be-characterized mechanism(s), to repress the production of Cish. Absence (or reduced expression) of Cish would thus result in the maintenance of dendritic cells in an immature phase and prevent the accumulation of an effective cytotoxic T-lymphocyte response, both of which would be beneficial for the persistence of the pathogen. The expression of both Ifitm6 and Csf1r was higher after infection with Mtb:Δ-sigH, compared with wild type (Figure 3B; P < .05). Ifitm6 belongs to the family of interferon-induced transmembrane proteins, and the higher levels of Ifitm6 found in Mtb:Δ-sigH–infected cells are a consequence of its more effective silencing. Csf1 and its receptor regulate key effector functions of macrophages and contribute to excessive inflammatory responses in sepsis [40]. Thus, we suggest that SigH may modulate Csf1r to regulate inflammation.
Our results showed that overexpression of type I Ifnab–inducible transcripts, caused by the downregulation of Il6, was concomitant with an increase in bacterial burden after 48 hours of silencing, indicating disease progression. Similarly, an in vivo study showed that M. tuberculosis CFUs in lungs from C57BL6/J mice treated with the soluble inhibitor of Il6 trans-signaling (sgp130Fctg) were slightly but significantly increased after 21 days, but not 14 days, compared with infected control mice [9]. The deleterious role of type I Ifns in the pathogenesis of tuberculosis is corroborated by a recent study that showed increased expression of type I Ifn–inducible transcripts in the blood of patients with active tuberculosis [28]. In addition, to further substantiate the correlation between disease severity and increased type I Ifn response, there have been reports of tuberculosis reactivation during Ifna treatment for hepatitis C and D virus infections [36, 41].
Modulation of proinflammatory responses is highly relevant to M. tuberculosis pathogenesis, and here we show that M. tuberculosis dampens Tlr2-dependent proinflammatory responses and suggest that the SigH regulon restricts the onset and magnitude of such responses by limiting Tlr2 activation in macrophages. In addition, microarray analysis revealed that Tlr2, Tlr3, and Tlr4 were upregulated after Il-6 silencing. Accordingly, many of the proinflammatory responses, including secretion of Il-6, are downstream of signaling through Tlrs. We therefore suggest that Tlrs are induced in a positive-feedback manner by the knockdown of Il6.
Both type I Ifns and Tlr induce Il15 through Myd88 [42]. In this study, we provide evidence that Il15 expression is under transcriptional control of type I interferons and Tlrs, which in turn are regulated by Il6. Exogenous Il15 increases natural killer (NK) cell lytic activity, and monocytes have shown to play a regulatory role in NK cell activation [43]. Our microarray experiments showed induction of Il15 after silencing of Il6, suggesting an indirect effect of Il6 in the activation of NK cells. This hypothesis requires further investigation.
Recently, studies have demonstrated a role for miRs in the regulation of inflammatory responses [44]. Here we used microarray technology to identify miRs that mediate modulation of Il6. It has been reported that let-7a and miR-142-3p directly inhibit Il6 expression [22–24]. In view of that, we found that the wild-type strain downregulated both miRs, whereas Mtb:Δ-sigH downregulated only miR-142-3p. Thus, we provided evidence that the downregulation of both miRs by wild type, but not Mtb:Δ-sigH, induced higher Il6 gene translation into protein. This posttranscriptional regulation explains the reduced IL6 protein levels presented by Mtb:Δ-sigH–infected cells, compared with M. tuberculosis–infected cells, and suggests compensation as a defense mechanism. Also, the delayed activation of macrophages infected with Mtb:Δ-sigH resulted in reduced subsequent inflammatory stimuli and improved silencing efficiency.
Thus, it appears that macrophages regulate Il6 production to inhibit type I Ifn signaling and, consequently, disease progression. Our data indicate that a SigH-dependent M. tuberculosis factor interacts with the host innate immune system to modulate the Il6 levels, leading to disease susceptibility. These findings have important implications for the better understanding of the mechanisms behind M. tuberculosis virulence and pathogenesis, although to fully understand the complex signaling network induced by the silencing of Il6, additional in vivo experiments are required. We anticipate that type I IFN acts by increasing the susceptibility of macrophage to apoptosis-inducing stimuli [25]. Hence, our data provides an initial road map to further investigate the mechanisms that may account for the deleterious effects of type I IFN in M. tuberculosis infection.
Notes
Acknowledgments.A. N. M. and D. K. were involved in study design; A. N. M. and S. M. were involved in research; A. N. M., S. M., and D. K were involved in data analysis; A. N. M. and D. K. were involved in writing; and D. K. was involved in funding.
Financial support. This work was supported by the National Institutes of Health (grants AI089323, HL106790, AI091457, RR026006, RR020159, RR000164/OD011104, and C06AI058609); the Howard Hughes Medical Institutes (Kwa-Zulu Natal Research Institute in TB and HIV-AIDS); the Louisiana Vaccine Center; the Tulane Research Enhancement Fund; the Tulane Center for Infectious Diseases; the Office of the Director, Tulane National Primate Research Center; and the Office of the Vice President for Research, Tulane University (bridge grant).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 2009;11:1170–8. doi: 10.1111/j.1462-5822.2009.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatt K, Salgame P. Host innate immune response to Mycobacterium tuberculosis. J Clin Immunol. 2007;27:347–62. doi: 10.1007/s10875-007-9084-0. [DOI] [PubMed] [Google Scholar]
- 3.Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–66. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 4.Flynn JL, Chan J. Immune evasion by Mycobacterium tuberculosis: living with the enemy. Curr Opin Immunol. 2003;15:450–5. doi: 10.1016/s0952-7915(03)00075-x. [DOI] [PubMed] [Google Scholar]
- 5.Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol. 2010;8:296–307. doi: 10.1038/nrmicro2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol. 2009;27:393–422. doi: 10.1146/annurev.immunol.021908.132703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy CR, Mocarski ES. Pathogen subversion of cell-intrinsic innate immunity. Nat. Immunol. 2007;8:1179–87. doi: 10.1038/ni1528. [DOI] [PubMed] [Google Scholar]
- 8.Dutta NK, Mehra S, Martinez AN, et al. The stress-response factor SigH modulates the interaction between Mycobacterium tuberculosis and host phagocytes. PLoS One. 2012;7:e28958. doi: 10.1371/journal.pone.0028958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sodenkamp J, Waetzig GH, Scheller J, et al. Therapeutic targeting of interleukin-6 trans-signaling does not affect the outcome of experimental tuberculosis. Immunobiology. 2012;217: 996:1004. doi: 10.1016/j.imbio.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Ladel CH, Blum C, Dreher A, Reifenberg K, Kopf M, Kaufmann SH. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 1997;65:4843. doi: 10.1128/iai.65.11.4843-4849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saunders BM, Frank AA, Orme IM, Cooper AM. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect Immun. 2000;68:3322. doi: 10.1128/iai.68.6.3322-3326.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagabhushanam V, Solache A, Ting LM, Escaron CJ, Zhang JY, Ernst JD. Innate inhibition of adaptive immunity: Mycobacterium tuberculosis-induced IL-6 inhibits macrophage responses to IFN-gamma. J Immunol. 2003;171:4750–7. doi: 10.4049/jimmunol.171.9.4750. [DOI] [PubMed] [Google Scholar]
- 13.Leal IS, Smedegard B, Andersen P, Appelberg R. Interleukin-6 and interleukin-12 participate in induction of a type 1 protective T-cell response during vaccination with a tuberculosis subunit vaccine. Infect Immun. 1999;67:5747. doi: 10.1128/iai.67.11.5747-5754.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaushal D, Schroeder BG, Tyagi S, et al. Reduced immunopathology and mortality despite tissue persistence in a Mycobacterium tuberculosis mutant lacking alternative sigma factor, SigH. Proc Natl Acad Sci U S A. 2002;11:8330–5. doi: 10.1073/pnas.102055799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehra S, Kaushal D. Functional genomics reveals extended roles of the Mycobacterium tuberculosis stress response factor sigmaH. J Bacteriol. 2009;191:3965–80. doi: 10.1128/JB.00064-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehra S, Dutta NK, Mollenkopf HJ, Kaushal D. Mycobacterium tuberculosis MT2816 encodes a key stress-response regulator. J Infect Dis. 2010;202:943–53. doi: 10.1086/654820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehra S, Pahar B, Dutta NK, et al. Transcriptional reprogramming in nonhuman primate (rhesus macaque) tuberculosis granulomas. PLoS One. 2010;5:e12266. doi: 10.1371/journal.pone.0012266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehra S, Golden NA, Stuckey K, et al. The Mycobacterium tuberculosis stress response factor SigH is required for bacterial burden as well as immunopathology in primate lungs. J Infect Dis. 2012;205:1203–13. doi: 10.1093/infdis/jis102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautam A, Dixit S, Philipp MT, et al. Interleukin-10 alters effector functions of multiple genes induced by Borrelia burgdorferi in macrophages to regulate Lyme disease inflammation. Infect Immun. 2011;79:4876–92. doi: 10.1128/IAI.05451-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao L, Hazari S, Mehra S, Kaushal D, Moroz K, Dash S. Increased expression of p-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol. 2012;180:2490–503. doi: 10.1016/j.ajpath.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madan-Lala R, Peixoto KV, Re F, Rengarajan J. Mycobacterium tuberculosis Hip1 dampens macrophage proinflammatory responses by limiting toll-like receptor 2 activation. Infect Immun. 2011;279:4828–38. doi: 10.1128/IAI.05574-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nam Y, Chen C, Gregory RI, Chou JJ, Sliz P. Molecular basis for interaction of let-7 microRNAs with Lin28. Cell. 2011;147:1080–91. doi: 10.1016/j.cell.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y, Varambally S, Maher CA, et al. Targeting of microRNA-142–3p in dendritic cells regulates endotoxin-induced mortality. Blood. 2011;117:6172–83. doi: 10.1182/blood-2010-12-325647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207:2053–63. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bamezai A. Mouse Ly-6 proteins and their extended family: markers of cell differentiation and regulators of cell signaling. Arch Immunol Ther Exp (Warsz) 2004;52:255–66. [PubMed] [Google Scholar]
- 27.Seo SU, Kwon HJ, Ko HJ, et al. Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathog. 2011;7:e1001304. doi: 10.1371/journal.ppat.1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berry MP, Graham CM, McNab FW, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–7. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colpitts SL, Stoklasek TA, Plumlee CR, Obar JJ, Guo C, Lefrançois L. Cutting edge: the role of IFN-α receptor and MyD88 signaling in induction of IL-15 expression in vivo. J Immunol. 2012;188:2483–7. doi: 10.4049/jimmunol.1103609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fortune SM, Solache A, Jaeger A, et al. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J Immunol. 2004;172:6272–80. doi: 10.4049/jimmunol.172.10.6272. [DOI] [PubMed] [Google Scholar]
- 31.Pai RK, Convery M, Hamilton TA, Boom WH, Harding CV. Inhibition of IFN-gamma-induced class II transactivator expression by a 19-kDa lipoprotein from Mycobacterium tuberculosis: a potential mechanism for immune evasion. J Immunol. 2003;171:175–84. doi: 10.4049/jimmunol.171.1.175. [DOI] [PubMed] [Google Scholar]
- 32.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 33.Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nature Rev Immunol. 2005;5:675–87. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- 34.Manca C, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res. 2005;25:694–701. doi: 10.1089/jir.2005.25.694. [DOI] [PubMed] [Google Scholar]
- 35.Ordway D, et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol. 2007;179:522–31. doi: 10.4049/jimmunol.179.1.522. [DOI] [PubMed] [Google Scholar]
- 36.Telesca C, Angelico M, Piccolo P, et al. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J Infect. 2007;54:e223–6. doi: 10.1016/j.jinf.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Essers MA, Offner S, Blanco-Bose WE, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–8. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 38.Khor CC, Vannberg FO, Chapman SJ, et al. CISH and susceptibility to infectious diseases. N Engl J Med. 2010;362:2092–101. doi: 10.1056/NEJMoa0905606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miah MA, Yoon CH, Kim J, Jang J, Seong YR, Bae YS. CISH is induced during DC development and regulates DC-mediated CTL activation. Eur J Immunol. 2012;42:58–68. doi: 10.1002/eji.201141846. [DOI] [PubMed] [Google Scholar]
- 40.Irvine KM, Burns CJ, Wilks AF, Su S, Hume DA, Sweet MJ. A CSF-1 receptor kinase inhibitor targets effector functions and inhibits pro-inflammatory cytokine production from murine macrophage populations. FASEB J. 2006;20:1921–3. doi: 10.1096/fj.06-5848fje. [DOI] [PubMed] [Google Scholar]
- 41.Sabbatani S, Manfredi R, Marinacci G, Pavoni M, Cristoni L, Chiodo F. Reactivation of severe, acute pulmonary tuberculosis during treatment with pegylated interferon-alpha and ribavirin for chronic HCV hepatitis. Scand J Infect Dis. 2006;38:205–8. doi: 10.1080/00365540500263268. [DOI] [PubMed] [Google Scholar]
- 42.Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol. 2001;167:1179–87. doi: 10.4049/jimmunol.167.3.1179. [DOI] [PubMed] [Google Scholar]
- 43.Schierloh P, Alemán M, Yokobori N, et al. NK cell activity in tuberculosis is associated with impaired CD11a and ICAM-1 expression: a regulatory role of monocytes in NK activation. Immunology. 2005;116:541–52. doi: 10.1111/j.1365-2567.2005.02259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10:111–22. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]