

Abstract
Francisella tularensis is a highly infectious bacterium whose virulence relies on its ability to rapidly reach the macrophage cytosol and extensively replicate in this compartment. We previously identified a novel Francisella virulence factor, DipA (FTT0369c), which is required for intramacrophage proliferation and survival, and virulence in mice. DipA is a 353 amino acid protein with a Sec-dependent signal peptide, four Sel1-like repeats (SLR), and a C-terminal coiled-coil (CC) domain. Here, we determined through biochemical and localization studies that DipA is a membrane-associated protein exposed on the surface of the prototypical F. tularensis subsp. tularensis strain SchuS4 during macrophage infection. Deletion and substitution mutagenesis showed that the CC domain, but not the SLR motifs, of DipA is required for surface exposure on SchuS4. Complementation of the dipA mutant with either DipA CC or SLR domain mutants did not restore intracellular growth of Francisella , indicating that proper localization and the SLR domains are required for DipA function. Co-immunoprecipitation studies revealed interactions with the Francisella outer membrane protein FopA, suggesting that DipA is part of a membrane-associated complex. Altogether, our findings indicate that DipA is positioned at the host–pathogen interface to influence the intracellular fate of this pathogen.
Introduction
The Gram-negative intracellular bacterium Francisella tularensis is the causative agent of tularemia, a potentially fatal zoonosis affecting a variety of mammals, including humans [1]. Human tularemia can be contracted through multiple routes with the most acute form of disease resulting from inhalation of as few as 10 organisms [2]. Of the three subspecies of F. tularensis, subsp. tularensis, holarctica, and mediasiatica, the former two are responsible for the majority of human infections and disease [2]. In addition, F . novicida , a closely related species, is considered non-pathogenic for immunocompetent humans, yet retains high virulence in mice and is widely used as a model organism [3].
Given its pathogenic potential and renewed concerns for its misuse as a bioweapon, much research has focused on understanding the mechanisms of virulence of F. tularensis. Although F. tularensis can infect a range of host cells that include hepatocytes, neutrophils, fibroblasts and endothelial cells, a key virulence trait is the ability of F. tularensis to reside within mononuclear phagocytes [4–7]. In particular, macrophages are considered an important target for infection within which F. tularensis demonstrates a multifaceted lifecycle that is essential to its pathogenesis [4,8]. Upon internalization, the bacterium transiently resides within a phagosome from which it rapidly escapes to reach the macrophage cytosol [9–15]. Cytosolic bacteria undergo extensive replication, induce macrophage apoptosis or pyroptosis, and eventual egress from infected cells [16–19]. A subset of post-replicative bacteria is re-captured into endocytic vacuoles in murine bone marrow-derived macrophages (BMMs) through an autophagy related process [11]. A number of Francisella factors that contribute to various aspects of its complex intracellular lifecycle have been identified (reviewed in 20,21). The most prominent virulence determinant is the Francisella pathogenicity island (FPI), a 30-kb locus that encodes components of a type VI secretion system (T6SS) [22,23]. Several non-FPI encoded factors have also been shown to contribute to Francisella pathogenesis, although many of these, as well as those encoded by genes within the FPI, show no homology to known bacterial proteins and thus, their specific functions remain unclear [10,24–31].
Previously, we identified a Francisella -specific locus that was transcriptionally upregulated in F. tularensis subsp. tularensis strain SchuS4 during the cytosolic replication stage of BMM infection [10]. Deletion of dipA in SchuS4 did not affect phagosomal escape, but impaired intracellular replication and survival in BMMs. Furthermore, the SchuS4ΔdipA mutant was defective for replication, dissemination and lethality in mice, demonstrating that dipA encodes a bona fide virulence factor [10]. The dipA locus encodes a novel protein predicted to contain several conserved domains, four Sel1-like repeats (SLRs) and a coiled-coil (CC) motif, that are implicated in protein–protein interactions. Aside from these domains, DipA has little similarity to other known proteins and is conserved among the subspecies, suggesting a potentially unique, Francisella -specific, molecular mechanism of virulence.
In the present study, we examined the biochemical and structural characteristics of DipA to gain insight into its role in F. tularensis pathogenesis. We report that DipA is a membrane-associated protein localized on the bacterial surface during macrophage infection, and show that the SLR and CC domains are functionally distinct. We also identified a Francisella outer membrane protein, FopA, that interacts with DipA, suggesting that DipA may be part of a membrane complex.
Results and Discussion
DipA is localized to the surface of F. tularensis subsp. tularensis SchuS4
To characterize the functional role of DipA, we first analyzed the translated amino acid sequence for conserved domains using protein structure prediction programs (SMART, Pfam, COILS, and Marcoil). DipA is a 353 amino acid protein predicted to possess a 20 amino acid Sec-dependent signal peptide, four Sel1-like repeat (SLR) domains, and a C-terminal coiled-coil (CC) domain (Figure 1A and 1C). The signal peptide suggests that DipA may be secreted in a Sec-dependent manner, while the SLR and CC domains are ubiquitous structural motifs known to facilitate protein–protein interactions. Immunoblot analysis of bacterial subcellular fractions revealed that DipA, although detected in all fractions, was predominantly localized to the inner and outer membrane enriched fractions of GFP-expressing SchuS4 (SchuS4 GFP) (Figure 2A), indicating an extracytoplasmic location as predicted by the signal peptide. Known outer membrane (FopA), inner membrane (PdpB), and soluble (GFP) proteins were detected only in the expected fractions, indicating effective fraction separation (Figure 2A) [28,32–36]. In addition, we examined the localization of IglA in SchuS4 GFP because IglA was included as a control throughout this study. In F . novicida , IglA has been detected in the cytoplasm, outer membrane and membrane insoluble fractions, and the ambiguity of its distribution has been ascribed to its association with a macromolecular structure spanning the periplasm from the inner membrane [36–38]. Consistent with this model, we detected IglA in the soluble and inner membrane fractions of SchuS4 GFP (Figure 2A).
Figure 1. Predicted structure of DipA.

(A) Schematic representations of DipA and domain deletion mutants generated in this study. The predicted N-terminal 20 amino acid signal peptide is denoted is red, the Sel1-like repeat domains are denoted in blue and the coiled-coil (CC) domain is denoted in green. Domain deletion mutants were designed to encompass deletion of Sel1a and Sel1b domains (DipAΔSel1ab), Sel1c and Sel1d domains (DipAΔSel1cd), and the CC domain (DipAΔCC). (B) Helical wheel representations of the CC domain corresponding to amino acid residues of DipA. Heptad-repeat positions are denoted a to g. Two DipA CC domain substitution mutants where three hydrophobic residues in core positions a and d were mutated to aspartate [DipACC(AIL 3D) and DipACC(LAL 3D)]. Mutations are indicated in red. (C) Three-dimensional ribbon model of DipA predicted by I-TASSER and visualized using Chimera software. The Sel1a domain corresponding to residues 96-132 is indicated in red, the Sel1b domain corresponding to residues 133-169 is indicated in blue, the Sel1c domain corresponding to residues 193-229 is indicated in green, the Sel1d domain corresponding to residues 231-262 is indicated in purple, and the CC domain corresponding to residues 311-343 is indicated in cyan.
Figure 2. DipA is a surface-exposed, membrane-associated protein.
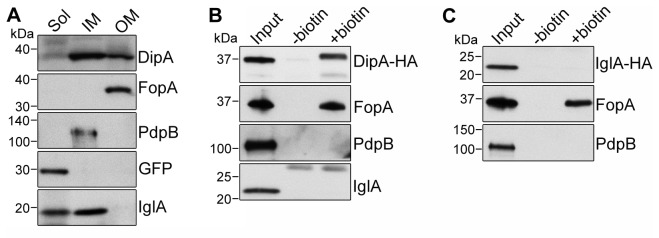
(A) Subcellular localization of DipA, FopA, PdpB, GFP, and IglA from GFP-expressing SchuS4. Soluble (Sol), inner membrane (IM), and outer membrane (OM) enriched fractions were separated based on Sarkosyl solubility and subjected to immunoblot analysis with antibodies against DipA, FopA, PdpB, GFP and IglA. Each fraction was concentrated to the same volume and equal volumes were loaded. GFP, PdpB and FopA were used as soluble, inner membrane and outer membrane markers, respectively. (B and C) Immunoblot analysis of purified surface biotinylated proteins from SchuS4ΔdipA(pdipA-HA) (B) or SchuS4(piglA-HA) (C) lysates. DipA-HA and IglA-HA were detected using anti-HA antibodies. FopA was used as a positive control; PdpB and IglA were used as negative controls. Input, untreated (-biotin) and biotinylated (+biotin) samples were processed for CFU enumeration and immunoblotting as described in Materials and Methods. Samples were loaded based on CFU equivalents as follows: 1x107 (Input) or 1x108 (-/+ biotin) for anti-DipA-HA analysis, 5x106 (Input) or 1x108 (-/+ biotin) for anti-FopA analysis, 1x107 (Input) or 5x108 (-/+ biotin) for anti-PdpB analysis, 1x107 (Input) or 5x108 (-/+ biotin) for anti-IglA analysis, 1x107 (Input) or 5x108 (-/+ biotin) for anti-IglA-HA analysis.
The presence of DipA in the bacterial outer membrane prompted us to ascertain whether DipA is localized to the bacterial surface. We used a SchuS4ΔdipA strain expressing DipA with a C-terminal tandem HA tag (DipA-HA) because of the higher sensitivity of the monoclonal anti-HA antibody. In surface protein biotinylation experiments, DipA-HA was detected in the biotinylated fraction (Figure 2B), revealing its exposure on the bacterial surface. FopA and PdpB served as positive and negative controls, respectively. Consistent with previous findings, FopA, but not PdpB, showed exposure on the bacterial surface (Figure 2B) [38,39]. IglA was previously detected on the surface of LVS and F . novicida strains over-expressing PdpD [38,39]. Contrary to these reports, but consistent with the fractionation properties reported here (Figure 2A) and by de Bruin et al. [37], IglA was not detected on the bacterial surface (Figure 2B). Surface biotinylation of IglA was also not detected in SchuS4 overexpressing IglA fused with an HA epitope arguing against the possibility of detection sensitivity of our methods, and ensuring that biotinylation of DipA-HA reflects true surface exposure and not an artefact of over-expression (Figure 2C). Furthermore, none of the proteins examined were detected in the absence of biotin labelling, confirming the specificity of streptavidin binding to biotinylated surface proteins (Figure 2B and 2C).
Since DipA is surface-exposed on bacteria grown in vitro, we next examined its localization in the context of a macrophage infection. Under conditions that do not permeabilize bacterial membranes, we detected DipA-HA on the surface of intracellular bacteria at 10 h post infection (p.i.) in BMMs by immunofluorescence labelling (Figure 3A). IglI-HA was similarly detected, consistent with previous reports of IglI secretion by F. tularensis during macrophage infection [35,40]. Co-staining with anti-LPS antibodies revealed co-localization of both DipA-HA and IglI-HA with bacterial surface structures (Figure 3A). Both DipA-HA and IglI-HA were only detected on the surface of a subset of bacteria either within BMMs (Figure 3A) or grown in vitro (data not shown). Since this variable staining pattern was not confined to an infection setting or to DipA only, it suggests a detection issue rather than differential surface expression within the bacterial population. In contrast, IglA-HA was not detected on the surface SchuS4 during BMM infection, corroborating our biochemical observations from in vitro grown bacteria. Lack of IglA-HA immunodetection was not due to lack of expression since DipA-HA, IglI-HA, and IglA-HA showed nearly equivalent levels of expression (Figure S1A).
Figure 3. DipA is exposed to the host cytosol during macrophage infection.

(A) Representative confocal micrographs of BMMs infected for 10 h with SchuS4ΔdipA(pdipA-HA), SchuS4(piglI-HA), or SchuS4(piglA-HA). Using conditions that permeabilize host plasma membranes but not bacterial membranes (described in Materials and Methods), samples were processed for immunofluorescence labelling of HA-tagged proteins (green) and bacterial LPS (red), and counterstained with DAPI to label DNA (blue). Magnified insets show single channel images of the boxed area. Scale bars, 10 or 2 µm. (B) Quantification of CCF2/AM cleavage in J774A.1 cells that were either uninfected or infected with SchuS4, or SchuS4 expressing C-terminal TEM1 fusions with IglI, IglA or DipA. After 16 h, infected macrophages were loaded with CCF2/AM and analyzed by live cell microscopy for blue fluorescence emission. At least 100 cells were scored per experiment. Data are means ± SD from a representative experiment performed in triplicate out of three independent repeats. Asterisks indicate statistically significant differences compared to uninfected, SchuS4-infected, and SchuS4 expressing IglA-TEM1-infected controls (* P < 0.05, 1-way ANOVA, Tukey’s post-test). (C) Representative fluorescence micrographs of J774A.1 cells that were either uninfected or infected for 16 h with SchuS4, SchuS4 expressing IglI-TEM1, SchuS4 expressing IglA-TEM1, and SchuS4 expressing DipA-TEM1. Cells emitting blue fluorescence indicate delivery of TEM1 β-lactamase fusions to the cytosol and CCF2/AM cleavage. Intact CCF2/AM, indicating the absence of TEM1 β-lactamase activity in the cytosol, results in green fluorescence emission. Scale bar, 50 µm.
To verify these observations, we used a fluorescence-based assay that relies on the accessibility of TEM1 β-lactamase protein fusions to the host cytosol to cleave and disrupt the fluorescence resonance energy transfer (FRET) of the β-lactamase-sensitive fluorescent substrate CCF2/AM. Expression of the TEM1-fusion proteins in SchuS4 was confirmed by immunoblot analysis and revealed comparable levels among the constructs (Figure S1B). At 16 h p.i., none of the uninfected and SchuS4-infected J774A.1 macrophage-like cells displayed CCF2/AM cleavage via blue fluorescence emission, indicating a low background level of intrinsic β-lactamase activity (Figure 3B and 3C), despite the presence of the blaA1 (FTT0681c) and blaB1 (FTT0611c) genes in the SchuS4 genome [41]. In contrast, a significantly higher percentage of J774A.1 cells emitting blue fluorescence were detected upon infection with SchuS4 strains expressing IglI-TEM1 (53.8 ± 17.0%; Figure 3B and C), in agreement with previous findings demonstrating IglI secretion via the FPI-encoded T6SS [35,40,42]. Infection with a SchuS4 strain expressing DipA-TEM1 also resulted in blue fluorescence emission (23.8 ± 2.9% of J774A.1 cells), indicating delivery of DipA-TEM1 to the host cytosol (Figure 3B and 3C). As a negative control, very few macrophages displaying CCF2/AM conversion were detected when infected with SchuS4 expressing IglA-TEM1 (2.7 ± 1.1%; Figure 3B and 3C). Because F. tularensis is a cytosolic pathogen, this assay does not discriminate between DipA secretion into the macrophage cytosol or exposure to the cytosol due to its localization on the bacterial surface. Altogether, our results show that DipA is a membrane-associated protein exposed both on the surface of in vitro grown Francisella and to the cytosol of infected macrophages.
The Sel1-like repeat domains are important for DipA function
Four regions of DipA were predicted to form putative SLR domains by SMART (residues 96-132, 133-169, 193-229, and 231-262) and Pfam (residues 100-132, 133-169, 196-229 and 235-262) (Figure 1A). SLRs are structural domains of paired anti-parallel α-helical repeats that provide a scaffold for protein–protein interactions [43]. SLR regions consist of variable sequence lengths spanning 36-44 amino acids with low sequence identity [43]. Because SLR sequences are highly divergent, we relied on homology model prediction by I-TASSER (C-Score -0.53, TM-Score 0.65 ± 0.13, RMSD 7.7 ± 4.3 Å) [44] to determine a mutagenesis strategy for the SLR regions of DipA (Figure 1C). The three dimensional structure prediction of DipA revealed a spatial proximity between the Sel1a and Sel1b helices, indicating potential interactions between these two SLR domains (Figure 1C). Similarly, the proximity of Sel1c and Sel1d domains suggests helical interactions between this pair of SLRs. Thus, we constructed two DipA SLR domain mutants according to this model: deletion of Sel1a and Sel1b between amino acid residues 95-170 (DipAΔSel1ab) and deletion of Sel1c and Sel1d between amino acid residues 192-263 (DipAΔSel1cd) (Figure 1A). HA-tagged versions of these DipA mutants were introduced into the SchuS4ΔdipA strain to evaluate the role of the SLR domains in DipA function. Expression of DipAΔSel1ab-HA and DipAΔSel1cd-HA mutants were similar to that of full-length DipA-HA (Figure S1A). In BMMs, SchuS4ΔdipA displayed limited intracellular growth whereas SchuS4 replicated 2.5 log over 16 h (Figure 4A). While the intracellular growth of SchuS4ΔdipA was restored to wild-type levels upon complementation with the full length DipA, neither DipAΔSel1ab nor DipAΔSel1cd significantly restored the ability of SchuS4ΔdipA to replicate in BMMs (Figure 4A). The DipAΔSel1ab and DipAΔSel1cd truncations partitioned like the full-length protein in the IM and OM fractions (Figure 4B), remained surface-exposed on SchuS4ΔdipA (Figure 4C), and were exposed to the macrophage cytosol during infection (Figure 4D and 4E). Thus, despite targeting to the correct location, the inability of either the DipAΔSel1ab or DipAΔSel1cd truncations to complement the SchuS4ΔdipA mutant growth defect indicates that the SLR domains are required for the biological function of DipA.
Figure 4. The SLR and CC domains of DipA are functionally distinct.

(A) Ability of DipA variants to complement the intracellular growth defect of SchuS4ΔdipA. Viable intracellular bacteria were enumerated at 1 h and 16 h p.i. from BMMs infected with SchuS4, SchuS4ΔdipA, or SchuS4ΔdipA expressing HA-tagged DipA variants (DipA-HA, DipAΔSel1ab-HA, DipAΔSel1cd-HA, DipAΔCC-HA, DipACC(AIL 3D)-HA, or DipACC(LAL 3D)-HA). Fold change in replication was calculated by comparing CFUs at 16 h p.i. versus 1 h p.i. Data are means ± SD from three independent experiments. Asterisks indicate statistically significant differences compared to SchuS4-infected and SchuS4ΔdipA expressing DipA-HA-infected macrophages (* P < 0.05, 1-way ANOVA, Tukey’s post-test). (B) Subcellular localization of HA-tagged DipA variants as described in (A). Soluble (Sol), inner membrane (IM), and outer membrane (OM) enriched fractions were separated based on Sarkosyl solubility and subjected to immunoblot analysis with antibodies against HA. Each fraction was concentrated to the same volume and equal volumes were loaded. (C) Immunoblot analysis of purified surface biotinylated proteins from lysates of SchuS4ΔdipA strains expressing HA-tagged DipA variants as described in (A). Input and biotinylated (surface) samples were processed for CFU enumeration and immunoblotting as described in Materials and Methods. Samples were loaded based on CFU equivalents as follows: 1x106 (Input) or 1x108 (surface). (D) Quantification of J774A.1 cells emitting blue fluorescence that were either uninfected or infected with SchuS4, or SchuS4 expressing C-terminal TEM1 fusions with IglI, IglA, DipA, DipAΔSel1ab, DipAΔSel1cd, DipAΔCC, DipACC(AIL 3D), or DipACC(LAL 3D). Infected cells were analyzed for CCF2/AM cleavage at 16 h pi. At least 100 cells were scored per experiment. Data are means ± SD from a representative experiment performed in triplicate out of three independent repeats. Asterisks indicate statistically significant differences compared to uninfected, SchuS4-infected, and SchuS4 expressing IglA-TEM1-infected controls (* P < 0.001, 1-way ANOVA, Tukey’s post-test). (E) Representative fluorescence micrographs of J774A.1 macrophages infected for 16 h with SchuS4 expressing either DipA-TEM1, DipAΔSel1ab-TEM1, DipAΔSel1cd-TEM1, DipAΔCC-TEM1, DipACC(AIL 3D)-TEM1, or DipACC(LAL 3D)-TEM1. Cells emitting blue fluorescence indicate delivery of TEM1 β-lactamase fusions to the cytosol to cleave the CCF2/AM substrate. Intact CCF2/AM, indicating the absence of TEM1 β-lactamase activity in the cytosol, results in green fluorescence emission. Scale bar, 50 µm.
The coiled-coil domain is required for DipA targeting to the bacterial surface
The putative CC domain of DipA was predicted by SMART (residues 311-343), COILS (residues 313-341) and Marcoil (residues 314-343) to form near the C-terminus (Figure 1A and 1C). CC domains are structural elements comprised of multiple amphipathic α-helices that wind around one another to generate distinct protein binding sites [45]. CC domain sequences characteristically contain a repetitive seven-residue pattern, represented as a-b-c-d-e-f-g, in which the residues at positions a and d form a hydrophobic core to drive inter-helical interactions [45]. The core residues of the DipA CC domain predicted by COILS to form the hydrophobic seam are L414, A317, L321, I324, and L331 (Figure 1B). Based on these predictions, we altered the putative CC domain by either in-frame deletion of the entire CC region between amino acid residues 311-343 (DipAΔCC, Figure 1A) or substitution of three key hydrophobic residues at position d of the α-helix interface to aspartate residues A317D, I324D, L331D [DipACC(AIL 3D)] or at positions a and d to aspartate residues L414D, A317D, L321D [DipACC(LAL 3D)] (Figure 1B). These three DipA CC mutants were compared to wild-type DipA in a SchuS4ΔdipA background to assess the role of the CC domain in DipA function. None of the DipA CC HA-tagged mutants were able to functionally complement the intracellular growth defect of SchuS4ΔdipA (Figure 4A), even though all three constructs were expressed at levels similar to full-length DipA-HA (Figure S1A). Although DipAΔCC-HA, DipACC(AIL 3D)-HA and DipACC(LAL 3D)-HA were detected in the IM and OM fractions of SchuS4ΔdipA, none were detected on the bacterial surface by biotinylation (Figure 4C). Moreover, the DipA CC mutants were not accessible to the macrophage cytosol as indicated by the low percentages of CCF2/AM conversion in macrophages infected with SchuS4 expressing DipAΔCC-TEM1 (5.1 ± 3.2%), DipACC(AIL 3D)-TEM1 (6.6 ± 1.4%) and DipACC(LAL 3D)-TEM1 (6.8 ± 1.1%), which were not significantly different from the negative controls (Figure 4D and 4E). Expression of these TEM1-fusions were also verified and showed little variation in expression levels compared with full length DipA-TEM1 (Figure S1B). Hence, the C-terminal coiled-coil domain of DipA is required for surface exposure of DipA. Moreover, these data indicate that proper surface localization of DipA is required to fulfill its function, although one cannot exclude that mutations of the CC domain functionally disable DipA independently of its localization.
DipA interacts with FopA
Having determined that DipA is exposed to the macrophage cytosol and contains a combination of domains known to mediate protein–protein interactions, we first hypothesized that DipA interacts with host factors to promote Francisella intracellular replication. To test this hypothesis, co-immunoprecipitation attempts using either ectopically expressed DipA-GFP in HeLa cells, pulldowns from HeLa and BMM cell lysates using purified recombinant DipA-GST, or co-immunoprecipitations from DipA-HA-expressing Schu S4-infected BMMs were performed, yet with no success (data not shown). We then hypothesized that DipA is part of a membrane-associated bacterial macromolecular complex and sought to identify potential bacterial interacting proteins by performing co-immunoprecipitation coupled to mass spectrometry. DipA-HA was immunoprecipitated from SchuS4ΔdipA expressing DipA-HA and additional proteins resolved by SDS-PAGE were identified by mass spectrometry. Peptides identified from an evident 40 kDa band co-immunoprecipitated with DipA-HA were compared to those identified from a faint corresponding band co-immunoprecipitated from SchuS4ΔdipA expressing non-tagged DipA to rule out non-specific candidates (Figure 5A). As expected, DipA was detected in the immunoprecipitated material from the lysate containing DipA-HA since it migrates at ~ 40 kDa. By this approach, we identified three F. tularensis proteins that were immunoprecipitated specifically along with DipA-HA: FTT1407c (16 matching peptides, 46% sequence coverage), a Francisella -specific putative membrane protein of unknown function; FTT1365c/FbaB (1 matching peptide, 3% sequence coverage), a fructose-1,6-bisphosphate aldolase (FBA) homolog; and FTT0583/FopA (1 matching peptide, 5% sequence coverage), an immunogenic outer membrane protein of unknown function (Figure 5A) [32,46]. Several glycolytic enzymes, including FBA, have been reported to localize to the surface of bacterial pathogens where they play a role in virulence [47–51]. Thus, the interacting protein candidates identified corroborate the localization profile of DipA as a surface-exposed membrane-associated protein.
Figure 5. Identification of DipA interacting partners.
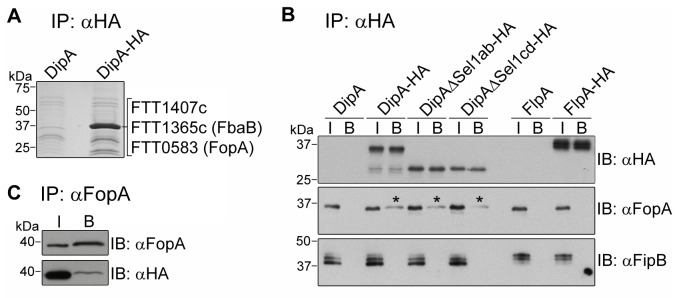
(A) Code blue stained gel of proteins immunoprecipitated with anti-HA conjugated beads from lysates of SchuS4ΔdipA expressing DipA [SchuS4ΔdipA(pdipA)] or DipA-HA [SchuS4ΔdipA(pdipA-HA)]. Samples were resolved by SDS-PAGE and stained with GelCode® Blue. Protein bands migrating at ~40 kDa were subjected to mass spectrometry-based identification. FTT1407c, FTT1365c and FTT0583 were specifically co-immunoprecipitated with DipA-HA. (B and C) Verification of FopA interaction with DipA by immunoblot analysis. (B) Lysates from SchuS4ΔdipA expressing DipA derivatives (DipA, Dip-HA, DipAΔSel1ab-HA, DipAΔSel1cd-HA), or SchuS4ΔflpA expressing FlpA derivatives (FlpA, FlpA-HA) were subjected to immunoprecipitation (IP) using anti-HA conjugated beads followed by immunoblot (IB) analysis with anti-HA, anti-FopA and anti-FipB antibodies. I denotes sample input; B denotes bound fraction. (C) Lysate from SchuS4ΔdipA expressing DipA-HA was subjected to immunoprecipitation (IP) using anti-FopA antibodies followed by immunoblot (IB) analysis with anti-FopA antibodies to detect FopA and anti-HA antibodies to detect Dip-HA. I denotes sample input; B denotes bound fraction.
Binding of DipA to FopA was confirmed by co-immunoprecipitation followed by immunoblot analysis with an anti-FopA antibody (Figure 5B). Due to a lack of reagents for FTT1407c and FbaB, we were unable to verify their interaction with DipA. FopA was specifically co-immunoprecipitated with DipA-HA, but not from control lysates of SchuS4ΔdipA expressing DipA lacking the HA-tag (Figure 5B). DipA interaction with FopA was confirmed by reciprocal co-immunoprecipitation (Figure 5C). To further examine the specificity of the DipA-FopA pull down, we tested whether FopA co-immunoprecipitated with FTT1676, a putative membrane lipoprotein [10]. Indeed, FTT1676 partitioned to the inner and outer membrane enriched fractions of GFP-expressing SchuS4 (Figure S2A), and was acylated in3 [H] palmitate incorporation experiments (Figure S2B), confirming the predicted membrane localization and lipoprotein nature of FTT1676. Hence, we renamed this protein FlpA for Francisella lipoprotein A. Unlike for DipA, FopA was not co-immunoprecipitated from lysates of SchuS4ΔflpA expressing either FlpA or HA-tagged FlpA, (Figure 5B). FipB, another Francisella lipoprotein, did not co-immunoprecipitate with DipA-HA, lending additional evidence to the specificity of FopA binding to DipA (Figure 5B) [30,31]. SLR domain mutations did not affect DipA interactions with FopA (Figure 5B), implying that other regions of DipA are responsible for this interaction. We were unable to assess the role of the CC domain in FopA binding because neither the HA-tagged nor FLAG-tagged versions of the DipA CC domain mutants could be efficiently immunoprecipitated with the corresponding antibodies (data not shown). In summary, these results demonstrate that DipA binds either directly or indirectly to FopA, an interaction that points toward a potential function of DipA as part of an outer membrane complex.
Deletion of fopA, but not of FTT1407c, reproduces the intracellular defects of the ∆dipA mutant
To extend our findings and explore the roles of the DipA interacting candidates in pathogenesis, we generated in-frame deletions of fopA and FTT1407c loci in SchuS4 and assessed the intracellular growth and in vivo virulence of the resulting mutants. Not surprisingly, since other enzymes in the glycolytic pathway are predicted to be essential [52], attempts to delete fbaB were unsuccessful, suggesting that this locus is indispensable for growth under in vitro conditions. Deletion of FTT1407c did not affect bacterial replication in BMMs over a period of 24 h (Figure 6A), whereas deletion of fopA resulted in impaired intracellular proliferation that mirrored the phenotype of SchuS4ΔdipA (Figure 6B) [10,53]. We then examined the intracellular trafficking of SchuS4ΔfopA, because phagosomal escape is a pre-requisite for cytosolic replication [9,10,26,54–56]. Using co-localization of bacteria with LAMP-1-positive membranes as an indicator of vacuolar or cytosolic location in BMMs, SchuS4ΔfopA showed phagosome escape kinetics similar to wild-type and SchuS4ΔdipA bacteria (Figure 6C). Thus, deletion of fopA specifically impairs cytosolic replication of SchuS4 in BMMs. Growth of the SchuS4ΔfopA mutant in modified Mueller-Hinton broth was not affected, excluding any physiological impairment (data not shown). In trans complementation with full-length fopA fully restored the ability of SchuS4ΔfopA to survive and grow in BMMs, confirming that the observed phenotypic defect was due to deletion of fopA (Figure 6B). Since SchuS4ΔdipA and SchuS4ΔfopA demonstrated similar intracellular growth defects, we examined the effect of a double deletion mutant of both loci on the ability of SchuS4 to replicate in BMMs, predicting that the combination of the two deletions should not have additive effects if both proteins participate in the same bacterial functions. SchuS4ΔdipAΔfopA bacteria showed the same intracellular defects as the single mutants (Figure 6B), further suggesting that DipA and FopA contribute to the same bacterial function that can be disabled by single deletion of either protein. We have shown previously that dipA is essential for SchuS4 virulence in BALB/cJ mice [10,57]. To test whether deletion of FTT1407c or fopA affects virulence of SchuS4, BALB/cJ mice were intranasally infected with 10 CFUs of each mutant strain. Similar to infection with wild-type SchuS4, all mice infected with the SchuS4ΔFTT1407c mutant had to be euthanized by 6 days p.i. (Figure 6D). In contrast, SchuS4ΔfopA displayed attenuated virulence, since 100% of intranasally infected animals survived up to 30 days p.i. (Figure 6D), consistent with the attenuation of the ΔdipA mutant [10].
Figure 6. FopA is required for SchuS4 intracellular growth in BMMs and virulence in mice, but not for DipA outer membrane and surface localization.

(A) Intracellular growth of SchuS4 and its isogenic ΔFTT1407c mutant in BMMs. BMMs were infected with either strain and CFUs were enumerated at various times p.i. Data are means ± SD from a representative experiment performed in triplicate out of two independent repeats. (B) Intracellular growth of SchuS4, its isogenic ΔfopA and ΔdipAΔfopA mutants and the complemented ΔfopA(pfopA-HA) strains in BMMs. BMMs were infected with either strain and CFUs were enumerated at various times p.i. Data are means ± SD from a representative experiment performed in triplicate out of two independent repeats. (C) Quantification of bacteria enclosed within LAMP-1-positive phagosomal membranes. BMMs were infected for 1 h with either SchuS4 or its isogenic ΔdipA, ΔfopA and ΔdipAΔfopA mutants. Samples were processed for immunofluorescence labelling of bacteria and LAMP-1-positive membranes. Infected BMMs were scored for number of infected cells with bacteria enclosed within LAMP-1-positive compartments. At least 100 bacteria per experiment were scored for each condition. Data are means ± SD from three independent experiments. (D) Survival curves of BALB/cJ mice infected with SchuS4, SchuS4ΔFTT1407c or SchuS4ΔfopA by intranasal inoculation. Intranasal inocula were 27 (SchuS4), 16 (ΔFTT1407c), and 15 (ΔfopA) CFUs. (E) Subcellular localization of DipA, FopA, and PdpB from SchuS4ΔfopA (top panels) or SchuS4ΔdipA (bottom panels). Soluble (Sol), inner membrane (IM), and outer membrane (OM) enriched fractions were separated based on Sarkosyl solubility and subjected to immunoblot analysis with antibodies against DipA, FopA, and PdpB. Each fraction was concentrated to the same volume and equal volumes were loaded. (F) Immunoblot analysis of purified surface biotinylated proteins from SchuS4ΔfopA(pdipA-HA) (top panels) or SchuS4ΔdipA (bottom panels) lysates. DipA-HA was detected using anti-HA antibodies; FopA was detected using anti-FopA antibodies. PdpB and was used as a negative control. Input, untreated (-biotin) and biotinylated (+biotin) samples were processed for CFU enumeration and immunoblotting as described in Materials and Methods. Samples were loaded based on CFU equivalents as follows: 2x106 (Input) or 1x108 (-/+ biotin) for anti-DipA-HA analysis, 5x106 (Input) or 1x108 (-/+ biotin) for anti-FopA analysis, 1x107 (Input) or 5x108 (-/+ biotin) for anti-PdpB analysis.
Because both DipA and FopA are exposed on the surface of SchuS4 and interact with each other, we examined whether the absence of one affects the localization of the other. DipA remained enriched to the inner and outer membranes (Figure 6E) and its HA-tagged version was detected via biotinylation on the surface of a SchuS4ΔfopA strain (Figure 6F), thus, the localization of DipA does not depend on FopA. Reciprocally, localization of FopA to the bacterial outer membrane and exposure on the bacterial surface remained unaffected in the absence of DipA (Figure 6E and 6F). Hence, outer membrane localization and surface exposure of DipA and FopA are independent of one another.
In this study, we have examined how the various structural domains of DipA contribute to its functions, in order to gain insights into its role in Francisella pathogenesis. We demonstrate that DipA is targeted to the surface of SchuS4 to fulfill its role as a Francisella virulence factor. Our data highlights the importance of the SLR and CC domains for DipA function. Among other functions, proteins containing SLR domains have been found to serve as adaptor proteins for the assembly of membrane-bound macromolecular complexes. For example, the yeast Hrd3 SLR protein is anchored to the ER membrane in a complex that functions as part of the ER-associated protein degradation mechanism [58]. In prokaryotes, the SLR protein MotX from Vibrio parahaemolyticus is anchored to the cytoplasmic membrane via peptidoglycan and complexes with flagellar proteins, MotY and PomB, and contributes to motor function [59,60]. The effector proteins LpnE, EnhC, and LidL secreted by the Legionella pneumophila Type IV secretion system are SLR-containing proteins that have been shown to contribute to the trafficking of the Legionella containing vacuole and establishment of an intracellular replicative niche [61–63]. Deletion of even a pair of the eight SLR regions rendered LpnE non-functional, emphasizing the importance of these domains [63]. Similarly, deletion of either pair of the SLR domains impaired the ability of DipA to complement the growth defect of the SchuS4ΔdipA mutant. The CC domain is also important for DipA function. A key characteristic of CC domains is the proclivity to form multimeric complexes, a feature driven by structural stability [64,65]. For example, the majority of the 200 coiled-coil interactions from Saccharomyces cerevisiae involve multiple heterotypic partners [66]. In bacteria, CC domains are found commonly in proteins associated with secretion systems [67]. The CC domain of EspA, while dispensable for its secretion, is essential for the multimeric assembly of the type III secretion apparatus on the surface of Enteropathogenic Escherichia coli [68]. In Salmonella enterica serovar typhimurium, CC domains of T3SS effectors are responsible for their targeting to host membranes where they interact with host factors [69]. Given their structural nature and propensity for oligomerization, the presence of these domains suggests that DipA may function as a structural scaffold to link various components of a multiprotein complex that imparts a unique mechanism of virulence to F. tularensis.
Of the three putative DipA interacting partners identified, FopA may shed some light on the function of DipA even though the biological significance of the interaction is currently unclear. FopA contains a conserved OmpA domain at its C-terminus, while the rest of the protein sequence does not display homology to any known domains. The function of FopA has not been directly addressed; however, other OmpA-like proteins serve a myriad of functions including well-established roles in bacterial adhesion, invasion and immune evasion [70–74]. For pathogens such as uropathogenic Escherichia coli [75], Neisseria gonorrhoeae [76], and Yersinia spp. [77], their respective OmpA homologs have been shown to enhance intracellular survival. The OmpA-like protein of Pseudomonas aeruginosa OprF is thought to act as a sensor for virulence factor activation upon host cell contact [74]. We have also demonstrated a requirement for FopA in intramacrophage growth and in vivo virulence of SchuS4 (Figure 6). Additionally, OmpA homologs are also known to function structurally and in adaptation to environmental stresses [74,78–81]. Based on the demonstrated roles of various OmpA homologs, DipA may function as part of a structural membrane complex with FopA that contributes to the intracellular adaptation of F. tularensis, ultimately allowing it to survive and proliferate within the macrophage cytosol. Consistent with its contribution to the virulence of F. tularensis, DipA possesses domains and interactions with proteins that are implicated in bacterial pathogenesis. Further examination of these interactions will be important in discerning the role of these factors in the virulence of F. tularensis.
Materials and Methods
Ethics Statement
All animal rearing, handling and experimental methods were conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals, 8th Edition, of the National Research Council under protocols approved by the Rocky Mountain Laboratories Institutional Animal Care and Use Committee (IACUC). The Rocky Mountain Laboratories is an AAALAC International-accredited institute.
Bacterial strains and culture conditions
The prototypic Type A virulent strain, F. tularensis subsp. tularensis SchuS4 was obtained from Rick Lyons (University of New Mexico, Albuquerque, NM, USA). GFP-expressing SchuS4 and SchuS4∆dipA (∆FTT0369c) have been described previously [10]. SchuS4∆FTT1407c, SchuS4∆fopA, and SchuS4∆dipA∆fopA mutants were generated as described below. SchuS4 and its derivatives were grown either on modified Mueller-Hinton (MMH) agar plates [Mueller-Hinton medium supplemented with 0.1% glucose (Sigma), 0.025% ferric pyrophosphate (Sigma Aldrich) and 2% IsoVitaleX (Becton Dickinson) or MMH supplemented with 10 µg/ml kanamycin (Sigma) for 3 days at 37°C under 7% CO2. When indicated, colonies from freshly streaked MMH plates were re-suspended in MMH broth or Tryptic-Soy broth supplemented with 0.1% cysteine (TSB-C) grown at 37 °C under agitation. MMH medium was supplemented with either 10 µg/ml kanamycin (Sigma) or 8% sucrose (Sigma) for allelic replacement. All manipulations of F. tularensis strain SchuS4 and its derivatives were performed in a Biosafety Level 3 facility according to standard operating procedures approved by the Rocky Mountain Laboratories Institutional Biosafety Committee and the CDC Division of Select Agents and Toxins regulations.
Sequence analysis and structural modelling
The translated amino acid sequence of DipA was analyzed using SMART (http://smart.embl-heidelberg.de/) and Pfam (http://pfam.sanger.ac.uk/) databases to identify conserved protein domains. SignalP (http://www.cbs.dtu.dk/services/SignalP/) and LipoP (http://www.cbs.dtu.dk/services/LipoP/) prediction programs were used to determine the signal peptidase cleavage site. COILS (http://embnet.vital-it.ch/software/COILS_form.html) [82] with windows of 28, 21 and 14 residues and Marcoil [83] were used in conjunction with SMART to predict the heptad repeat of the coiled-coil domain. A three-dimensional structure of DipA was built by the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) [44,84] and visualized using the UCSF Chimera package (http://www.cgl.ucsf.edu/chimera) [85]. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). The model includes amino acid residues 21-353 of DipA.
Plasmids and molecular techniques
Plasmids and primers used in this study are described in Tables S1 and S2, respectively. Plasmids used in this study were derived from pFNLTP6 [86]. pFNLTP6omp26 (pJC900) was generated by amplification of the omp26 promoter region from SchuS4 genomic DNA with primers JC801 and JC823; the amplified fragment was subcloned into the TA cloning vector pCR2.1 (Life Technologies) its sequence confirmed, and cloned into the EcoRI-digested vector pFNLTP6.
HA fusions were generated using primers described in Table S2. To tag DipA, FlpA, IglI, IglA, and FopA with double-HA, their open reading frames were PCR amplified using SchuS4 genomic DNA as template. The DNA fragment encoding the two tandem repeats of the HA tag was synthesized and used as a template, along with the PCR amplified open reading frames, in overlap extension PCR to fuse the two fragments. The dipA-HA and flpA-HA amplified fusions were sub-cloned into the TA cloning vector pCR2.1 and subsequently sequenced. The fusions were excised from pCR2.1 and ligated into the NheI/XhoI digested pFNLTP6 and pFNLTP6omp26, respectively. The iglA-HA and fopA-HA amplified fusions were cloned into the NheI/XhoI sites and the iglI-HA fusion was cloned into the NotI/XhoI sites of pFNLTP6omp26 using the In-Fusion® PCR Cloning System. The Sel1 domain deletion mutants DipAΔSel1ab-HA and DipAΔSel1cd-HA were generated using the pFNLTP6-dipA-HA construct as template. To delete the first pair of SLR regions (DipAΔSel1ab), 300 bp upstream of the start codon, the start codon, and the first 285 bp of the dipA open reading frame were amplified with oligonucleotides JC943 and JC956; 552 bp of the dipA open reading frame downstream of the Sel1ab domain and the tandem HA tag were amplified with oligonucleotides JC955 and JC957. The two fragments were fused by overlap extension PCR and cloned into EcoRI- and XhoI-digested pFNLTP6 using In-Fusion® PCR Cloning System. To delete the second pair of SLR regions (DipAΔSel1cd), 300 bp upstream of the start codon, start codon, and first 573 bp of the DipA open reading frame were amplified with oligonucleotides JC943 and JC958; 273 bp of the dipA open reading frame downstream of the Sel1cd domain and the tandem HA tag were amplified with oligonucleotides JC955 and JC959. The two fragments were fused by overlap extension PCR and cloned into EcoRI- and XhoI-digested pFNLTP6 using In-Fusion® PCR Cloning System. To generate a CC deletion mutant of DipA with a tandem HA-tag, 300 bp upstream of the start codon, start codon, and first 927 bp of the dipA open reading frame were amplified with oligonucleotides JC943 and JC960 using pFNLTP6-dipA-HA as template. The 30 bp downstream of the CC domain and HA tag were amplified with oligonucleotides JC955 and JC961. The two fragments were fused by overlap extension PCR and cloned into EcoRI- and XhoI-digested pFNLTP6 using In-Fusion® PCR Cloning System. The CC domain substitution mutants, DipACC(AIL 3D)-HA and DipACC(LAL 3D)-HA, were generated by introducing the mutations within the primer sets used for overlap extension PCR and the pFNLTP6-dipA-HA construct as template. The resultant fragments were cloned into EcoRI- and XhoI-digested pFNLTP6 using In-Fusion® PCR Cloning System. The identity and orientation of all constructs generated by PCR were confirmed by restriction digest analysis, sequencing and protein expression (Figure S1A).
To tag DipA and IglI with TEM1, the open reading frames of these genes were PCR amplified using SchuS4 genomic DNA as template. The TEM1 tag was PCR amplified from template pCX340 [87]. The fragments were fused by overlap extension PCR and sub-cloned into pCR2.1. Following sequencing, dipA-TEM1 was excised and cloned into pFNLTP6omp26 at NheI and XhoI sites. The iglI-TEM1 insert was excised and cloned into pFNLTP6omp26 at NotI and XhoI sites. The iglA-TEM1 insert was cloned directly into pFNLTP6omp26 at NheI and XhoI sites by In-Fusion® PCR Cloning System. The DipA domain deletions were TEM1 tagged by PCR-amplifying the domain deletions using previously constructed pFNLTP6 HA-tagged constructs, and fused with TEM1 by overlap extension using primers described in Table S2. The identity and orientation of all constructs generated by PCR were confirmed by restriction digest analysis, sequencing and protein expression (Figure S1B).
Construction of SchuS4 deletion mutants
SchuS4 in-frame deletion mutants were generated using the SacB-assisted allelic replacement suicide vector pJC84, as described previously [10]. Deletions of either the fopA, FTT1407c or fbaB loci were designed to preserve the integrity of downstream genes and avoid any polar effects. To engineer an in-frame deletion of fopA, a 5’-fragment containing 1253 bp upstream of the start codon of fopA (FTT0583), its start and the first codon was generated by PCR amplification using the primers TW202 and TW203, and a 3’ fragment containing 971 bp downstream of the stop codon and the last 7 codons of fopA using primers TW204 and TW205 (Table S2). The fragments were fused by overlap extension PCR using primers TW202 and TW205. The resulting fragment was cloned into the SalI and BglII sites of pJC84 using the In-Fusion® PCR Cloning System (Clontech) and was fully sequenced. To engineer an in-frame deletion of FTT1407c, a 5’-fragment containing 969 bp upstream of the start codon of FTT1407c, its start and the first codon was generated by PCR amplification using the primers TW151 and TW152, and a 3’ fragment containing 1116 bp downstream of the stop codon and the last 6 codons of FTT1407c using primers TW153 and TW154 (Table S2). The fragments were fused by overlap extension PCR using primers TW151 and TW154. The resulting fragment was cloned into the BamHI and SalI sites of pJC84 using the In-Fusion® PCR Cloning System (Clontech) and was fully sequenced. To engineer an in-frame deletion of fbaB, a 5’-fragment containing 1476 bp upstream of the start codon of fbaB (FTT1365c), its start and the first 3 codons was generated by PCR amplification using the primers RC407 and RC408, and a 3’ fragment containing 1385 bp downstream of the stop codon and the last 8 codons of fopA using primers RC409 and RC410 (Table S2). The fragments were fused by overlap extension PCR using primers RC407 and RC410. The resulting fragment was cloned into the BamHI and SalI sites of pJC84 using the In-Fusion® PCR Cloning System (Clontech) and was fully sequenced.
To perform allelic replacement in the chromosome of Schu S4, electrocompetent bacteria were prepared and electroporated with recombinant pJC84 plasmid DNA as previously described [10]. Kanamycin-resistant merodiploid colonies were tested for integration of the allelic replacement plasmid, using colony PCR with primers JC420 and JC427 or JC589 and JC428 (Table S2). Independent clones were then subjected to sucrose counter selection to isolate clones that have undergone allelic replacement as previously described [10]. Absence of the targeted allele and allelic replacement within the correct chromosomal locus were verified by PCR using primers listed in Table S2. Independent clones with the correct in-frame deletion were isolated and used for further studies. Allelic replacement was performed on the single deletion mutant ΔdipA to generate the double deletion mutant ΔdipAΔfopA.
Antibody generation
The rat polyclonal anti-DipA and anti-FlpA antisera used in this study were generated by Aldevron (Freiburg, Germany) via genetic immunization using codon-optimized FTT0369c and FTT1676 open reading frames. Rabbit polyclonal antisera against PdpB and IglA were generated by New England Peptide, Inc. by immunizing New-Zealand rabbits with PdpB (KTDDRWESKDFSKPEC) and IglA (CSVDAKKEFADREVRR) peptides.
Immunoblot analysis
Samples were resolved on 12% SDS-polyacrylamide gels, and transferred to Hybond-ECL nitrocellulose membranes (Amersham Biosciences) as previously described [9]. Primary antibodies used for immunoblot analysis were: mouse monoclonal anti-HA (clone 16B12, Covance Research Products), rat anti-DipA (this study), rat anti-FopA (gift from Jason Huntley, University of Toledo, Toledo, Ohio), rabbit anti-PdpB (this study), rabbit anti-IglA (this study), rabbit anti-GFP (Life Technologies), rabbit anti-FipB [31], rat anti-FlpA (this study). Secondary antibodies used were: HRP-conjugated anti-rabbit IgG, anti-rat IgG or anti-mouse IgG (Cell Signaling Technology), and HRP-conjugated light chain specific anti-rat IgG or anti-mouse IgG (Jackson ImmunoResearch). Immunodetection was performed using SNAP i.d. (Millipore). Signals were visualized with ECL Advance™ immunoblotting detection kit (Amersham Biosciences) followed by autoradiography and scanning using a Kodak Image Station 4000MM Pro and assembled using Adobe Photoshop CS3.
Fractionation
For fractionation of SchuS4 strains, ~2x1011 CFU were harvested from freshly streaked MMH plates, washed once with PBS, and resuspended in ice cold lysis buffer [PBS supplemented with EDTA-free cOmplete protease inhibitor tablet (Roche), 0.6 µg/mL DNase I (Sigma) and 0.6 µg/mL RNase (Roche)]. Aliquots of these suspensions were taken for plating to determine the actual CFU. Bacteria were resuspended into Lysing Matrix B tubes containing 0.1 mm silica spheres (MP Biomedicals) and disrupted for 9-cycles of 45 s/cycle at maximum speed in a FastPrep FP120 Cell Disrupter (ThermoSavant). Tubes were incubated on ice for 1.5 min between cycles. Samples were taken for plating to determine lysis. Lysates were clarified by two rounds of centrifugation at 8000 x g at 4 °C for 10 min followed by ultracentrifugation for 1.5 h at 100 000 x g at 4 °C. Supernatants were saved as soluble protein fractions. Membrane pellets were resuspended in 1% Sarkosyl (Sigma) and subjected to ultracentrifugation for 1.5 h at 100 000 x g at 4 °C to separate the Sarkosyl-soluble (inner membrane) and Sarkosyl-insoluble (outer membrane) fractions. Each fraction was ultracentrifuged twice to ensure efficient separation. The pelleted outer membrane fractions were resuspended in PBS. Soluble and inner membrane fractions were concentrated using 10 kDa cut-off Amicon ultrafilters (Millipore) to the same volume as the outer membrane fractions. Equal volumes from each fraction were loaded and separated by SDS-PAGE followed by immunoblotting analysis.
Biotinylation of SchuS4 surface proteins
SchuS4 strains were harvested from freshly streak MMH plates, washed three times by centrifugation at 10,000 x g at 4 °C for 5 min, and resuspended in cold PBS at ~1x109 CFU/mL. Aliquots (0.5 mL) were incubated with 0.25 mL of EZ-Link sulfo NHS-LC-LC-biotin (15 mg/mL, Pierce) for 0.5 h at room temperature to label surface proteins. Biotinylated suspensions of intact bacteria were centrifuged at 8000 x g at 4 °C for 4 min, washed with 1 mL of a salt solution (50 mM Tris, 300 mM NaCl, pH 7.5) and two 1 mL washes with cold PBS. Aliquots were taken for plating to determine the actual CFU. After the final wash, bacteria were resuspended in 50 µL of PBS, lysed with 500 µL of B-PERII (Pierce), aliquots plated to ensure complete lysis, and centrifuged at 15,200 x g at 4 °C for 1 min to clarify the lysate. Supernatants were transferred to new tubes to which 0.2 mL of Ultralink immobilized NeutrAvidin beads (Pierce) were added. Samples were incubated for 30 min at room temperature with gentle rocking. The bead complexes were washed thrice in 1 mL of high salt buffer [500 mM NaCl, 25 mM Tris, pH 7.5, 0.2% Tween 20 (v/v)] followed by two more washes in 1 mL of low salt buffer [50 mM NaCl, 25 mM Tris, pH 7.5, 0.2% Tween 20 (v/v)] using centrifugation conditions of 1000 x g for 1 min at room temperature. To recover biotinylated proteins, pelleted NeutrAvidin beads were resuspended in 40 µL of standard SDS-PAGE sample buffer, boiled at 95 °C for 15 min, centrifuged at 500 x g for 1 min to separate supernatants containing biotinylated proteins from the beads. The supernatants were normalized to CFU equivalents resolved by SDS-PAGE and subjected to immunoblot analysis.
Macrophage culture, infection and bacterial CFU determination
Murine BMMs harvested from C57BL/6J (Jackson Laboratories) were differentiated as described previously [9]. J774A.1 macrophage-like cells (ATCC, TIB-67) were cultured and maintained in 4.5 g/L glucose Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies) supplemented with 10% fetal bovine serum (FBS, Life Technologies) and 4 mM L-glutamine (Life Technologies). Two days prior to infection, J774A.1 cells were plated onto 96-well flat cell culture-treated plates at a density of 1x104 macrophages/well. Immediately prior to infection of macrophages, a few colonies from a freshly streaked plate were resuspended in MMH broth and the OD600nm was measured to estimate bacterial numbers. Bacteria were diluted to the appropriate multiplicity of infection (MOI) in BMM media and 0.5 ml of bacterial suspension was added to chilled cells. Macrophage were infected at an applied MOI of 50 (BMM) or 100 (J774A.1) as previously described [9]. Intracellular growth was monitored by determining the number of colony-forming units (CFU) recovered from lysed BMMs as previously described [9]. At 1 h and 16 h p.i., the numbers of viable intracellular bacteria from lysed BMMs were determined from triplicate wells.
Immunofluorescence microscopy
BMMs on 12 mm glass coverslips were infected as described previously [9]. To ensure only BMM plasma membranes were permeabilized, infected BMMs were washed three times with PBS, fixed for 10 min at 37 °C in 1% paraformaldehyde, washed three times with PBS, and incubated for 10 min in 50 mM NH4Cl in PBS to quench free aldehyde groups. Samples were blocked and permeabilized in blocking buffer (10% horse serum, 0.05% saponin in PBS) for 30 min at room temperature. Samples were labelled by incubating inverted coverslips on drops of primary antibodies diluted in blocking buffer for 1 h at room temperature. Primary antibodies used were mouse monoclonal anti-F. tularensis LPS (US Biologicals), rat monoclonal anti-HA (clone 3F10, Life Technologies) and rabbit anti-GFP (Life Technologies). Bound antibodies were detected by incubation of coverslips on drops of secondary antibodies diluted in blocking buffer for 1 h at room temperature. Secondary antibodies used were Alexa Fluor™ 568-conjugated goat anti-mouse (Life Technologies), Alexa Fluor™ 568-conjugated goat anti-rabbit (Life Technologies) and Alexa Fluor™ 488-conjugated goat anti-rabbit (Life Technologies). Macrophage nuclei were counterstained with DAPI (Life Technologies) diluted in ddH 20 for 10 min at room temperature after incubation with secondary antibodies. Samples were washed twice with 0.05% saponin in PBS, once in PBS and once in ddH 20 and then mounted in Mowiol 4-88 mounting medium (Calbiochem). When permeabilized and stained under these conditions, antibody labelling of GFP was detected in fewer than 5% of BMMs infected with GFP-expressing SchuS4 (data not shown). Labelling of GFP by antibodies was readily detected in BMMs infected with GFP-expressing SchuS4 when permeabilized with 0.5% Triton X-100 (Sigma). In vitro grown bacteria were immunostained in suspension as described above and mounted on slides in Mowiol 4-88. Confocal images of 1024x1024 pixels were acquired using Carl Zeiss LSM 710 confocal laser scanning microscope for image acquisition and assembled using Adobe Photoshop CS3.
TEM1 β-lactamase assay
TEM1 fusions described in Table S1 were electroporated into SchuS4 and expression verified by immunoblot analysis with an anti-β-lactamase antibody (QED Bioscience Inc). J774.A1 cells were then infected with SchuS4 strains harboring the TEM1 fusions at an MOI of 100 as previously described [9]. At 16 h pi, cells were washed twice with PBS and loaded with the fluorescent substrate CCF2/AM (LiveBLAzer-FRET B/G loading kit, Life Technologies) in the β-lactamase loading solution supplemented with 15 mM Probenecid (Life Technologies). Cells were incubated in the dark for 90 min at room temperature and then observed under epifluorescence using a Carl Zeiss Axiovert 200M fitted with a β-lactamase Blue/Aqua 41031 filterset (Chroma Technology Corp.). At least 300 cells were counted in triplicate wells to determine the percentage of cells emitting blue fluorescence (TEM1-positive). Data are means + SD from three independent experiments performed in triplicate. Following quantification, macrophages were then fixed and immunostained for Francisella with a mouse monoclonal anti-F. tularensis LPS (US Biologicals) antibody to verify that > 95% cells were infected, ensuring that the CCF2/AM cleavage occurred in infected cells.
Co-immunoprecipitations
Co-immunoprecipitations to identify potential DipA interacting protein partners were performed with agarose beads conjugated to monoclonal anti-HA antibody (Clone HA-7, Sigma). Approximately 10 OD600 of SchuS4ΔdipA expressing either DipA [SchuS4ΔdipA(pFNLTP6dipA)] or DipA-HA [SchuS4ΔdipA(pFNLTP6dipA-HA)] were harvested from overnight MMH broth cultures. The cultures were washed twice with cold PBS and lysed with 1 mL/5 OD600 of B-PERII lysis reagent (Pierce). The lysates were then clarified by three centrifugations at 16,100 x g at 18 °C for 10 min, then the supernatants were incubated with anti-HA conjugated beads for 3 h at room temperature with gentle rotation. The bead complexes were washed three times with B-PERII then twice with wash buffer (0.2% NP40, 150 mM NaCl, 1 mM MgCl2, 20 mM Tris, pH 7.5) in which the mixtures were centrifuged at 8,200 x g for 1 min. Immunoprecipitated proteins were recovered from the beads with three rounds of 50 µL elution buffer (0.1 M glycine, pH 2.5, 0.2% NP40), neutralized with 15 µL Tris pH 9.5, and concentrated with Amicon Ultrafilters (10 kDa cut-off, Millipore). Immunoprecipitated proteins were resolved by SDS-PAGE and visualized with GelCode® Blue Stain Reagent (Thermo Scientific). A prominent band of proteins with apparent molecular weight of 40 kDa co-immunoprecipitated from SchuS4ΔdipA(pFNLTP6dipA-HA) lysate was excised and analyzed by mass spectrometry by the Taplin Biological Mass Spectrometry Facility (Harvard Medical School, Boston, MA). A corresponding band of proteins co-immunoprecipitated from SchuS4ΔdipA(pFNLTP6dipA) lysate was similarly analyzed to serve as a background control. Only peptides identified uniquely by co-immunoprecipitation from SchuS4ΔdipA(pFNLTP6dipA-HA) lysate were considered potential DipA interacting candidates; common peptides identified from both co-immunoprecipitated samples were disregarded.
FopA interaction with DipA was verified by co-immunoprecipitation using the bead conjugated anti-HA antibody as described above using 1 OD600 of SchuS4ΔdipA(pFNLTP6dipA), SchuS4ΔdipA(pFNLTP6dipA-HA), SchuS4ΔdipA(pFNLTP6dipAΔSel1ab-HA), SchuS4ΔdipA(pFNLTP6dipAΔSel1cd-HA), SchuS4ΔflpA (pFNLTP6omp26-flpA), or SchuS4ΔflpA (pFNLTP6omp26-flpA-HA) cultures. Immunoprecipitated material was resolved by SDS-PAGE and analyzed by immunoblotting for HA-tagged, FopA and FipB proteins.
FopA interaction with DipA was also confirmed by reverse co-immunoprecipitation using antibodies against FopA (gift from Jason Huntley) coupled to Dynabeads® Protein G (Life Technologies). To couple antibody to beads, 2% anti-FopA antibodies (v/v) in PBS were incubated with Dynabeads® Protein G for 1 h at room temperature with gentle agitation, washed three times with PBS, and resuspended in 1 mL of PBS. Approximately 1 OD600 of SchuS4ΔdipA(pFNLTP6dipAHA) lysate, prepared for co-immunoprecipitation as described above, was incubated with anti-FopA antibody-coupled protein G beads for 3 h at room temperature. The bead complexes were washed five times in PBS using Dynal® MPCTM magnet (Life Technologies). Co-immunoprecipitated proteins were separated from the beads by boiling for 10 min in SDS-PAGE loading buffer and centrifugation at 16,000 x g for 5 min. The supernatants containing co-immunoprecipitated proteins were resolved by SDS-PAGE and analyzed by immunoblotting for FopA and HA-tagged DipA. Light chain specific HRP conjugated secondary antibodies (Jackson ImmunoResearch) were used for protein detection to minimize interference from the precipitated primary antibody.
To [3H] palmityolate and immunoprecipitate FlpA, a fresh overnight culture of SchuS4 was subcultured into TSB-C broth and incubated until 0.3 OD600 at which point either 200 µCi/mL of [3H] palmitic acid or an equivalent volume of ethanol was added and incubated overnight. Approximately 1 OD600 of radiolabelled or unlabeled SchuS4 culture was harvested, washed once with PBS, and lysed with 0.5 mL of RIPA buffer (Sigma). The clarified lysates were agitated overnight at 4 °C with rat anti-FlpA coupled Dynabeads® Protein G (Life Technologies). Antiserum coupled-beads were prepared as described above. The bead complexes were washed five times in PBS using Dynal® MPCTM magnet (Life Technologies). Co-immunoprecipitated proteins were separated from the beads by boiling for 5 min in SDS-PAGE loading buffer and centrifugation at 16,1000 x g for 5 min. Supernatants containing co-immunoprecipitated proteins were resolved by SDS-PAGE, transferred onto Immobilon™-P PVDF membrane (Sigma-Aldrich), and analyzed either by autoradiography or by immunoblot analysis with anti-FlpA antibodies.
Animal infections
Groups of ten 6-8 week old BALB/cJ mice were intranasally infected with the indicated wild-type or mutant SchuS4 strains for survival studies as previously described [10]. Briefly, mice were anesthetized intraperitoneally with 12.5 mg/mL ketamine and 3.8 mg/mL xylazine immediately prior to infection. Bacteria were diluted in PBS and inoculated into the nares of each mouse in 25 µL total volume. Actual doses were determined by plating onto MMH agar plates. All infections were performed in an Animal Biosafety Level 3 (ABSL3) facility according to protocols reviewed and approved by the Rocky Mountain Laboratories Institutional Biosafety Committee and the RML Institutional Animal Care and Use Committee (IACUC), in compliance with the CDC Division of Select Agents and Toxins regulations.
Supporting Information
(A) Immnoblot analysis of C-terminally tagged HA-fusion proteins. Sample loading was normalized to 1x106 CFU/lane from lysates of SchuS4 expressing IglA-HA or IglI-HA, SchuS4ΔdipA expressing DipA-HA, DipAΔSel1ab-HA, DipAΔSel1cd-HA, DipAΔCC-HA, DipA-CC(AIL 3D)-HA, or DipA-CC(LAL 3D)-HA, SchuS4ΔflpA expressing FlpA-HA, and SchuS4ΔfopA expressing FopA-HA. (B) Immnoblot analysis of C-terminally tagged TEM1-fusion proteins. Sample loading was normalized to 5x106 CFU/lane from lysates of SchuS4 and SchuS4 expressing DipA-TEM1, DipAΔSel1ab-TEM1, DipAΔSel1cd-TEM1, DipA-CC(AIL 3D)-TEM1, DipA-CC(LAL 3D)-TEM1, DipAΔCC-TEM1, IglA-TEM1 or IglI-TEM1. Samples were resolved by SDS-PAGE and analyzed by immunoblot analysis with anti-HA (A) or anti-TEM1 β-lactamase (B) antibodies.
(TIF)
(A) Subcellular localization of FlpA from GFP-expressing SchuS4. Soluble (Sol), inner membrane (IM), and outer membrane (OM) enriched fractions were separated based on Sarkosyl solubility and subjected to immunoblot analysis with antibodies against FlpA. Each fraction was concentrated to the same volume and equal volumes were loaded. (B) Autoradiograph of [3H] palmitate radiolabeled SchuS4 lysate (lane 1), and unlabeled (lane 2) or [3H] palmitate radiolabeled (lane 3) lysates that were subjected to immunoprecipitation with anti-FlpA antibodies. Samples were separated by SDS-PAGE, and analyzed by autoradiography. (C) Immunoblot analysis of the same samples as (B) probed with anti-FlpA antibodies. FlpA, IgG heavy (IgG Hc) and light (IgG Lc) chain bands are indicated by arrows.
(TIF)
(DOCX)
(DOCX)
Acknowledgments
We are grateful to Ross Tomaino at the Taplin Biological Mass Spectrometry Facility for his assistance with the identification of DipA-interacting proteins, to Elizabeth Case for critical reading of the manuscript and helpful suggestions.
Funding Statement
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases, project number AI000953-01. The funders have no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Pechous RD, McCarthy TR, Zahrt TC (2009) Working toward the future: insights into Francisella tularensis pathogenesis and vaccine development. Microbiol Mol Biol Rev 73: 684-711. doi:10.1128/MMBR.00028-09. PubMed: 19946137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oyston PC, Sjostedt A, Titball RW (2004) Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol 2: 967-978. doi:10.1038/nrmicro1045. PubMed: 15550942. [DOI] [PubMed] [Google Scholar]
- 3. Keim P, Johansson A, Wagner DM (2007) Molecular epidemiology, evolution, and ecology of Francisella. Ann N Y Acad Sci 1105: 30-66. doi:10.1196/annals.1409.011. PubMed: 17435120. [DOI] [PubMed] [Google Scholar]
- 4. Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S et al. (2008) Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun 76: 5843-5852. doi:10.1128/IAI.01176-08. PubMed: 18852251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anthony LD, Burke RD, Nano FE (1991) Growth of Francisella spp. in rodent macrophages. Infect Immun 59: 3291-3296. PubMed: 1879943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Forestal CA, Benach JL, Carbonara C, Italo JK, Lisinski TJ et al. (2003) Francisella tularensis selectively induces proinflammatory changes in endothelial cells. J Immunol 171: 2563-2570. PubMed: 12928407. [DOI] [PubMed] [Google Scholar]
- 7. Balagopal A, MacFarlane AS, Mohapatra N, Soni S, Gunn JS et al. (2006) Characterization of the receptor-ligand pathways important for entry and survival of Francisella tularensis in human macrophages. Infect Immun 74: 5114-5125. doi:10.1128/IAI.00795-06. PubMed: 16926403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fortier AH, Green SJ, Polsinelli T, Jones TR, Crawford RM et al. (1994) Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol Ser 60: 349-361. PubMed: 8251580. [PubMed] [Google Scholar]
- 9. Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR et al. (2008) The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun 76: 5488-5499. doi:10.1128/IAI.00682-08. PubMed: 18852245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wehrly TD, Chong A, Virtaneva K, Sturdevant DE, Child R et al. (2009) Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell Microbiol 11: 1128-1150. doi:10.1111/j.1462-5822.2009.01316.x. PubMed: 19388904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J (2006) Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A 103: 14578-14583. doi:10.1073/pnas.0601838103. PubMed: 16983090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clemens DL, Lee BY, Horwitz MA (2009) Francisella tularensis phagosomal escape does not require acidification of the phagosome. Infect Immun 77: 1757-1773. doi:10.1128/IAI.01485-08. PubMed: 19237528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clemens DL, Lee BY, Horwitz MA (2004) Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun 72: 3204-3217. doi:10.1128/IAI.72.6.3204-3217.2004. PubMed: 15155622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santic M, Molmeret M, Abu Kwaik Y (2005) Modulation of biogenesis of the Francisella tularensis subsp. novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN-gamma. Cell Microbiol 7: 957-967. doi:10.1111/j.1462-5822.2005.00529.x. PubMed: 15953028. [DOI] [PubMed] [Google Scholar]
- 15. Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjöstedt A (2003) An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun 71: 5940-5950. doi:10.1128/IAI.71.10.5940-5950.2003. PubMed: 14500514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lai XH, Golovliov I, Sjöstedt A (2001) Francisella tularensis induces cytopathogenicity and apoptosis in murine macrophages via a mechanism that requires intracellular bacterial multiplication. Infect Immun 69: 4691-4694. doi:10.1128/IAI.69.7.4691-4694.2001. PubMed: 11402018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lai XH, Sjöstedt A (2003) Delineation of the molecular mechanisms of Francisella tularensis-induced apoptosis in murine macrophages. Infect Immun 71: 4642-4646. doi:10.1128/IAI.71.8.4642-4646.2003. PubMed: 12874344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Santic M, Pavokovic G, Jones S, Asare R, Kwaik YA (2010) Regulation of apoptosis and anti-apoptosis signalling by Francisella tularensis. Microbes Infect 12: 126-134. doi:10.1016/j.micinf.2009.11.003. PubMed: 19925880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mariathasan S, Weiss DS, Dixit VM, Monack DM (2005) Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med 202: 1043-1049. doi:10.1084/jem.20050977. PubMed: 16230474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chong A, Celli J (2010) The Francisella intracellular life cycle: toward molecular mechanisms of intracellular survival and proliferation. Front Microbiol 1: 138 PubMed: 21687806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meibom KL, Charbit A (2010) The unraveling panoply of Francisella tularensis virulence attributes. Curr Opin Microbiol 13: 11-17. doi:10.1016/j.mib.2009.11.007. PubMed: 20034843. [DOI] [PubMed] [Google Scholar]
- 22. Nano FE, Zhang N, Cowley SC, Klose KE, Cheung KK et al. (2004) A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol 186: 6430-6436. doi:10.1128/JB.186.19.6430-6436.2004. PubMed: 15375123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gray CG, Cowley SC, Cheung KK, Nano FE (2002) The identification of five genetic loci of Francisella novicida associated with intracellular growth. FEMS Microbiol Lett 215: 53-56. doi:10.1111/j.1574-6968.2002.tb11369.x. PubMed: 12393200. [DOI] [PubMed] [Google Scholar]
- 24. Brotcke A, Weiss DS, Kim CC, Chain P, Malfatti S et al. (2006) Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect Immun 74: 6642-6655. doi:10.1128/IAI.01250-06. PubMed: 17000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K et al. (2007) In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A 104: 6037-6042. doi:10.1073/pnas.0609675104. PubMed: 17389372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindgren H, Golovliov I, Baranov V, Ernst RK, Telepnev M et al. (2004) Factors affecting the escape of Francisella tularensis from the phagolysosome. J Med Microbiol 53: 953-958. doi:10.1099/jmm.0.45685-0. PubMed: 15358816. [DOI] [PubMed] [Google Scholar]
- 27. Twine S, Byström M, Chen W, Forsman M, Golovliov I et al. (2005) A mutant of Francisella tularensis strain SCHU S4 lacking the ability to express a 58-kilodalton protein is attenuated for virulence and is an effective live vaccine. Infect Immun 73: 8345-8352. doi:10.1128/IAI.73.12.8345-8352.2005. PubMed: 16299332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fuller JR, Craven RR, Hall JD, Kijek TM, Taft-Benz S et al. (2008) RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect Immun 76: 4934-4943. doi:10.1128/IAI.00475-08. PubMed: 18765722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qin A, Mann BJ (2006) Identification of transposon insertion mutants of Francisella tularensis tularensis strain Schu S4 deficient in intracellular replication in the hepatic cell line HepG2. BMC Microbiol 6: 69. doi:10.1186/1471-2180-6-69. PubMed: 16879747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qin A, Scott DW, Thompson JA, Mann BJ (2009) Identification of an essential Francisella tularensis subsp. tularensis virulence factor. Infect Immun 77: 152-161. doi:10.1128/IAI.01113-08. PubMed: 18981253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qin A, Scott DW, Rabideau MM, Moore EA, Mann BJ (2011) Requirement of the CXXC motif of novel Francisella infectivity potentiator protein B FipB, and FipA in virulence of F. tularensis subsp. tularensis. PLOS ONE 6: e24611. doi:10.1371/journal.pone.0024611. PubMed: 21931773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nano FE (1988) Identification of a heat-modifiable protein of Francisella tularensis and molecular cloning of the encoding gene. Microb Pathog 5: 109-119. doi:10.1016/0882-4010(88)90013-7. PubMed: 3237052. [DOI] [PubMed] [Google Scholar]
- 33. Huntley JF, Conley PG, Hagman KE, Norgard MV (2007) Characterization of Francisella tularensis outer membrane proteins. J Bacteriol 189: 561-574. doi:10.1128/JB.01505-06. PubMed: 17114266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schmerk CL, Duplantis BN, Wang D, Burke RD, Chou AY et al. (2009) Characterization of the pathogenicity island protein PdpA and its role in the virulence of Francisella novicida. Microbiology 155: 1489-1497. doi:10.1099/mic.0.025379-0. PubMed: 19372153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bröms JE, Lavander M, Meyer L, Sjöstedt A (2011) IglG and IglI of the Francisella pathogenicity island are important virulence determinants of Francisella tularensis LVS. Infect Immun 79: 3683-3696. doi:10.1128/IAI.01344-10. PubMed: 21690239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Bruin OM, Duplantis BN, Ludu JS, Hare RF, Nix EB et al. (2011) The biochemical properties of the Francisella pathogenicity island (FPI)-encoded proteins IglA, IglB, IglC, PdpB and DotU suggest roles in type VI secretion. Microbiology 157: 3483-3491. doi:10.1099/mic.0.052308-0. PubMed: 21980115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Bruin OM, Ludu JS, Nano FE (2007) The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol 7: 1. doi:10.1186/1471-2180-7-1. PubMed: 17233889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ludu JS, de Bruin OM, Duplantis BN, Schmerk CL, Chou AY et al. (2008) The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J Bacteriol 190: 4584-4595. doi:10.1128/JB.00198-08. PubMed: 18469101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Melillo A, Sledjeski DD, Lipski S, Wooten RM, Basrur V et al. (2006) Identification of a Francisella tularensis LVS outer membrane protein that confers adherence to A549 human lung cells. FEMS Microbiol Lett 263: 102-108. doi:10.1111/j.1574-6968.2006.00413.x. PubMed: 16958857. [DOI] [PubMed] [Google Scholar]
- 40. Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA et al. (2009) The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol 74: 1459-1470. doi:10.1111/j.1365-2958.2009.06947.x. PubMed: 20054881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Larsson P, Oyston PC, Chain P, Chu MC, Duffield M et al. (2005) The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat Genet 37: 153-159. doi:10.1038/ng1499. PubMed: 15640799. [DOI] [PubMed] [Google Scholar]
- 42. Bröms JE, Meyer L, Sun K, Lavander M, Sjöstedt A (2012) Unique substrates secreted by the type VI secretion system of Francisella tularensis during intramacrophage infection. PLOS ONE 7: e50473. doi:10.1371/journal.pone.0050473. PubMed: 23185631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mittl PR, Schneider-Brachert W (2007) Sel1-like repeat proteins in signal transduction. Cell Signal 19: 20-31. doi:10.1016/j.cellsig.2006.05.034. PubMed: 16870393. [DOI] [PubMed] [Google Scholar]
- 44. Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5: 725-738. doi:10.1038/nprot.2010.5. PubMed: 20360767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lupas AN, Gruber M (2005) The structure of alpha-helical coiled coils. Adv Protein Chem 70: 37-78. doi:10.1016/S0065-3233(05)70003-6. PubMed: 15837513. [DOI] [PubMed] [Google Scholar]
- 46. Fulop M, Manchee R, Titball R (1996) Role of two outer membrane antigens in the induction of protective immunity against Francisella tularensis strains of different virulence. FEMS Immunol Med Microbiol 13: 245-247. doi:10.1111/j.1574-695X.1996.tb00245.x. PubMed: 8861037. [DOI] [PubMed] [Google Scholar]
- 47. Blau K, Portnoi M, Shagan M, Kaganovich A, Rom S et al. (2007) Flamingo cadherin: a putative host receptor for Streptococcus pneumoniae. J Infect Dis 195: 1828-1837. doi:10.1086/518038. PubMed: 17492599. [DOI] [PubMed] [Google Scholar]
- 48. Tunio SA, Oldfield NJ, Berry A, Ala’Aldeen DA, Wooldridge KG et al. (2010) The moonlighting protein fructose-1, 6-bisphosphate aldolase of Neisseria meningitidis: surface localization and role in host cell adhesion. Mol Microbiol 76: 605-615. doi:10.1111/j.1365-2958.2010.07098.x. PubMed: 20199602. [DOI] [PubMed] [Google Scholar]
- 49. de la Paz Santangelo M, Gest PM, Guerin ME, Coinçon M, Pham H et al. (2011) Glycolytic and non-glycolytic functions of Mycobacterium tuberculosis fructose-1,6-bisphosphate aldolase, an essential enzyme produced by replicating and non-replicating bacilli. J Biol Chem 286: 40219-40231. doi:10.1074/jbc.M111.259440. PubMed: 21949126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ling E, Feldman G, Portnoi M, Dagan R, Overweg K et al. (2004) Glycolytic enzymes associated with the cell surface of Streptococcus pneumoniae are antigenic in humans and elicit protective immune responses in the mouse. Clin Exp Immunol 138: 290-298. doi:10.1111/j.1365-2249.2004.02628.x. PubMed: 15498039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Agarwal S, Kulshreshtha P, Bambah Mukku D, Bhatnagar R (2008) alpha-Enolase binds to human plasminogen on the surface of Bacillus anthracis. Biochim Biophys Acta 1784: 986-994. doi:10.1016/j.bbapap.2008.03.017. PubMed: 18456007. [DOI] [PubMed] [Google Scholar]
- 52. Gallagher LA, Ramage E, Jacobs MA, Kaul R, Brittnacher M et al. (2007) A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc Natl Acad Sci U S A 104: 1009-1014. doi:10.1073/pnas.0606713104. PubMed: 17215359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chong A, Wehrly TD, Child R, Hansen B, Hwang S et al. (2012) Cytosolic clearance of replication-deficient mutants reveals Francisella tularensis interactions with the autophagic pathway. Autophagy 8: 0--1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bönquist L, Lindgren H, Golovliov I, Guina T, Sjöstedt A (2008) MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect Immun 76: 3502-3510. doi:10.1128/IAI.00226-08. PubMed: 18474647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Santic M, Molmeret M, Klose KE, Jones S, Kwaik YA (2005) The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol 7: 969-979. doi:10.1111/j.1462-5822.2005.00526.x. PubMed: 15953029. [DOI] [PubMed] [Google Scholar]
- 56. Schmerk CL, Duplantis BN, Howard PL, Nano FE (2009) A Francisella novicida pdpA mutant exhibits limited intracellular replication and remains associated with the lysosomal marker LAMP-1. Microbiology 155: 1498-1504. doi:10.1099/mic.0.025445-0. PubMed: 19372155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rockx-Brouwer D, Chong A, Wehrly TD, Child R, Crane DD et al. (2012) Low dose vaccination with attenuated Francisella tularensis strain SchuS4 mutants protects against tularemia independent of the route of vaccination. PLOS ONE 7: e37752. doi:10.1371/journal.pone.0037752. PubMed: 22662210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS et al. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101: 249-258. doi:10.1016/S0092-8674(00)80835-1. PubMed: 10847680. [DOI] [PubMed] [Google Scholar]
- 59. Okabe M, Yakushi T, Homma M (2005) Interactions of MotX with MotY and with the PomA/PomB sodium ion channel complex of the Vibrio alginolyticus polar flagellum. J Biol Chem 280: 25659-25664. doi:10.1074/jbc.M500263200. PubMed: 15866878. [DOI] [PubMed] [Google Scholar]
- 60. McCarter LL (1994) MotX, the channel component of the sodium-type flagellar motor. J Bacteriol 176: 5988-5998. PubMed: 7928960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Conover GM, Derré I, Vogel JP, Isberg RR (2003) The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol Microbiol 48: 305-321. doi:10.1046/j.1365-2958.2003.03400.x. PubMed: 12675793. [DOI] [PubMed] [Google Scholar]
- 62. Cirillo SL, Lum J, Cirillo JD (2000) Identification of novel loci involved in entry by Legionella pneumophila. Microbiology 146(6): 1345-1359. PubMed: 10846213. [DOI] [PubMed] [Google Scholar]
- 63. Newton HJ, Sansom FM, Dao J, McAlister AD, Sloan J et al. (2007) Sel1 repeat protein LpnE is a Legionella pneumophila virulence determinant that influences vacuolar trafficking. Infect Immun 75: 5575-5585. doi:10.1128/IAI.00443-07. PubMed: 17893138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Engel J, Kammerer RA (2000) What are oligomerization domains good for? Matrix Biol 19: 283-288. doi:10.1016/S0945-053X(00)00075-5. PubMed: 10963988. [DOI] [PubMed] [Google Scholar]
- 65. Uversky VN, Gillespie JR, Fink AL (2000) Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 41: 415-427. doi:10.1002/1097-0134(20001115)41:3. PubMed: 11025552. [DOI] [PubMed] [Google Scholar]
- 66. Newman JR, Wolf E, Kim PS (2000) A computationally directed screen identifying interacting coiled coils from Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 97: 13203-13208. doi:10.1073/pnas.97.24.13203. PubMed: 11087867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gazi AD, Charova SN, Panopoulos NJ, Kokkinidis M (2009) Coiled-coils in type III secretion systems: structural flexibility, disorder and biological implications. Cell Microbiol 11: 719-729. doi:10.1111/j.1462-5822.2009.01297.x. PubMed: 19215225. [DOI] [PubMed] [Google Scholar]
- 68. Delahay RM, Knutton S, Shaw RK, Hartland EL, Pallen MJ et al. (1999) The coiled-coil domain of EspA is essential for the assembly of the type III secretion translocon on the surface of enteropathogenic Escherichia coli. J Biol Chem 274: 35969-35974. doi:10.1074/jbc.274.50.35969. PubMed: 10585486. [DOI] [PubMed] [Google Scholar]
- 69. Knodler LA, Ibarra JA, Pérez-Rueda E, Yip CK, Steele-Mortimer O (2011) Coiled-coil domains enhance the membrane association of Salmonella type III effectors. Cell Microbiol 13: 1497-1517. doi:10.1111/j.1462-5822.2011.01635.x. PubMed: 21679290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sukumaran SK, Shimada H, Prasadarao NV (2003) Entry and intracellular replication of Escherichia coli K1 in macrophages require expression of outer membrane protein A. Infect Immun 71: 5951-5961. doi:10.1128/IAI.71.10.5951-5961.2003. PubMed: 14500515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dabo SM, Confer A, Montelongo M, York P, Wyckoff JH 3rd (2008) Vaccination with Pasteurella multocida recombinant OmpA induces strong but non-protective and deleterious Th2-type immune response in mice. Vaccine 26: 4345-4351. doi:10.1016/j.vaccine.2008.06.029. PubMed: 18598730. [DOI] [PubMed] [Google Scholar]
- 72. Kim SW, Choi CH, Moon DC, Jin JS, Lee JH et al. (2009) Serum resistance of Acinetobacter baumannii through the binding of factor H to outer membrane proteins. FEMS Microbiol Lett 301: 224-231. doi:10.1111/j.1574-6968.2009.01820.x. PubMed: 19878322. [DOI] [PubMed] [Google Scholar]
- 73. Torres AG, Kaper JB (2003) Multiple elements controlling adherence of enterohemorrhagic Escherichia coli O157:H7 to HeLa cells. Infect Immun 71: 4985-4995. doi:10.1128/IAI.71.9.4985-4995.2003. PubMed: 12933841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fito-Boncompte L, Chapalain A, Bouffartigues E, Chaker H, Lesouhaitier O et al. (2011) Full virulence of Pseudomonas aeruginosa requires OprF. Infect Immun 79: 1176-1186. doi:10.1128/IAI.00850-10. PubMed: 21189321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nicholson TF, Watts KM, Hunstad DA (2009) OmpA of uropathogenic Escherichia coli promotes postinvasion pathogenesis of cystitis. Infect Immun 77: 5245-5251. doi:10.1128/IAI.00670-09. PubMed: 19797074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Serino L, Nesta B, Leuzzi R, Fontana MR, Monaci E et al. (2007) Identification of a new OmpA-like protein in Neisseria gonorrhoeae involved in the binding to human epithelial cells and in vivo colonization. Mol Microbiol 64: 1391-1403. doi:10.1111/j.1365-2958.2007.05745.x. PubMed: 17542928. [DOI] [PubMed] [Google Scholar]
- 77. Bartra SS, Gong X, Lorica CD, Jain C, Nair MK et al. (2012) The outer membrane protein A (OmpA) of Yersinia pestis promotes intracellular survival and virulence in mice. Microb Pathog 52: 41-46. doi:10.1016/j.micpath.2011.09.009. PubMed: 22023991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang Y (2002) The function of OmpA in Escherichia coli. Biochem Biophys Res Commun 292: 396-401. doi:10.1006/bbrc.2002.6657. PubMed: 11906175. [DOI] [PubMed] [Google Scholar]
- 79. Sonntag I, Schwarz H, Hirota Y, Henning U (1978) Cell envelope and shape of Escherichia coli: multiple mutants missing the outer membrane lipoprotein and other major outer membrane proteins. J Bacteriol 136: 280-285. PubMed: 361695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sugawara E, Nikaido H (2012) OmpA is the principal nonspecific slow porin of Acinetobacter baumannii. J Bacteriol 194: 4089-4096. doi:10.1128/JB.00435-12. PubMed: 22636785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nestorovich EM, Sugawara E, Nikaido H, Bezrukov SM (2006) Pseudomonas aeruginosa porin OprF: properties of the channel. J Biol Chem 281: 16230-16237. doi:10.1074/jbc.M600650200. PubMed: 16617058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lupas A (1996) Prediction and analysis of coiled-coil structures. Methods Enzymol 266: 513-525. doi:10.1016/S0076-6879(96)66032-7. PubMed: 8743703. [DOI] [PubMed] [Google Scholar]
- 83. Delorenzi M, Speed T (2002) An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18: 617-625. doi:10.1093/bioinformatics/18.4.617. PubMed: 12016059. [DOI] [PubMed] [Google Scholar]
- 84. Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9: 40. doi:10.1186/1471-2105-9-40. PubMed: 18215316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM et al. (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25: 1605-1612. doi:10.1002/jcc.20084. PubMed: 15264254. [DOI] [PubMed] [Google Scholar]
- 86. Maier TM, Havig A, Casey M, Nano FE, Frank DW et al. (2004) Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl Environ Microbiol 70: 7511-7519. doi:10.1128/AEM.70.12.7511-7519.2004. PubMed: 15574954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Charpentier X, Oswald E (2004) Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 beta-lactamase as a new fluorescence-based reporter. J Bacteriol 186: 5486-5495. doi:10.1128/JB.186.16.5486-5495.2004. PubMed: 15292151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Immnoblot analysis of C-terminally tagged HA-fusion proteins. Sample loading was normalized to 1x106 CFU/lane from lysates of SchuS4 expressing IglA-HA or IglI-HA, SchuS4ΔdipA expressing DipA-HA, DipAΔSel1ab-HA, DipAΔSel1cd-HA, DipAΔCC-HA, DipA-CC(AIL 3D)-HA, or DipA-CC(LAL 3D)-HA, SchuS4ΔflpA expressing FlpA-HA, and SchuS4ΔfopA expressing FopA-HA. (B) Immnoblot analysis of C-terminally tagged TEM1-fusion proteins. Sample loading was normalized to 5x106 CFU/lane from lysates of SchuS4 and SchuS4 expressing DipA-TEM1, DipAΔSel1ab-TEM1, DipAΔSel1cd-TEM1, DipA-CC(AIL 3D)-TEM1, DipA-CC(LAL 3D)-TEM1, DipAΔCC-TEM1, IglA-TEM1 or IglI-TEM1. Samples were resolved by SDS-PAGE and analyzed by immunoblot analysis with anti-HA (A) or anti-TEM1 β-lactamase (B) antibodies.
(TIF)
(A) Subcellular localization of FlpA from GFP-expressing SchuS4. Soluble (Sol), inner membrane (IM), and outer membrane (OM) enriched fractions were separated based on Sarkosyl solubility and subjected to immunoblot analysis with antibodies against FlpA. Each fraction was concentrated to the same volume and equal volumes were loaded. (B) Autoradiograph of [3H] palmitate radiolabeled SchuS4 lysate (lane 1), and unlabeled (lane 2) or [3H] palmitate radiolabeled (lane 3) lysates that were subjected to immunoprecipitation with anti-FlpA antibodies. Samples were separated by SDS-PAGE, and analyzed by autoradiography. (C) Immunoblot analysis of the same samples as (B) probed with anti-FlpA antibodies. FlpA, IgG heavy (IgG Hc) and light (IgG Lc) chain bands are indicated by arrows.
(TIF)
(DOCX)
(DOCX)