

Abstract
Motivation: High-spatial resolution imaging datasets of mammalian brains have recently become available in unprecedented amounts. Images now reveal highly complex patterns of gene expression varying on multiple scales. The challenge in analyzing these images is both in extracting the patterns that are most relevant functionally and in providing a meaningful representation that allows neuroscientists to interpret the extracted patterns.
Results: Here, we present FuncISH—a method to learn functional representations of neural in situ hybridization (ISH) images. We represent images using a histogram of local descriptors in several scales, and we use this representation to learn detectors of functional (GO) categories for every image. As a result, each image is represented as a point in a low-dimensional space whose axes correspond to meaningful functional annotations. The resulting representations define similarities between ISH images that can be easily explained by functional categories. We applied our method to the genomic set of mouse neural ISH images available at the Allen Brain Atlas, finding that most neural biological processes can be inferred from spatial expression patterns with high accuracy. Using functional representations, we predict several gene interaction properties, such as protein–protein interactions and cell-type specificity, more accurately than competing methods based on global correlations. We used FuncISH to identify similar expression patterns of GABAergic neuronal markers that were not previously identified and to infer new gene function based on image–image similarities.
Contact: noalis@gmail.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
In recent years, high-resolution expression data measured in mammalian brains became available in quantities and qualities never witnessed before (Henry and Hohmann, 2012; Lein et al., 2007; Ng et al., 2009), calling for new ways to analyze neural gene expression images. Most existing methods for bio-imaging analysis were developed to handle data with different characteristics, like Drosophila embryos (Frise et al., 2010; Peng et al., 2007; Pruteanu-Malinici et al., 2011) or cellular imagery (Coelho et al., 2010; Peng et al., 2010). The mammalian brain, composed of billions of neurons and glia, is organized in highly complex anatomical structures and poses new challenges for analysis. Current approaches for analyzing brain images are based on smooth non-linear transformations to a reference atlas (Davis and Eddy, 2009; Hawrylycz et al., 2011) and may be insensitive to fine local patterns like those emerging from the layered structure of the cerebellum or the spatial distribution of cortical interneurons.
Another challenge for automatic analysis of biological images lies in providing human interpretable analysis. Most machine-vision approaches are developed for tasks in analysis of natural images, like object recognition. In such tasks, humans can understand the scene effortlessly and infer complex relations between objects easily. In bio-imaging, however, the goal of image analysis is often to reveal features and structures that are hardly seen even by experts. It is, therefore, important that an image analysis approach provides meaningful interpretation to any patterns or structures that it detects.
Here, we develop a method to learn functional representations of expression images by using predefined functional ontologies. This approach has two main advantages, accuracy and interpretability, and it builds on a growing body of work in object recognition in natural images, showing how images can be represented using the activations of a large set of detectors (Deng et al., 2011; Li et al., 2010a, b; Malisiewicz, 2012; Malisiewicz et al., 2011; Torresani et al., 2010). For object recognition, the detectors may include common objects, like a detector for the presence of a chair, a mug or a door. Here, we show how to adapt this idea to represent gene expression images, by training a large set of detectors, each corresponding to a known functional category, like axon guidance or glutamatergic receptors. Once this representation is trained, every gene is represented as a point in a low-dimensional space whose axes correspond to functional meaningful categories.
We describe in Section 2.2 how to learn functional representations in a discriminative way and demonstrate the effectiveness of the approach on in situ hybridization (ISH) gene expression images of the adult mouse brain collected by the Allen Institute for Brain Science (Lein et al., 2007). ISH image analysis has been used in the past to infer gene biological functions from spatial co-expression in non-neural tissues (Frise et al., 2010). However, inferring functions based on gene expression patterns in the brain is believed to be hard, as several studies found very low variability between transcriptomic patterns of different brain regions, sometimes even lower than between-subject variability for the same area (Khaitovich et al., 2004, 2005). Neural expression patterns are usually studied using methods that average expression values over a brain region, and this averaging removes fine-resolution spatial information that may differentiate between brain regions. Here, we analyze high-resolution ISH images at several scales, taking into account subtle, even cellular resolution, information for functional inference.
We find that gene function can indeed be inferred from neural ISH images, particularly in biological processes that are related to neural activities. Our approach detects related genes with better accuracy based on the similarity of their functional representations. Furthermore, these similarities can be explained and interpreted using semantic terms.
2 METHODS
2.1 The data
We used whole-brain, expression-masked images of gene expression measured using ISH, publicly available at the Allen Brain Atlas (www.brain-map.org, also see Supplementary Material). Expression was measured for the entire mouse genome. For each gene, a different adult mouse brain was sliced into 100 -µm thick slices, mRNA abundance was measured and the slice was imaged. The database holds image series for >20 K transcripts. Most genes have one corresponding image series, containing ∼25 imaged brain slices. Some genes were imaged more than once and have several associated image series. In our analysis, we used the most medial slice for each image series, yielding a typical image size of 8 K × 16 K pixels. In all, 4823 of the available 21 174 images showed no expression in the brain and were ignored in subsequent analysis, leaving 16 351 images representing 15 612 genes. We also tested our approach on a larger image set constructed by taking three images for each gene: the medial slice, and lateral slices at 30% and 50% of brain size (from one hemisphere). The results with this three-image set were mixed, and all results reported later in the text are for the one-slice dataset (Supplementary Material). Figure 1 shows examples of images, demonstrating the complexity of neural expression patterns across brain regions and multiple scales. The images analyzed in our study were in gray scale but are shown here as color-coded by expression intensity for better visualization.
Fig. 1.

The raw data. ISH image for the gene Tuba1 shown (A) at different scales and (B) in three different regions
2.2 A functional representation of images
We present a method to identify similarities between neural ISH images and to explain these similarities in functional terms.
Our method consists of a visual phase, where we transform the raw pixel images into a robust visual representation, and a semantic phase, where we transform that visual representation using a set of 2081 gene-function detectors. The output of these detectors comprises a higher-order semantic representation of the images in a gene-functional space (Fig. 2). Similar two-phase systems have recently been proposed and applied successfully for tasks, such as cross-domain image similarity and object detection in natural images (Deng et al., 2011; Li et al., 2010a, b; Malisiewicz, 2012; Malisiewicz et al., 2011; Torresani et al., 2010).
Fig. 2.

Illustration of the image processing pipeline. (A) Original image in pixel grayscale indicating level of gene expression. (B) Local SIFT descriptors are extracted from image at 4 resolutions. (C) Descriptors from all 16351 images are clustered into 500 representative ‘visual words' for each resolution level using k-Means. (D) Each image is represented as a histogram counting the occurrences of visual words. (E) L2-regularized logistic regression classifiers are applied for 2081 GO categories. (F) The final 2081 dimensional image representation
For the first, visual, phase, we first represent each image as a collection of local descriptors using SIFT features (Lowe, 2004). This step aims to address the problem that ISH brain images of the same gene vary significantly in shape and size when measured in different brains (Kirsch et al., 2012). SIFT features are histograms of oriented gradients on a small grid. The resulting image-patch SIFT descriptor is invariant to small rotation and illumination (but not to scale), making imaged-slices from different brains more comparable. We computed SIFT descriptors of dimension 128 extracted on a dense grid spanning the full image (Bosch et al., 2006, 2007; Csurka and Dance, 2004), at four spatial resolutions. In ISH images, different information lies in different descriptor sizes, and we wish that the representation captures spatial patterns both at the level of single cells, micro-circuitry and at the coarser level of distribution of expression across brain layers. To capture information at multiple scales, we used the VLFeat implementation of SIFT (Vedaldi and Fulkerson, 2010), where scale-invariance is not incorporated automatically. Specifically, each image is represented as a collection of ∼1 M SIFT descriptors, computed by down sampling each image at a factor of 1, 2, 4 and 8. As the descriptors were extracted from high-resolution images, which are mostly dark, many descriptors were completely dark and were discarded.
Next, to achieve a compact non-linear representation of each image, we aggregate the descriptors from all images for a given resolution level and cluster them to form a dictionary of distinct ‘visual words’ per each resolution level. We used the original Lloyd optimization for k-Means with L2 distance, initializing the centroids by randomly sampling data points. The clustering procedure was repeated multiple times (n = 3), and the solution with the lowest energy was used. We tested four different dictionary sizes (k = 100, 200, 500 and 1000), all yielding similar results (Supplementary Material), and we report later in the text results for k = 500, which obtained slightly higher accuracies. Next, we construct a standard ‘bag-of-words’20,21 description of each image. As a result of this process, each image is described by four concatenated 500-dimensional vectors counting how many times each ‘visual word’ appeared in it at a given resolution level. We also added a count of the number of zero descriptors per resolution level, ending up with a 2004-dimensional vector describing each image. Using this approach, similar spatial information from different brain regions is preserved, as opposed to using global correlation-based approaches.
We then turn to the second, ‘semantic’, phase, and represent each image by a set of functional descriptors. Given a set of predefined Gene Ontology (GO) annotations of each gene, we train one separate classifier for each known biological annotation category, using the SIFT bag-of-words representation as an input vector. Specifically, here, we trained a set of 2081 L2-regularized logistic regression classifiers [using LIBLINEAR (Fan et al., 2008)] corresponding to biological-processes GO classes that have 15–500 annotated genes (Supplementary Material). We trained the classifiers using two layers of 5-fold cross-validation, performed as follows: the full set of 16 351 gene images was split into five non-overlapping equal sets (without controlling for the number of positives in each split), training the classifiers on four of them and testing performance on the fifth unseen test set of images. This procedure was repeated five times, each time with a different set acting as the test set. All accuracy and other results later in the text are reported for a held-out test set that was not used during training.
To tune the logistic regression regularization hyperparameter, we used a second layer of cross-validation. We repeated the splitting procedure within each of the five training sets, splitting each of them again into five subsets of images, using four for training and the fifth as a validation set. The regularization hyperparameter was selected from the values (0.001, 0.01, 0.1, 1, 10 and 100). At the end of this process, each gene is then represented as a vector of ‘activations’, corresponding to the likelihood that the gene belongs to one functional category, such as ‘forebrain development’ or ‘regulation of fatty acid transport’.
The representation described earlier in the text removes important information about global location in the brain. We, therefore, also tested an approach using spatial pyramids (Lazebnik et al., 2006), where descriptor histograms are computed separately for different parts of the image. Unfortunately, this approach results in feature vectors whose dimensionality was too high for the current dataset and yielded poor classification results (Supplementary Material).
2.3 Similarity between functional profiles
We use two gene–gene similarity measures in this work, taking each gene as a vector of functional category activations. The first, flat-sim, is simply the linear correlation of two functional category activation vectors. The second, GO-sim, takes into account the known directed acyclic graph (DAG) structure among the functional categories of the GO annotation.
Formally, the flat-sim score between a pair of 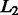 -normalized feature vectors
-normalized feature vectors 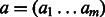 and
and 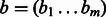 is given by their dot product flat-sim
is given by their dot product flat-sim
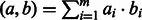 . This additive similarity measure allows assessing the contribution of each individual feature to the overall similarity score, by setting the contribution of the feature i (corresponding to GO category i) to
. This additive similarity measure allows assessing the contribution of each individual feature to the overall similarity score, by setting the contribution of the feature i (corresponding to GO category i) to 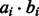 . Thus, for each pair of similar images, we can sort the GO categories by order of their contribution to the similarity, providing a semantic interpretation of the correlation.
. Thus, for each pair of similar images, we can sort the GO categories by order of their contribution to the similarity, providing a semantic interpretation of the correlation.
However, flat-sim does not take into account that the activation of some functional categories can be far more informative than others. For example, two genes that share a specific function like ‘negative regulation of systemic arterial blood pressure’ are much more likely to be functionally similar than a pair of genes sharing a more general category like ‘metabolism’. We address this issue by adapting a functional similarity measure between gene products developed by (Schlicker et al., 2006), which we refer to as GO-sim. GO-sim is designed to give high similarity scores to gene pairs that share many specific and similar functional categories. We treat our model’s functional activations as binary annotations (using a threshold of 0.5) and calculate GO-sim as follows.
For each GO category i, we calculate its information content (IC) as 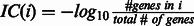 , which measures the specificity of each category. For each pair of categories i and j, we consider the set of their common ancestors
, which measures the specificity of each category. For each pair of categories i and j, we consider the set of their common ancestors 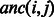 and define
and define 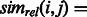
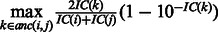 . The measure
. The measure  is symmetric, bounded between 0 and 1, and attains larger values for pairs of categories that are both specific and close to each other in the GO graph.
is symmetric, bounded between 0 and 1, and attains larger values for pairs of categories that are both specific and close to each other in the GO graph.
In our method, each gene is annotated with multiple categories. Naïvely, we could calculate the mean  measure between all pairs of categories, but calculating this mean could give weight to many irrelevant categories and be sensitive to the addition of extra annotations to a gene. Instead, we use a more robust method to measure similarity between two sets of function annotations, developed by (Schlicker et al., 2006). This method relies on the most similar gene pairs, instead of all the pairs. For two binary activation vectors
measure between all pairs of categories, but calculating this mean could give weight to many irrelevant categories and be sensitive to the addition of extra annotations to a gene. Instead, we use a more robust method to measure similarity between two sets of function annotations, developed by (Schlicker et al., 2006). This method relies on the most similar gene pairs, instead of all the pairs. For two binary activation vectors 
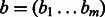 define a matrix
define a matrix 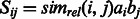 . Then we define
. Then we define 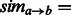
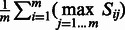 that measures for each annotation of
that measures for each annotation of  its most similar annotation in
its most similar annotation in 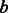 and averages across all of
and averages across all of  ‘s annotations. We similarly define
‘s annotations. We similarly define 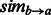 with the roles of
with the roles of  and
and 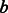 switched, and use it to define GO-sim
switched, and use it to define GO-sim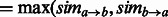 ). To assess the contribution of individual gene functional annotations to the GO-sim measure, we look at the category pairs (i,j) corresponding to the highest values of
). To assess the contribution of individual gene functional annotations to the GO-sim measure, we look at the category pairs (i,j) corresponding to the highest values of 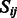 . Each such pair also has its ‘most informative common ancestor’
. Each such pair also has its ‘most informative common ancestor’ 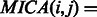
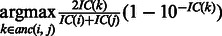 . These ancestor functional categories give a succinct interpretation of the similarity between genes
. These ancestor functional categories give a succinct interpretation of the similarity between genes  and
and 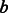 .
.
Computing GO-sim for n = 16 351 genes, each with m functional annotations, is computationally burdensome, requiring O(n2m2) operations. In this study, we, therefore, use only 164 brain-related categories of the 2081 functional categories for calculating GO-sim.
3 RESULTS
We start with evaluating the quality of the low-dimensional semantic representation that we learned in two aspects: the classification accuracy for individual semantic terms and the precision of our gene–gene similarity measure compared with a spatial correlation-based method. We then take a closer look at discriminative spatial patterns, mapping them back onto raw images. Finally, we use the geometry of the low-dimensional semantic space to infer new gene functions via gene similarities and their interpretations.
3.1 Predicting functional annotations using brain ISH images
We applied FuncISH to 16 K ISH images of 15 K genes, and we mapped each image to a vector corresponding to 2000 GO categories as functional features. We used the area under the ROC curve (AUC) as a measure of classification accuracy. All evaluations were performed on a separate held-out test set. We find that 37% of the GO categories tested yielded a test set AUC value that was significantly above random (permutation test, P < 0.05). This was encouraging, as the variability of expression between brain regions was previously shown to be very low (Khaitovich et al., 2004, 2005). This suggests that fine spatial resolution in neural tissues can reveal highly meaningful expression patterns.
Which functional categories can be best predicted by ISH images? Table 1 lists the top 15 GO categories that achieved the best test-set AUC classification scores. Interestingly, these include mostly biosynthesis/metabolism processes and neural processes. To further test whether neural categories achieve higher classification values based on neural expression patterns, Figure 3 compares the AUC scores of 164 categories related to the nervous system with the AUC scores of the remaining categories. As expected, neural GO categories receive significantly higher AUCs (Wilcoxon, P < 10−38), with 69% of categories yielding significantly above random AUC values.
Table 1.
The GO categories classified with highest test-set AUC values
| GO ID | GO category name | No. of genes | AUC |
|---|---|---|---|
| GO:0060311 | Negative regulation of elastin catabolic process | 17 | 1 |
| GO:0042759 | Long-chain fatty acid biosynthetic process | 23 | 0.98 |
| GO:0009449 | γ-Aminobutyric acid biosynthetic process | 20 | 0.96 |
| GO:0009448 | γ-Aminobutyric acid metabolic process | 23 | 0.96 |
| GO:0032348 | Negative reg. of aldosterone biosynthetic process | 21 | 0.94 |
| GO:2000065 | Negative regulation of cortisol biosynthetic process | 21 | 0.94 |
| GO:0043206 | Fibril organization | 23 | 0.94 |
| GO:0031947 | Negative reg. of glucocorticoid biosynthetic process | 22 | 0.94 |
| GO:0042136 | Neurotransmitter biosynthetic process | 23 | 0.94 |
| GO:0022010 | Central nervous system myelination | 29 | 0.89 |
| GO:0008038 | Neuron recognition | 20 | 0.87 |
| GO:0042220 | Response to cocaine | 30 | 0.87 |
| GO:0050919 | Negative chemotaxis | 16 | 0.86 |
| GO:0042274 | Ribosomal small subunit biogenesis | 15 | 0.86 |
| GO:0016486 | Peptide hormone processing | 17 | 0.85 |
Fig. 3.

AUC scores for GO categories related to the nervous system (dashed, red) and the remaining categories (solid, blue). AUC scores are significantly higher for neural categories (Wilcoxon test, p < 10−38). The red and blue ticks indicate the median of each set
These AUC values suggest that when a gene is represented as a feature vector of classifiers activations, many of the features carry a meaningful signal. The axes of the new low-dimensional representation correspond to functional properties of each gene, linking functions of the genes to the geometry of the space in which they are embedded.
3.2 Comparison with Neuroblast, the ABA image-correlation tool
How well does FuncISH compare with other methods suggested for finding similarity between these images? We compared our results with NeuroBlast, a method to detect image–image similarities available on the ABA website (Hawrylycz et al., 2011). This method uses a non-linear mapping of the images to a reference anatomical atlas to apply voxel–voxel correlation between the images.
To evaluate the quality of the similarity measure, we used three sets of pairwise relations as evidence of gene relatedness: (i) markers of known cell types (Cahoy et al., 2008), such as astrocytes or oligodendrocytes; (ii) occurrence in the same KEGG pathway (Kanehisa, 2002); and (iii) a set of known protein–protein interactions taken from IntAct (Kerrien et al., 2012). For each of the 16 531 genes, we ranked the 100 most similar genes according to four different similarity measures: (i) FuncISH GO-sim, (ii) FuncISH flat-sim, (iii) cosine similarity between the SIFT bag-of-words representations (Fig. 2D) and (iv) the ABA NeuroBlast tool. For each of the pairwise relations (cell-type markers, KEGG pathway and PPIs), we plot the mean fraction of relations retrieved at the top-K most similar genes (precision-at-k), a standard method in information retrieval (Manning and Raghavan, 2009). Figure 4 shows that for all three validation labels, FuncISH GO-sim provides superior precision for the top 10 ranked similar genes. The superior precision of GO-sim over flat-sim is presumably because GO-sim weighs categories more correctly and also possibly because GO-sim was limited to brain-related categories that tend to be more accurately predicted (Fig. 3). On the other hand, we see that NeuroBlast outperforms flat-sim in most cases.
Fig. 4.

Precision at top-K for similarity defined by (A) cell type marker (B) KEGG pathways (C) protein–protein interaction. Precision was measured using functional representations (FuncISH, purple lines for GO-sim, orange for flat-sim), SIFT (red) and NeuroBlast (blue)
3.3 Identifying and explaining similarities between GABAergic neuron markers
We now turn to a deeper look into the similarity predictions. Interestingly, the highest classification scores were achieved for the neural-related categories GABA biosynthetic process and GABA metabolic process (shown in Table 1), implying that our algorithm can identify spatial patterns of GABAergic neurons. A prominent member of the GABAergic neuron marker family is parvalbumin B (Pvalb), which encodes for a calcium-binding protein. We examined the genes that are most similar to Pvalb, and we found that another GABAergic neuronal marker and a calcium-binding protein, calbindin D28K (Calb1), is at the top 15 most similar gene lists for all associated image series. Pvalb and Calb1 belong to a family of cellular Ca2+ buffers in GABAergic interneurons. The third member in this family is calretinin (Calb2). Looking at the similarity rank of Calb1 and Calb2, Calb2 ranks at the top 2 percentile (of 16 351 images in the dataset) at 16 of 17 cases. Similarities between these three genes were not identified by NeuroBlast. This may be because NeuroBlast uses spatial correlation measures that produce results heavily reliant on the spatial location of expression, whereas FuncISH can identify patterns that can appear in different regions of the brain. A major benefit of representing genes in the functional embedding space is that similarities between genes can be ‘explained’ in functional terms. Calb1, Pvalb and Calb2 are all involved in regulation of synaptic plasticity (Schwaller, 2012). When looking at the semantic interpretations explaining the similarities between the genes, 6 of the top 10 GO categories are indeed directly related to synaptic plasticity, such as ‘synaptic transmission’, ‘regulation of synaptic plasticity’ and ‘learning’.
3.4 Finding important spatial patterns in different scales using SIFT ‘visual words’
A major advantage of representing ISH images with SIFT descriptors is the ability to point directly to spatial patterns in these complex images. Although their name suggest differently, SIFT descriptors at several scales capture different types of patterns. Figure 5 shows three visual words for each of the four scales, selected as the visual words that contributed most to classification. Scale invariance is often assumed when analyzing natural images, as objects are photographed at varying distances. ISH images, however, contain distinctive information in the different scales. As Figure 5 demonstrates, the four sizes of visual words correspond to grids capturing different neural entities. The smallest descriptors cover an actual area of 36 × 36 µm2 and capture fine-scaled information, such as cell shapes and cell densities; the medium-size discriminative descriptors of 72 × 72 µm2 tend to trace thinner cell layers; larger descriptor sizes of 144 × 144 µm2 and 288 × 288 µm2 can cover large and intricate patterns of a mixture of cells and cell types in a tissue. Interestingly, the four visual words with the highest contribution to classification were the words counting the zero descriptors in each scale. This means that the highest information content lies in ‘least informative’ descriptors, and that overall expression levels (‘sparseness’ of expression) are important factors in functional prediction of genes based on their spatial expression. Our method presents a new representation of ISH imagery as SIFT descriptors, and using multiple scales allows revealing the multi-resolution nature of the images.
Fig. 5.

Representing ISH images with visual words. (A, B, C, D) The three visual words with highest absolute weight (averaged over all categories) at each scale. The SIFT descriptors (red grid) are plotted on top of each panel. The histogram of oriented gradients used in the SIFT descriptor is plotted in the center of each element of the grid,as a set of red lines, where the length of the line correspond to the magnitude of the gradient in its direction. (E) Mean absolute weight for the four scales of visual words calculated over classifiers for all categories
Which scale carries the most meaningful signal for functional prediction? Figure 5E shows the mean absolute value of visual words weights in every scale for all GO categories, showing that all scales contribute significantly to the scores, with the medium contributing most.
Figure 5A–D shows descriptors that contributed to classification of all the categories. Furthermore, each GO category has its own visual words that are important to its classification, and looking into their details reveals spatial properties that are unique to specific biological processes.
As an interesting example of this effect, we considered the gene adducin β (Add2). Add2 is annotated to several GO categories, including ‘positive regulation of protein binding’ and ‘actin filament bundle assembly’. Figure 6 overlays the top weighted visual words of the two categories over the Add2 ISH image. It is easy to see that the descriptors important for classification of ‘actin filament bundle assembly’ are much smaller than those important for classification of the more general category ‘positive regulation of protein binding’ (t-test, P < 10−17). This implies that small-scaled features, such as specific cell shapes, are important to identify genes related to actin filament bundle assembly processes. Actin assemblies are important for the navigation of neural growth cones, by re-orienting growth cones away from inhibitory cues (Challacombe et al., 1996). Representing the images with histograms of oriented gradients could capture tiny differences in cell shapes that are in the process of synapse formation, a developmental process occurring continuously throughout adulthood (Vidal-Sanz et al., 1987).
Fig. 6.

The visual words important in classifying Add2 GO categories are overlaid on the Add2 ISH image. Larger descriptors are needed for the classification of ‘regulation of protein binding’ (A), while the discriminative visual words for ‘actin filament bundle assembly’ (B) are much smaller, capturing properties such as cell shapes. The descriptors are color-coded by their importance in classification, highest importance is in bright yellow
3.4 Inferring new gene functions via explainable similarities
We now demonstrate how the semantic representation learned by FuncISH can be used to propose new gene functional annotations. Consider as an example the gene synaptopodin 2 (Synpo2) that is known to bind actin, but otherwise has little known associated information. FuncISH can be used to propose functional annotations for synpo2 by looking at the genes that are similar to Synpo2 and considering both the GO functions that contribute to this similarity and the spatial pattern of expression.
First, we find that Synpo2 is similar to two other genes Npepps and Rasa4, but for different reasons (the list of top five semantic explanations for these similarities is shown in Table 2). Npepps is an aminopeptidase that is active specifically in the brain (Hui, 2007), and the similarity between Synpo2 and Npepps is explained by processes related to protein processing, such as ubiquitination and protein proteolysis. At the same time, Rasa4 is a GTPase-activating protein that suppresses the Ras/mitogen-activated protein kinase pathway in response to Ca2+ (Vigil et al., 2010), and the similarity between Synpo2 and Rasa4 is explained by high-level neural processes, such as axon guidance or synaptic transmission.
Table 2.
Top 10 GO annotations explaining the similarities between the gene Synpo2 and Npepps (left column) and Rasa4 (right column)
|
Synpo2–Npepps |
Synpo2–Rasa4 |
||
|---|---|---|---|
| GO ID | GO name | GO ID | GO name |
| GO:0070646 | Protein modification by small protein removal | GO:0006836 | Neurotransmitter transport |
| GO:0006412 | Translation | GO:0051970 | Negative regulation of transmission of nerve impulse |
| GO:0016567 | Protein ubiquitination | GO:0050805 | Negative regulation of synaptic transmission |
| GO:0051603 | Proteolysis involved in cellular protein catabolic process | GO:0007411 | Axon guidance |
| GO:0032446 | Protein modification by small protein conjugation | GO:0031645 | Negative regulation of neurological system process |
Interestingly, Synpo2 and Rasa4 are expressed in different brain regions: looking at their spatial expression patterns reveals that Synpo2 is expressed exclusively in the thalamus, whereas Rasa4 is expressed in olfactory areas. Therefore, their similarity is not in their global expression patterns across regions, but rather in local spatial patterns. This could reflect expression in similar cell types or tissues that exhibit similar spatial distribution at different brain regions. Npepps is more ubiquitously expressed in the brain, and it is located in the thalamic area where synpo2 is expressed. The co-location of Synpo2 and Npepps suggests they could be participating in similar biological processes in these areas, possibly in protein-modification processes as suggested by the list of top explanations for the similarity.
4 SUMMARY
We present FuncISH—a method to learn functional representations of neural ISH images, yielding an interpretable measure of similarity between complex images that are difficult to analyze and interpret. Using FuncISH, we successfully infer ∼700 functional annotations from neural ISH images, and we use them to detect gene–gene similarities. This approach reveals similarities that are not captured by previous global correlation-based methods, but it also ignores important global location information. Combining local and global patterns of expression is, therefore, an important topic for further research, as well as the use of more sophisticated non-linear classifiers, such as kernel-SVM, for creating better representations. Importantly, FuncISH provides semantic interpretations for similarity, enabling the inference of new gene functions from spatial co-expression.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the Allen Institute for Brain Science for making their data available to the scientific community and for helping us with any questions. They are also grateful to Lior Kirsch for valuable discussions and technical help. They thank the reviewers for their helpful comments and suggestions.
Funding: Israeli Science Foundation (1008/09 and 1090/12 to G.C.); Marie Curie Reintegration Grant (PIRG06-GA-2009-256566 to G.C.); Google Europe Fellowship in Machine Learning (to U.S.).
Conflict of Interest: none declared.
References
- Bosch A, et al. Scene classification via pLSA. Computer Vision-ECCV 2006. 2006;3954:517–530. [Google Scholar]
- Bosch A, et al. Image classification using random forests and ferns. IEEE 11th Int. Conf. Comput. Vis. 2007;23:1–8. [Google Scholar]
- Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challacombe JF, et al. Actin filament bundles are required for microtubule reorientation during growth cone turning to avoid an inhibitory guidance cue. J. Cell Sci. 1996;109(Pt 8):2031–2040. doi: 10.1242/jcs.109.8.2031. [DOI] [PubMed] [Google Scholar]
- Coelho LP, et al. Quantifying the distribution of probes between subcellular locations using unsupervised pattern unmixing. Bioinformatics. 2010;26:i7–i12. doi: 10.1093/bioinformatics/btq220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csurka G, Dance C. Workshop on Statistical Learning in Computer Vision, ECCV. 2004. Visual categorization with bags of keypoints. Vol. 1, p. 22. [Google Scholar]
- Davis FP, Eddy SR. A tool for identification of genes expressed in patterns of interest using the Allen Brain Atlas. Bioinformatics. 2009;25:1647–1654. doi: 10.1093/bioinformatics/btp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, et al. Hierarchical semantic indexing for large scale image retrieval. CVPR 2011. 2011:785–792. [Google Scholar]
- Fan, et al. LIBLINEAR: a library for large linear classification. J. Mach. Learn. Res. 2008;9:1871–1874. [Google Scholar]
- Frise E, et al. Systematic image-driven analysis of the spatial Drosophila embryonic expression landscape. Mol. Syst. Biol. 2010;6:345. doi: 10.1038/msb.2009.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz M, et al. Multi-scale correlation structure of gene expression in the brain. Neural Netw. 2011;24:933–942. doi: 10.1016/j.neunet.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Henry AM, Hohmann JG. High-resolution gene expression atlases for adult and developing mouse brain and spinal cord. Mamm. Genome. 2012;23:539–549. doi: 10.1007/s00335-012-9406-2. [DOI] [PubMed] [Google Scholar]
- Hui K-S. Brain-specific aminopeptidase: from enkephalinase to protector against neurodegeneration. Neurochem. Res. 2007;32:2062–2071. doi: 10.1007/s11064-007-9356-3. [DOI] [PubMed] [Google Scholar]
- Kanehisa M. The KEGG database. Novartis Found. Symp. 2002;247:91–101. discussion 101–103, 119–128, 244–252. [PubMed] [Google Scholar]
- Kerrien S, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40:D841–D846. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, et al. Regional patterns of gene expression in human and chimpanzee brains. Genome Res. 2004;14:1462–1473. doi: 10.1101/gr.2538704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, et al. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science. 2005;309:1850–1854. doi: 10.1126/science.1108296. [DOI] [PubMed] [Google Scholar]
- Kirsch L, et al. Localizing genes to cerebellar layers by classifying ISH images. PLoS Comput. Biol. 2012;8:e1002790. doi: 10.1371/journal.pcbi.1002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazebnik S, et al. Beyond bags of features: spatial pyramid matching for recognizing natural scene categories. IEEE Conf. Comput. Vis. and Pattern Recognition. Vol. 2 CVPR06. 2006;2:2169–2178. [Google Scholar]
- Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li L, et al. Object bank: a high-level image representation for scene classification and semantic feature sparsification. Proc. Neural Inf. Process. Syst. 2010. 2010a:1–9. [Google Scholar]
- Li L, et al. Trends and Topics in Computer Vision. Berlin Heidelberg: Springer; 2012. Objects as attributes for scene classification; pp. 57–69. [Google Scholar]
- Lowe DG. Distinctive image features from scale-invariant keypoints. Int. J. Comput. Vis. 2004;60:91–110. [Google Scholar]
- Malisiewicz T. Exemplar-based representations for object detection, association and beyond. 2012 PhD Thesis. [Google Scholar]
- Malisiewicz T, et al. Ensemble of exemplar-SVMs for object detection and beyond. 2011 International Conference on Computer Vision. 2011:89–96. [Google Scholar]
- Manning CD, Raghavan P. Introduction to Information Retrieval. Vol. 1. Cambridge: Cambridge University Press; 2008. [Google Scholar]
- Ng L, et al. An anatomic gene expression atlas of the adult mouse brain. Nat. Neurosci. 2009;12:356–362. doi: 10.1038/nn.2281. [DOI] [PubMed] [Google Scholar]
- Peng H, et al. Automatic image analysis for gene expression patterns of fly embryos. BMC Cell Biol. 2007;8:S7. doi: 10.1186/1471-2121-8-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, et al. Determining the distribution of probes between different subcellular locations through automated unmixing of subcellular patterns. Proc. Natl. Acad. Sci. USA. 2010;107:2944–2949. doi: 10.1073/pnas.0912090107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruteanu-Malinici I, et al. Automatic annotation of spatial expression patterns via sparse Bayesian factor models. PLoS Comput. Biol. 2011;7:e1002098. doi: 10.1371/journal.pcbi.1002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker A, et al. A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics. 2006;7:302. doi: 10.1186/1471-2105-7-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B. The use of transgenic mouse models to reveal the functions of Ca2+ buffer proteins in excitable cells. Biochim. Biophys. Acta. 2012;1820:1294–1303. doi: 10.1016/j.bbagen.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Torresani L, et al. Efficient object category recognition using classemes. Comput. Vis.–ECCV 2010. 2010:776–789. [Google Scholar]
- Vedaldi A, Fulkerson B. VLFeat—an open and portable library of computer vision algorithms. Design. 2010;3:1–4. [Google Scholar]
- Vidal-Sanz M, et al. Axonal regeneration and synapse formation in the superior colliculus by retinal ganglion cells in the adult rat. J. Neurosci. 1987;7:2894–2909. doi: 10.1523/JNEUROSCI.07-09-02894.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil D, et al. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat. Rev. Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.