

Abstract
Motivation: An important aspect of infectious disease research involves understanding the differences and commonalities in the infection mechanisms underlying various diseases. Systems biology-based approaches study infectious diseases by analyzing the interactions between the host species and the pathogen organisms. This work aims to combine the knowledge from experimental studies of host–pathogen interactions in several diseases to build stronger predictive models. Our approach is based on a formalism from machine learning called ‘multitask learning’, which considers the problem of building models across tasks that are related to each other. A ‘task’ in our scenario is the set of host–pathogen protein interactions involved in one disease. To integrate interactions from several tasks (i.e. diseases), our method exploits the similarity in the infection process across the diseases. In particular, we use the biological hypothesis that similar pathogens target the same critical biological processes in the host, in defining a common structure across the tasks.
Results: Our current work on host–pathogen protein interaction prediction focuses on human as the host, and four bacterial species as pathogens. The multitask learning technique we develop uses a task-based regularization approach. We find that the resulting optimization problem is a difference of convex (DC) functions. To optimize, we implement a Convex–Concave procedure-based algorithm. We compare our integrative approach to baseline methods that build models on a single host–pathogen protein interaction dataset. Our results show that our approach outperforms the baselines on the training data. We further analyze the protein interaction predictions generated by the models, and find some interesting insights.
Availability: The predictions and code are available at: http://www.cs.cmu.edu/∼mkshirsa/ismb2013_paper320.html
Contact: j.klein-seetharaman@warwick.ac.uk
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Infectious diseases are a major health concern worldwide, causing millions of illnesses and deaths each year. Newly emerging viral diseases, such as swine H1N1 influenza, severe acute respiratory syndrome (SARS) and bacterial infections, such as the recurrent Salmonella and Escherichia coli outbreaks not only lead to wide-spread loss of life and health, but also result in heavy economic losses. To better navigate this landscape of infectious diseases, it is important to not only understand the mechanisms of individual diseases, but also the commonalities between them. Combining knowledge from related diseases will give us deeper insights into infection and host immune response, will enhance our ability to comprehend new diseases and lead to efficient development of therapeutics.
Key to the infection process are host–pathogen interactions at the molecular level, where pathogen proteins physically bind with human proteins. Via these protein interactions, the pathogen manipulates important biological processes in the host cell, evades host immune response and multiplies within the host. Interactions between host and pathogen proteins can be studied using small-scale biochemical, biophysical and genetic experiments or large-scale high-throughput screening methods like yeast two-hybrid (Y2H) assays. Databases like PHI-base (Winnenburg et al., 2008), PIG (Driscoll et al., 2009), HPIDB (Kumar and Nanduri, 2010), PHISTO (Tekir et al., 2012) aggregate host–pathogen protein interactions from several small-scale and high-throughput experiments via manual literature curation. These databases are valuable sources of information for developing models of the modus operandi of pathogens.
However, interaction datasets from these databases are not only small but are available for only a few well-studied pathogens. For example, the PHI-base (Winnenburg et al., 2008) database covers 64 diseases but has only 1335 interactions, PIG (Driscoll et al., 2009) covers only 12 pathogens. Computationally, this calls for techniques that combine datasets and build joint models across several pathogens, which can then be used to analyze the commonalities in the pathogens and also to predict plausible interactions that are biased by this joint understanding.
In our work, we study host–pathogen protein–protein interaction (PPI) where the host is fixed and the pathogens are various bacterial species (Fig. 1A). The host species we consider is human and the bacterial species are Yersinia pestis, Francisella tularensis, Salmonella and Bacillus anthracis, which cause the diseases bubonic plague, acute pneumonia, typhoid and anthrax, respectively.
Fig. 1.
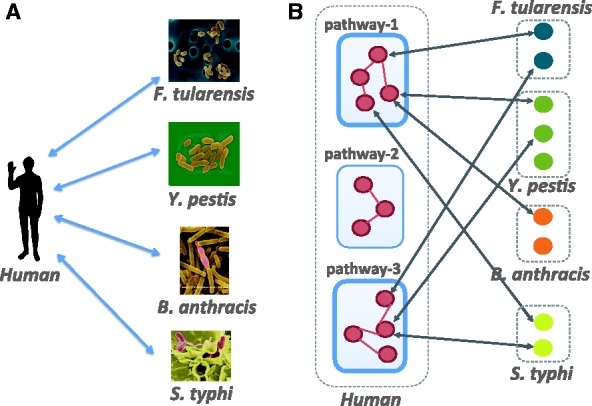
(A) Host–pathogen PPI prediction where the host is human and the pathogens are bacteria. (B) An example depicting the commonality in the bacterial attack of human proteins. Pathway-1 and pathway-3 (highlighted) represent critical processes targeted by all bacterial species
Some recent work on infectious diseases has alluded to the hypothesis that different pathogens target essentially the same critical biological processes in the human body. The analysis by Chen et al. (2012) suggests that HIV infection shares common molecular mechanisms with certain signaling pathways and cancers. Dyer et al. (2008) study bacterial and viral interactions with human genes and find infection mechanisms common to multiple pathogens. Experiments by Jubelin et al. (2010) show how various bacterial cyclomodulins target the host cell cycle. The study by Mukhtar et al. (2011) on plant pathogens, in particular, Arabidopsis concludes that pathogens from different kingdoms deploy independently evolved virulence proteins that interact with a limited set of highly connected cellular hubs to facilitate their diverse life cycle strategies. Figure 1B illustrates an example depicting the commonality in various bacterial species, where they are targeting the same biological pathways in their human host.
This biological hypothesis, which we henceforth call the commonality hypothesis, is exploited here to jointly learn PPI models for multiple bacterial species. We translate the hypothesis into a prior that will bias the learned models. We use a multitask learning–based approach, where each ‘task’ represents the interaction of one bacterial species with human. The prior is represented in the form of a regularizer that penalizes models to the degree that the above hypothesis is violated.
2 BACKGROUND
The most reliable experimental methods for studying PPI are often time-consuming and expensive, making it hard to investigate the prohibitively large set of possible host–pathogen interactions—for example, the bacterium B.anthracis, which causes anthrax, has about 2321 proteins, which when coupled with the 25 000 or so human proteins gives approximately 60 million protein pairs to test, experimentally. Computational techniques complement laboratory-based methods by predicting highly probable PPIs—thereby enabling experimental biologists to focus on fewer interactions and ruling out the vast majority of unlikely ones.
In particular, supervised machine learning–based methods use the few experimentally discovered interactions as training data and formulate the interaction prediction problem in a classification setting, with target classes: ‘interacting’ or ‘non-interacting’. Features are derived for each host–pathogen protein pair using various attributes of the two proteins such as protein sequence, gene expression, gene ontology (GO) etc. The general outline of the supervised PPI prediction procedure is illustrated in Supplementary Figure S1.
Most of the prior work in PPI prediction has focussed on building models separately for individual organisms (Chen and Liu, 2005; Qi et al., 2006; Singh et al., 2006; Wu et al., 2006) or on building a model specific to a disease in the case of host–pathogen PPI prediction (Dyer et al., 2007; Kshirsagar et al., 2012; Qi et al., 2009; Tastan et al., 2009). The use of PPI data from several organisms has predominantly been in the form of (i) features derived from various PPI datasets, (ii) use of common structural properties of proteins across organisms (Wang et al., 2007) or (iii) methods that narrow down predicted interactions in the organism of interest (Garcia et al., 2010). Some of these methods use the concepts of ‘homologs’, ‘orthologs’ and ‘interologs’ to define a similarity measure between PPIs from various organisms (Garcia et al., 2010).
There has been little work on combining PPI datasets with the goal of improving prediction performance for multiple organisms. Qi et al. (2010) proposed a semi-supervised multitask framework to predict PPIs from partially labeled reference sets. The basic idea is to perform multitask learning on a supervised classification task and a semi-supervised auxiliary task via a regularization term. Another line of work in PPI prediction (Xu et al., 2010) uses the Collective Matrix Factorization (CMF) approach proposed by Singh and Gordon (2008). The CMF method learns models for multiple networks by simultaneously factorizing several adjacency matrices and sharing parameters amongst the factors. Xu et al. (2010) use these ideas in their transfer learning setting, where the source network is a relatively dense interaction network of proteins and the objective is to infer PPI edges in a relatively sparse target network. To compute similarities between the nodes in the source and target networks, they use protein sequences and the topological structures of the interaction networks.
3 APPROACH
Multitask learning is a family of machine learning methods that addresses the issue of building models using data from multiple problem domains (i.e. ‘tasks’) by exploiting the similarity between them. The goal is to achieve performance benefits for all the tasks involved. This paradigm of building joint models has been applied successfully in many areas including text mining, computer vision, etc. Because bioinformatics datasets often represent an organism, a natural notion of a ‘task’ is an ‘organism’—for example, the work by Widmer et al. (2010) uses a multitask learning approach for splice-site prediction across many organisms. They use phylogenetic trees to incorporate similarity between organisms (i.e. tasks). For a survey of multitask learning in computational biology, see Xu and Yang (2011).
Our multitask learning method is based on the task regularization framework, which formulates the multitask learning problem as an objective function with two terms: an empirical loss term on the training data of all tasks, and a regularization term that encodes the relationships between tasks. Equation (1) shows the general form of such an objective, the term R being the regularizer raised to the power p and with a q-norm. The work by Evgeniou and Pontil (2004) is one of the early few to develop this general approach. The function in Equation (1) represents a simple multitask objective with a single regularizer R; many of the formulations often involve a summation over multiple terms.
| (1) |
We optimize this function by modifying the regularizer R to encode the biological hypothesis. Our approach differs greatly from prior work because we propose a technique to translate a problem-relevant biological hypothesis into a task regularization–based approach rather than applying existing general formalisms on a dataset. Our tasks try to capture a naturally occurring phenomenon. While our framework is developed in the context of a specific hypothesis, we also illustrate the incorporation of other hypotheses with an example. The key contributions of our work are as follows:
we present a novel way of combining experimental PPI data coming from several organisms
we incorporate domain knowledge in designing a prior that causes the learned models to exhibit the requisite common structure across the tasks
to optimize the resulting non-convex objective function, we implement a concave convex procedure (CCCP)-based method
In the Methods section (Section 4), we describe details of the PPI datasets and our multitask learning framework. The evaluation metrics and description of experiments is in Section 6, results and analysis in Section 7.
4 METHODS
4.1 Multitask pathway–based learning
In this section, we describe how we incorporate the commonality hypothesis into our multitask classification framework formulating it as an optimization problem.
We consider each human-bacteria PPI prediction problem as one task. The predicton problem is posed as a binary classification task, with each instance 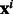 being a pair of proteins
being a pair of proteins 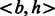 , where one protein is the bacterial protein ‘b’ (e.g. Y. pestis) and the other ‘h’ is the host protein (i.e. human). The class-label
, where one protein is the bacterial protein ‘b’ (e.g. Y. pestis) and the other ‘h’ is the host protein (i.e. human). The class-label 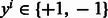 represents interacting and non-interacting proteins respectively. Features are defined for every protein-pair using various properties of the individual proteins and combining them all into a single feature vector. The positive class in our training data comprises the known human-bacterial PPI which are obtained from databases like PHISTO (Tekir et al., 2012). The construction of the negative-class data is explained in Section 5.
represents interacting and non-interacting proteins respectively. Features are defined for every protein-pair using various properties of the individual proteins and combining them all into a single feature vector. The positive class in our training data comprises the known human-bacterial PPI which are obtained from databases like PHISTO (Tekir et al., 2012). The construction of the negative-class data is explained in Section 5.
Our objective is to minimize the empirical error on the training data while favoring models that are biased toward the commonality hypothesis. To achieve this, we use a bias term in the form of a regularizer in our objective function. For brevity and without loss of generality, we will henceforth refer to each human–bacteria PPI prediction problem as a ‘task’ (We will also refer to a task by the name of the bacterial species only, as the host species, i.e. human, is common across all tasks).
Our method first combines all tasks in a pairwise manner, and finally aggregates the output from the pairwise models. Let 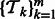 be the set of tasks to be combined, where m is the number of tasks. Consider two tasks
be the set of tasks to be combined, where m is the number of tasks. Consider two tasks 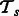 and
and 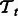 . Let the training data for the task
. Let the training data for the task 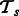 be
be 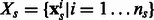 where each example
where each example 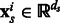 . Similarly, the training data for
. Similarly, the training data for 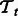 is
is 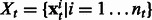 where
where 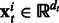 . ns and nt are the number of training examples and ds and dt denote the number of features in the two tasks. Let
. ns and nt are the number of training examples and ds and dt denote the number of features in the two tasks. Let 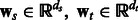 represent the parameter vectors, i.e. the models for the two tasks. We now describe how we combine these two tasks. Section 4.3 will show how such pairwise models are aggregated.
represent the parameter vectors, i.e. the models for the two tasks. We now describe how we combine these two tasks. Section 4.3 will show how such pairwise models are aggregated.
The pathway-based objective
Biologists often represent the set of human proteins involved in a particular biological process by a graph called a ‘biological pathway’. One such example, the ‘glucose transport pathway’ in human is shown in the Supplementary. To use this pathway construct, we revise our hypothesis to ‘proteins from different bacterial species are likely to interact with human proteins from the same biological pathway’. Figure 1B illustrates an example where this hypothesis holds. The pathway information for each human protein can be obtained from pathway databases like Reactome (Matthews et al., 2009) and PID (Schaefer et al., 2009). While pathways are generally represented as graphs, for our current work we do not use the edges. We treat a pathway as a set of proteins—a human protein h can be a member of several pathways depending on the biological processes it is involved in. Let N be the total number of pathways in human. For a protein pair 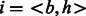 , let
, let 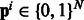 be the binary ‘pathway vector’ indicating the pathway membership of h.
be the binary ‘pathway vector’ indicating the pathway membership of h.
The commonality hypothesis suggests that the pathway memberships of human proteins from interactions should be similar across tasks. We define a pathway-summary function S, which aggregates all pathway vectors for a given task 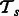 . Because our hypothesis is about interactions, we only consider pathway vectors of positive examples. Let
. Because our hypothesis is about interactions, we only consider pathway vectors of positive examples. Let 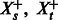 represent the set of positive examples from tasks
represent the set of positive examples from tasks 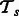 and
and 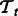 ; let
; let 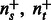 be their sizes. In Figure 2, we depict the aggregation done by S. Mathematically, we have
be their sizes. In Figure 2, we depict the aggregation done by S. Mathematically, we have
| (2) |
where 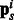 is the pathway vector for example i, and
is the pathway vector for example i, and 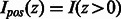 . S sums up the pathway vectors of examples predicted to be positive. We normalize using
. S sums up the pathway vectors of examples predicted to be positive. We normalize using 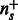 to compensate for the different dataset sizes across tasks.
to compensate for the different dataset sizes across tasks.
Fig. 2.
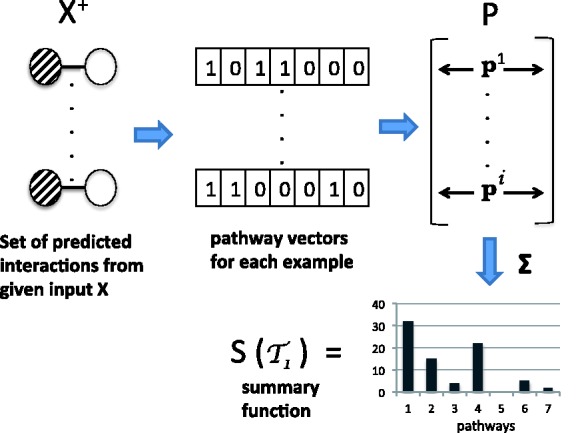
A schematic illustrating the pathway summarizing function S for a task  . On the left are the examples from the input predicted to be positive, indicated by
. On the left are the examples from the input predicted to be positive, indicated by  . The matrix P has the pathway vectors for each example in
. The matrix P has the pathway vectors for each example in  . The summary function aggregates the pathway vectors to get the distribution
. The summary function aggregates the pathway vectors to get the distribution
Let 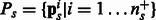 be a matrix containing all pathway vectors for positive examples from task
be a matrix containing all pathway vectors for positive examples from task 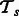 . Analogously,
. Analogously, 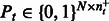 is a matrix for the positive examples from task
is a matrix for the positive examples from task 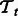 . Matrices Ps and Pt are constant matrices and are known a priori. Let
. Matrices Ps and Pt are constant matrices and are known a priori. Let 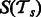 and
and 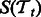 be the pathway summaries of the tasks. We want to penalize the dissimilarity between these summaries. Our objective function thus has the following general form:
be the pathway summaries of the tasks. We want to penalize the dissimilarity between these summaries. Our objective function thus has the following general form:
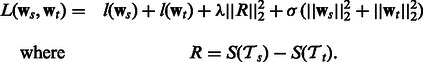 |
(3) |
Here 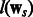 and
and 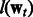 can be any convex loss functions computed over the two tasks. We use logistic loss in our work based on prior experience with PPI datasets. The last two
can be any convex loss functions computed over the two tasks. We use logistic loss in our work based on prior experience with PPI datasets. The last two 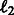 norms over the parameter vectors
norms over the parameter vectors 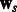 and
and 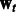 control overfitting. The parameters λ and σ take positive values.
control overfitting. The parameters λ and σ take positive values.
The indicator function Ipos is non-differentiable. So we approximate Ipos with the exponential function, which is a convex upper bound of the indicator function and will make optimization easier. Let 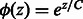 , where C is a positive constant. This function, for various values of C has been plot in Figure 3. Small positive values of
, where C is a positive constant. This function, for various values of C has been plot in Figure 3. Small positive values of 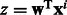 indicate positive-class predictions that are closer to the decision boundary of the classifier. Examples predicted to be positive with a high confidence have a large z. With varying values of C, the function
indicate positive-class predictions that are closer to the decision boundary of the classifier. Examples predicted to be positive with a high confidence have a large z. With varying values of C, the function 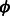 gives varying importance to predictions based on their classifier confidence ‘z’. Negative values of z, which correspond to examples predicted to be negative, are given close to zero importance by
gives varying importance to predictions based on their classifier confidence ‘z’. Negative values of z, which correspond to examples predicted to be negative, are given close to zero importance by 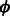 . The choice of an appropriate C is important so as to ensure the proper behavior of the summary function S. A steeply increasing curve (C = 1) is undesirable as it will assign too much weight to the summary of only some of the examples. We chose a moderate value of C = 30 for our experiments.
. The choice of an appropriate C is important so as to ensure the proper behavior of the summary function S. A steeply increasing curve (C = 1) is undesirable as it will assign too much weight to the summary of only some of the examples. We chose a moderate value of C = 30 for our experiments.
Fig. 3.
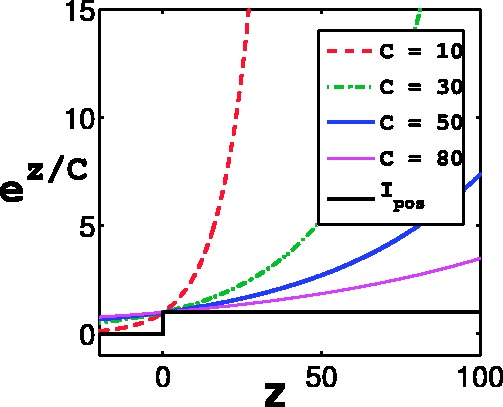
The exponential function 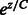 for different values of C
for different values of C
Replacing Ipos by 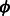 in Equation (2), our summary function S becomes
in Equation (2), our summary function S becomes 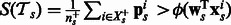 . Putting everything together, our objective with the logistic loss terms, the pathway summary function and the
. Putting everything together, our objective with the logistic loss terms, the pathway summary function and the 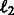 regularizer terms has the following form:
regularizer terms has the following form:
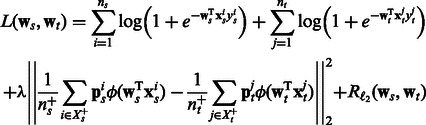 |
(3) |
where 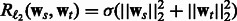
The objective in Equation (3) is non-convex, and with some algebraic simplifications we can reduce it to a difference of convex functions (DC). To optimize this function, we implement the CCCP algorithm, which was originally introduced by Yuille and Rangarajan (2003).
4.2 Solving the optimization problem
The objective in Equation (3) is non-convex in the shown form. We tried to optimize it directly using L-BFGS, but found that the objective does not decrease consistently. Below, we show that (3) is a DC functions. The first two log-loss terms [we abbreviate them henceforth as 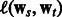 ] and the last
] and the last 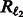 term are all convex and do not pose any problem with optimization.
term are all convex and do not pose any problem with optimization.
Proposition 1 —
The objective (3) is a DC functions.
(4)
Proof —
Expanding the pathway vectors
and
and rewriting Equation (3) we get
(5) Note that fk and gk are non-negative convex functions. This follows because
is a positive convex function and the matrices Ps and Pt are non-negative by construction. fk and gk are both thus positive linear combinations of convex functions and hence convex. We now decompose the squared term in Equation (5) as follows.
(6) We further observe that
is convex. To derive this, we use the following proposition: a composition of a monotonically increasing convex function and a convex function is still convex. The square function
is a monotonically increasing function for
, thus the composition with fk (i.e.
) is also convex by the positivity of fk. Analogously,
is also convex. Further,
is also convex by the same argument. Substituting (6) back into Equation (5) we get our result.
(7) To optimize this function, we use a CCCP algorithm (Yuille and Rangarajan, 2003). Our approach is inspired by the work from Yu and Joachims (2009) on learning structural SVMs. The idea is to compute a local upper bound on the concave function (
) and instead of optimizing L from Equation (4) directly, use an approximation based on the upper bound of
. Equation (7) shows this function Lapprox. Let w represent the concatenation of the two parameter vectors
and
. Let
be the k-th iterate. We have from Taylor’s first order approximation that
for all w. This allows us to obtain the following approximation, which we get by substituting the above bound in place of
in Equation (4):
(7) since
is a constant. The optimization problem in Equation (7) is now convex and can be solved using conventional techniques like L-BFGS, conjugate gradient, etc. The outline of our CCCP-based procedure is shown in Listing 1.
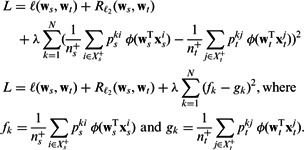
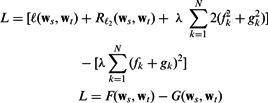
Algorithm 1 CCCP procedure.
1: Initialize 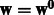
2: repeat
3: Compute 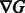 using
using 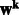
4: Compute current value Lapprox
5: Solve 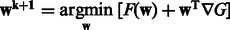
6: Set 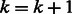
7: Compute new value 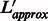
8: 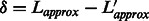
9: until
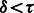
Yuille and Rangarajan (2003) show that such a CCCP-based algorithm is guaranteed to decrease the objective function at every iteration and to converge to a local minimum or saddle point. We observe a similar behavior in our experiments. Computationally, this algorithm is efficient because the regularizer works on a subset of the data—only the positive examples, which are a small fraction of the complete training data.
Stopping criteria
The convergence criterion for algorithm 1 is 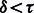 , where τ is a threshold. We used
, where τ is a threshold. We used 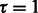 in our experiments. Smaller values required a long time to convergence. The inner optimization (line # 5), which uses L-BFGS, had a convergence threshold of 0.0001. This step took more iterations initially and fewer iterations getting closer to convergence.
in our experiments. Smaller values required a long time to convergence. The inner optimization (line # 5), which uses L-BFGS, had a convergence threshold of 0.0001. This step took more iterations initially and fewer iterations getting closer to convergence.
4.3 Combining pairwise models
In the previous sections, we described how we combine two tasks. In particular, Equation (3) involves pairwise learning, which results in two models 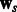 and
and 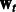 . Because our current framework can combine only two tasks at a time, for m tasks we perform
. Because our current framework can combine only two tasks at a time, for m tasks we perform 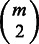 pairwise learning experiments and then combine their outputs. Each task will thus have m − 1 models as a result of pairing up with each of the other tasks. Let the set of models for task
pairwise learning experiments and then combine their outputs. Each task will thus have m − 1 models as a result of pairing up with each of the other tasks. Let the set of models for task 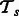 be
be 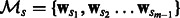 . We treat
. We treat 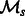 as an ensemble of models for this task and aggregate the output labels from all models to get the final labels on the test data. Let the output labels from each model for a given test instance x be
as an ensemble of models for this task and aggregate the output labels from all models to get the final labels on the test data. Let the output labels from each model for a given test instance x be 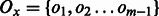 . Then the final output label y is computed by taking a vote and checking if it crosses a threshold:
. Then the final output label y is computed by taking a vote and checking if it crosses a threshold:
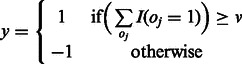 |
(8) |
where v is a vote threshold that should be crossed in order for the label to be positive. In our experiments, we found that the predictions for  from all models in
from all models in  overlapped greatly. Hence, we used v = 1, which implies that x is an interaction if any one of our four tasks labels it as such.
overlapped greatly. Hence, we used v = 1, which implies that x is an interaction if any one of our four tasks labels it as such.
5 DATASET AND FEATURES
For Salmonella typhi, we used the list of 62 interacting protein pairs reported in Schleker et al. (2012), which were obtained by the authors by manual literature curation. These interactions come from small-scale experiments. The other three PPI interaction datasets were obtained from the PHISTO database. Most of the reported interactions for these three bacterial species come from a single high-throughput experimental study reported in Dyer et al. (2010). While F.tularensis, S.typhi and Y.pestis are gram-negative gamma-protobacteria, B.anthracis is a gram-positive bacteria. The number of unique proteins in each bacterial species, the sizes of all datasets and the number of all possible host–pathogen protein pairs are listed in Table 1.
Table 1.
Characteristics of all four interaction datasets used
| B.anthracis | F.tularensis | Y.pestis | S.typhi | |
|---|---|---|---|---|
| Total no. of bacterial proteins (‘reviewed’ protein set from UniprotKB) | 2321 | 1086 | 4600 | 3592 |
| Total no. of human–bacteria protein pairs | 59.4 M | 27.8 M | 117.7 M | 87.7 M |
| No. of known interactions | 3073 | 1383 | 4059 | 62 |
| No. of interactions with no missing features | 655 | 491 | 839 | 62 |
| Size of training data with 1:100 class ratio | 66 155 | 49 591 | 84 739 | 6262 |
| No. of unique features in the training data | 69 4715 | 468 955 | 886 480 | 349 155 |
Note: Total no. of human proteins: 25 596; M, million. For each host–pathogen PPI dataset, the number of pathogen proteins, the size of the dataset and other such statistics are shown.
5.1 Feature set
For each protein pair, we compute features similar to the work in Kshirsagar et al. (2012). Some features use both proteins in the pair, while some others are based on either the host protein or the pathogen protein. While the features used for S.typhi were obtained directly from the authors, those for the other three datasets were derived from the following attributes of proteins available in public databases: protein sequences from Uniprot (UniProt Consortium, 2011), gene ontology from GO database (Ashburner et al., 2000), gene expression from GEO (Barrett et al., 2011), properties of human proteins in the human PPI network. Owing to the lack of space, we briefly mention only some of the prominent features here, and encourage the readers to refer to the supplementary for details. The sequence features count the frequency of amino acid–based n-grams or n-mers (for n = 2, 3, 4, 5) in the protein sequence. The GO features count the co-occurrence of host–pathogen GO term combinations. The human PPI network-based features compute various graph properties like node-degree, betweenness-centrality, clustering coefficient of the human protein.
Our features define a high-dimensional and sparse space (the number of features is listed in Table 1). Because our features are derived by integrating several databases, some of which are not complete, there are many examples and features with missing values. In our current work, we eliminate all examples with >10% missing features. For the rest, we use mean value–based feature imputation. Handling missing data effectively is an important aspect of the PPI prediction problem; however, it is not the focus of this work. The remaining examples after elimination and imputation are also shown in Table 1.
5.2 Negative class examples
The interactions listed in the table form the positive class. Because there is no experimental evidence about proteins that do not interact, we construct the ‘non-interacting’ (i.e. negative) class using a technique commonly used in PPI prediction literature. We use random pairs of proteins sampled from the set of all possible bacteria–human protein pairs. The number of random pairs chosen as the negative class is decided by what we expect the interaction ratio to be. We chose a ratio of 1:100 meaning that we expect 1 in every 100 random bacteria–human protein pairs to interact with each other. In general, there is no basis for choosing a more meaningful ratio, as there are few known interactions. We rely on previous work on better-studied organisms, where a ratio of 1:100 was used, based on the number of known interactions. Further, prior studies (Dyer et al., 2007; Tastan et al., 2009) also use a similar ratio. This random selection strategy is likely to introduce ∼1% false negatives into the training set.
5.3 Analyzing the known interactions
We analyze the known host–pathogen interactions from our datasets. This analysis also motivates our choice of a multitask approach that uses a pathway-based similarity across tasks. The known PPIs are compared across datasets in two ways: (i) pathway enrichment and (ii) presence of interologs.
The human proteins involved in each interaction dataset are used to obtain the human pathways that are enriched. We use Fisher’s test (based on the hypergeometric distribution) to compute the P-value of each pathway. We plot these P-values for each pathway, and for each dataset in the form of a heatmap shown in Figure 4. The heatmap shows how there are several commonly enriched pathways across the datasets (the black vertical lines spanning all four rows). It also shows the difference in the enrichment for the S.typhi dataset, which comes from small-scale PPI experiments.
We analyze the similarity between the PPIs from various datasets. A natural way to determine similarity is to check if proteins known to interact in one dataset have homologous proteins that are also interacting in another dataset. Such pairs of proteins, also called ‘interologs’, are defined as a quadruple of proteins
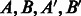 , where
, where 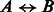 (interaction) and
(interaction) and 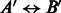 . Further, A,
. Further, A, 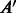 are homologs and B,
are homologs and B, 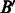 are also homologs. The number of such interologs existing between the four datasets is shown in Table 2. To compute homologs of a protein, we used BLASTP sequence alignment with an e-value cutoff of 0.1. As evident from Table 2, there are few interologs across the bacterial PPIs. None of the high-throughput datasets have an interolog in the small-scale S.typhi dataset. This seems to indicate that interolog-based approaches to compute task similarity are not relevant here. The phenomenon governing the similarity of these host–pathogen interactions is probably at a much higher level, rather than at the level of individual proteins. We explore one such possibility—the ‘commonality hypothesis’.
are also homologs. The number of such interologs existing between the four datasets is shown in Table 2. To compute homologs of a protein, we used BLASTP sequence alignment with an e-value cutoff of 0.1. As evident from Table 2, there are few interologs across the bacterial PPIs. None of the high-throughput datasets have an interolog in the small-scale S.typhi dataset. This seems to indicate that interolog-based approaches to compute task similarity are not relevant here. The phenomenon governing the similarity of these host–pathogen interactions is probably at a much higher level, rather than at the level of individual proteins. We explore one such possibility—the ‘commonality hypothesis’.
Fig. 4.
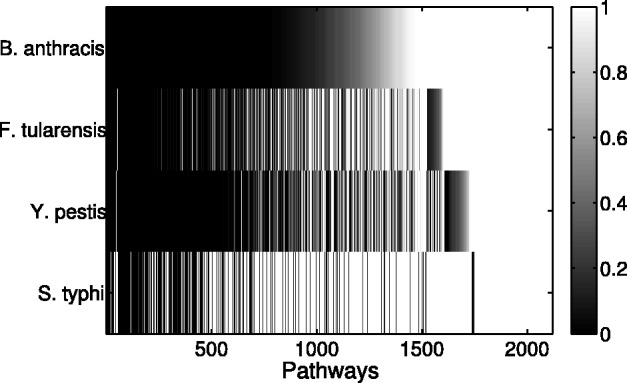
Heatmap showing pathways enriched in each bacterial–human PPI interactions dataset. The horizontal axis represents the pathways (about 2100 of them) and the vertical axis represents the four datasets. Each entry in the heatmap represents the P-value of a pathway w.r.t one dataset. Darker values represent more enrichment. The black columns that span across all four rows show the commonly enriched pathways
Table 2.
Conserved interactions in the form of interologs across the various host–bacterial datasets
| Human–bacteria PPI datasets compared | H-B versus H-F | H-B versus H-Y | H-B versus H-S | H-F versus H-Y | H-F versus H-S | H-Y versus H-S |
|---|---|---|---|---|---|---|
| Number of Interologs | 2 | 3 | 0 | 3 | 0 | 0 |
Note: H-X: stands for human–pathogen where the pathogen ‘X’ can be B, F, Y and S referring to B.anthracis, F.tularensis, Y.pestis and S.typhi., respectively. The non-zero entry ‘2’ for ‘H-B versus H-F’ means there are two PPIs in the H-B dataset that have interologs in the H-F dataset.
6 EXPERIMENTS
We use 10-fold cross validation (CV) to evaluate the performance of all algorithms. Our evaluation criteria do not use accuracy, which measures performance on both the classes. Because our datasets are highly imbalanced with a large number of negative samples, a naïve classifier that always says ‘no’ would still have a high accuracy. We instead use precision and recall computed on the interacting pairs (positive class) because they can deal with class imbalance.
 |
The baselines that we compare against are briefly described below.
Independent models (Indep.): We train models independently on each task using two standard classifiers: Support Vector Machines and Logistic regression with 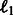 and
and 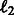 regularization. We used LibLinear (Fan et al., 2008) for these experiments and found that logistic regression with
regularization. We used LibLinear (Fan et al., 2008) for these experiments and found that logistic regression with  regularization performs the best across all tasks. For conciseness, we report only the best model’s performance.
regularization performs the best across all tasks. For conciseness, we report only the best model’s performance.
Coupled models: This baseline was implemented so as to couple the regularizer parameter across two tasks, thereby keeping the basic framework similar to that in our technique. To achieve this we optimize the function in Equation (9) and use the L-BFGS implementation from Mallet. Note that the previous baseline has separate regularization parameters for each task.
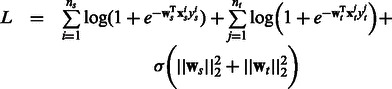 |
(9) |
Independent models with pathway features (Indep. Path.): This baseline incorporates the pathway information from the pathway vectors 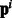 as features. For each example i, the feature vector is appended by the pathway vector
as features. For each example i, the feature vector is appended by the pathway vector 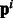 . While our method uses the pathway vectors only for the positive class examples (via the matrices Ps and Pt), this baseline uses the pathway information for all examples via features. The performance of this baseline will indicate whether using raw pathway information without incorporating any biologically relevant coupling does well. We learn independent models for each task as before, and find that logistic regression with
. While our method uses the pathway vectors only for the positive class examples (via the matrices Ps and Pt), this baseline uses the pathway information for all examples via features. The performance of this baseline will indicate whether using raw pathway information without incorporating any biologically relevant coupling does well. We learn independent models for each task as before, and find that logistic regression with 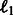 regularization does the best (only these results are reported).
regularization does the best (only these results are reported).
Mean Multi-task Learning (Mean MTL): This is a logistic regression-based implementation of the multitask SVM model proposed by (Evgeniou and Pontil, 2004). The important feature of this work is the use of a regularizer that penalizes the difference between a model and the ‘mean’ model formed by averaging over models from all m tasks. In the original paper, the loss functions 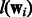 were all hinge loss. Because we find that logistic regression does better on our datasets, we replaced the original hinge loss function by logistic loss. The objective we use is shown in Equation (10).
were all hinge loss. Because we find that logistic regression does better on our datasets, we replaced the original hinge loss function by logistic loss. The objective we use is shown in Equation (10).
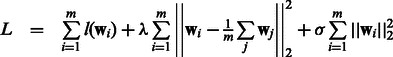 |
(10) |
Multitask pathway-based learning: This refers to our technique, which minimizes the sum of logistic loss over the two tasks with an 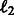 regularization penalizing the difference between the pathway summaries. We train two tasks at a time and compute the performance for each task. Because we have four tasks, there are six such pairwise learning experiments in all. While evaluating performance during 10-fold CV, we obtain the F1 on 1-fold of a task
regularization penalizing the difference between the pathway summaries. We train two tasks at a time and compute the performance for each task. Because we have four tasks, there are six such pairwise learning experiments in all. While evaluating performance during 10-fold CV, we obtain the F1 on 1-fold of a task 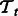 by averaging the F1 across all pairwise learning experiments that involve
by averaging the F1 across all pairwise learning experiments that involve 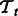 (see Section 4.3 for details). The final CV performance reported in our results is an average over 10-folds.
(see Section 4.3 for details). The final CV performance reported in our results is an average over 10-folds.
6.1 Parameter tuning
We followed an identical procedure for all algorithms. For the 10-fold CV experiments we train on 8-folds, use 1-fold as held-out and another as test. The optimal parameters (i.e. the best model) were obtained by parameter tuning on the held-out fold. The test fold was used to evaluate this best model—these results are reported in Section 7. The range of values we tried during the tuning of the regularization parameter (λ) were 150–10−4. For σ, the parameter controlling overfitting in multitask pathway–based learning (MTPL), we used a fixed value of 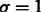 . For Mean MTL, we tune both λ and σ. To handle the high-class imbalance in our data, we used a weight-parameter Wpos to increase the weight of the positive examples in the logistic loss terms of our function. We tried three values and found
. For Mean MTL, we tune both λ and σ. To handle the high-class imbalance in our data, we used a weight-parameter Wpos to increase the weight of the positive examples in the logistic loss terms of our function. We tried three values and found 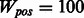 performed the best on training data.
performed the best on training data.
7 RESULTS AND DISCUSSION
7.1 Overall performance
Table 3 reports for each bacterial species, the average F1 along with the standard deviation for the 10-fold CV experiments. The performance of all baselines is similar, and our method outperforms the best of the baselines by a margin of 4 points for B.anthracis, 3.4 points for F.tularensis and 3.2 points for Y.pestis and 3.3 for S.typhi. The overall performance of all methods on this dataset is twice as good as that on the others. We believe that the difference in the nature of the datasets might explain the above observations. While the S.typhi dataset comprises small-scale interaction studies, the other datasets come from high-throughput experiments. Owing to its smaller size, it has less variance making it an easier task. This dataset is also likely to be a biased sample of interactions, as it comes from focussed studies targeting select proteins.
Table 3.
Averaged 10-fold CV performance for all methods for a positive:negative class ratio of 1:100
| Method | B.anthracis | F.tularensis | Y.pestis | S.typhi |
|---|---|---|---|---|
| Indep. | 27.8 ± 4 | 25.7 ± 5.4 | 28.8 ± 4 | 72.5 ± 11.4 |
| Coupled | 27 ± 3.9 | 25.5 ± 5 | 27.9 ± 3.4 | 69.8 ± 12.4 |
| Indep. Path. | 26.5 ± 4.7 | 26.1 ± 6.9 | 26.7 ± 4.3 | 69.1 ± 12.7 |
| Mean MTL | 25.2 ± 4.9 | 26.7 ± 4 | 27.5 ± 6.3 | 69.4 ± 12.1 |
| MTPL | 31.8 ± 3.9 | 30.1 ± 5.8 | 32.1 ± 2.5 | 75.8 ± 12.1 |
Note: Accuracy is reported as the F1 measure computed on the positive class. The standard deviation over the 10-folds is also reported. Bold values indicate the highest F1 value for each column (i.e. for that PPI dataset).
The coupled learner (Coupled) performs slightly worse than Indep. This is explained by the fact that Indep. has more flexibility in setting the regularization parameter for each task separately, which is not the case in Coupled. It is interesting to note that the independent models that use the pathway matrices Ps and Pt as features (i.e. Indep-Path) show a slightly worse performance than the Indep. models that do not use them. This seems to suggest that the cross-task pathway similarity structure that we enforce using our regularizer has more information than simply the pathway membership of proteins used as features. Precision-Recall curves: We also plot the P-R curves for MTPL. Please see the Supplementary Section 3.
7.2 Paired t-tests for statistical significance
Given two paired sets of k measured values, the paired t-test determines whether they differ from each other in a significant way. We compare MTPL with Indep.—the best baseline from the 10-fold CV results. Because the 10-fold CV results from the previous section give insufficient samples (i.e. only 10 samples), we instead use 50 bootstrap sampling experiments and use the results to compute the P-values. Each bootstrap sampling experiment consists of the following procedure: we first make two random splits of 80 and 20% of the data, such that the class ratio of 1:100 is maintained in both. The training set is then constructed using a bootstrap sample from the 80% split and the test data from the 20% split. A total of 50 models are thus trained and evaluated. We do not tune parameters again for each model and instead use the optimal setting of parameter values from our 10-fold CV experiments. The F1 is computed for each experiment thereby giving us 50 values, which will be our samples for the hypothesis test.
Because t-tests assume a normal distribution of the samples, we first did a normality test on each set of 50 F1 values. We performed the Shapiro–Wilk test with a significance level of 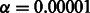 and found that our samples satisfy normality.
and found that our samples satisfy normality.
The averaged F1 over the 50 bootstrap experiments for the four tasks with MTPL and Indep. has been tabulated in the Supplementary Table S1. We observe that MTPL does better than Indep. for the three high-throughput datasets and marginally underperforms for the S.typhi. dataset. Table 4 shows the P-values on applying the paired t-tests to the 50 F1 values. For three of the four tasks, the performance improvement by MTPL is clearly statistically significant. For the fourth task, which involves S.typhi., the baseline has a slightly better averaged performance but the P-value does not indicate statistical significance. Hence we can say that the performance of MTPL and Indep. is similar for this task.
Table 4.
P-values from pairwise t-tests of statistical significance
Note: We compare MTPL with the best baseline ‘Indep.’, using results from 50 bootstrap sampling experiments. The null hypothesis is ‘there is no significant difference between the performance of MTPL and Indep.’. Null hypothesis: MTPL = Indep. aAlt. hypothesis: MTPL > Indep. bAlt. hypothesis: MTPL < Indep.
7.3 Pairwise performance of tasks in MTPL
The previous section gave a summary of the aggregated performance of MTPL for every task. Here we present the performance of every pairwise learning experiment of MTPL in Table 5. This gives an idea of how various tasks benefit from being paired up with other tasks. For each task, we check the task-pairing that gave the best performance (best F1 for each task is shown in bold). For instance, the best F1 of 32.3 for Y.pestis was obtained in the pairwise model learned with S.typhi. It is evident that coupling a model with one additional task seems to improve the performance over the baseline.
Table 5.
Pairwise model performance of MTPL
| Pairwise tasks | F1 |
|
|---|---|---|
| Task-1, Task-2 | Task-1 | Task-2 |
| B.anthracis, F.tularensis | 31.4 | 30.1 |
| B.anthracis, S.typhi | 32 | 76.3 |
| B. anthracis, Y.pestis | 31.6 | 32 |
| F.tularensis, S.typhi | 30.3 | 73 |
| F.tularensis, Y.pestis | 30 | 32.1 |
| S.typhi, Y.pestis | 74.2 | 32.3 |
Note: F1 computed during 10-fold CV of various pairwise models from MTPL. Positive: negative class ratio was 1:100. The best F1 achieved for each task (i.e. for each bacterial species) is shown in bold. For example, B.anthracis has the best performance of 32 when it is coupled with S.typhi.
7.4 Feature importance across tasks
To get an understanding of inter-task model similarity, we compared the parameter vectors ‘w’ of all tasks with each other (each w was learned on the entire training data). Because the number of features is large, we computed the cosine similarity between them. Note that we only use features that are common across tasks for this comparison. Gene expression features for instance were not used as they vary with regard to the number of expression time points, the experiment protocol, etc.
We found that the feature weights vary greatly across models—the cosine similarity ranges between 0.1 and 0.13. We also analyzed which features had the highest absolute weight. We found that the node-degree feature (computed using the human PPI graph) has a high positive weight across all tasks. Gene expression features have large negative weights across all tasks. In general, the GO and protein sequence–based n-gram features have different weights across tasks.
This seems to imply that having similar parameter values across models is not particularly important for this multitask problem. This explains why one of our baselines: the Mean MTL method, which penalizes differences between parameter vectors, does not perform well. Instead, regularization using the pathway summaries seems key in giving a better performance.
Sparsity of weights: We use 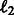 regularization in our optimization function, which does not produce sparse weight vectors. We observe that ∼50% of the features have 0 weight in all tasks. About 75–80% of the features have small weights in the range of (0.001 to −0.001).
regularization in our optimization function, which does not produce sparse weight vectors. We observe that ∼50% of the features have 0 weight in all tasks. About 75–80% of the features have small weights in the range of (0.001 to −0.001).
7.5 Analysis of predictions
The F1 measure gave us a quantitative idea of the performance of each method on training data. In this section, we present a qualitative analysis of the new interactions that our models predict. We first construct, for each task ‘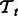 ’, a random set Rt of protein pairs that is disjoint from the training dataset. We train the pairwise models on the training data and obtain predictions on Rt. The method described in Section 4.3 is used to aggregate predictions from all pairwise models. The subset of Rt labeled as ‘positive’ is used for the analysis described below.
’, a random set Rt of protein pairs that is disjoint from the training dataset. We train the pairwise models on the training data and obtain predictions on Rt. The method described in Section 4.3 is used to aggregate predictions from all pairwise models. The subset of Rt labeled as ‘positive’ is used for the analysis described below.
Enriched human pathways
We perform enrichment analysis on the human pathways from the positive predictions of MTPL. We use Fisher’s exact test with the hypergeometric distribution. We intersect the top enriched pathways that satisfy P-value 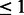 e-07 from each task to get the commonly enriched pathways. The sizes of the various intersections are shown in Figure 5. Seventeen pathways are commonly enriched across all four tasks. One hundred four pathways are enriched across the three high-throughput datasets, which is a significant fraction of the total number of pathways considered. This result indicates that the bias produced by our regularizer does produce predictions satisfying the commonality hypothesis.
e-07 from each task to get the commonly enriched pathways. The sizes of the various intersections are shown in Figure 5. Seventeen pathways are commonly enriched across all four tasks. One hundred four pathways are enriched across the three high-throughput datasets, which is a significant fraction of the total number of pathways considered. This result indicates that the bias produced by our regularizer does produce predictions satisfying the commonality hypothesis.
Fig. 5.

The intersection of enriched human pathways from predicted interactions. The total number of enriched pathways for each bacterial species are B.anthracis: 250, F.tularensis: 164, Y.pestis: 400 and S.typhi: 40. The size of the intersection between all tasks’ enriched pathways is 17. The size of this intersection for the high-throughput datasets (excluding S.typhi) is much larger: 104
Table 6 shows some of the common enriched pathways. The ‘Integrin alpha IIb beta3 (αIIb 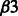 ) signaling’ pathway is enriched only in B.anthracis and Y.pestis in the training data. However, in the predictions it is enriched in all four bacterial datasets. Integrin-αIIb
) signaling’ pathway is enriched only in B.anthracis and Y.pestis in the training data. However, in the predictions it is enriched in all four bacterial datasets. Integrin-αIIb 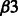 is a transmembrane receptor expressed in mast cells and plays an important role in innate immune responses against pathogens.
is a transmembrane receptor expressed in mast cells and plays an important role in innate immune responses against pathogens.
Table 6.
Five of the 17 commonly enriched pathways in the predicted interactions from MTPL
| Platelet activation, signaling and aggregation | |
| Integrin alpha IIb beta3 signaling | |
| Stabilization & expansion of E-cadherin adherens junction | |
| Post-translational regulation of adherens junction stability & disassembly | |
| Signaling by NGF |
We also analyze the overlap between the pathways enriched in the gold-standard positives and those enriched in the predictions. Please see the Supplementary Section 5 for details.
7.6 Incorporating other biological hypotheses
The regularizer in Equation (3) uses the pathway information matrix to enforce pathway-level similarity. The matrix can be used to represent any other common structure. For example, consider the hypothesis ‘all pathogens target hub proteins in the host’, which implies that bacterial proteins are often found to interact with host proteins that have a high node degree in the PPI network of the host. We tried two variants to incorporate this hypothesis—(i) we identify ‘hubs’ in the human PPI graph and use the binary vectors 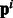 as an indicator of the ‘hub’ protein targeted by the bacterial protein, (ii) instead of a discrete ‘hub’/‘not hub’ indicator we use
as an indicator of the ‘hub’ protein targeted by the bacterial protein, (ii) instead of a discrete ‘hub’/‘not hub’ indicator we use 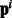 to represent the node degree [each component of
to represent the node degree [each component of 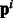 represents one node-degree bin say (10–20)]. We found that using (i) gives us an improvement of upto 2.5 F points over the baseline methods.
represents one node-degree bin say (10–20)]. We found that using (i) gives us an improvement of upto 2.5 F points over the baseline methods.
8 CONCLUSION
We presented a method that uses biological knowledge in jointly learning multiple PPI prediction tasks. Using a task regularization–based multitask learning technique, we were able to encode a biological hypothesis into the optimization framework effectively, thus enabling the commonality hypothesis to be tested. Our results indicate that the tasks benefit from this joint learning and we see an improvement of 4 F points over the baseline methods.
While our current results were presented on four bacterial species, we plan to extend our analysis to several other pathogens. Another direction to explore is the case where the pathogen is fixed and the hosts are different.
Our current approach integrates multiple tasks in a pairwise manner, which is inefficient because it does not scale well while integrating several PPI datasets. The most straightforward way of extending Equation (3) to learning m tasks simultaneously involves loss terms for each of the tasks and 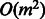 pairwise regularization terms, which unfortunately makes the optimization problem more complex and inefficient. A more promising and efficient direction would be to consider model selection at the task level where only the most relevant and useful tasks are used for multitask learning.
pairwise regularization terms, which unfortunately makes the optimization problem more complex and inefficient. A more promising and efficient direction would be to consider model selection at the task level where only the most relevant and useful tasks are used for multitask learning.
Supplementary Material
ACKNOWLEDGEMENT
We would like to thank Kevin Gimpel for a helpful discussion of DC programming and the reviewers for their excellent feedback.
Funding: The work has been supported in part by the Richard King Mellon Foundation, the EraSysBio+ grant funds from the European Union and BMBF to the Salmonella Host Interactions Project European Consortium, SHIPREC, as well as NIH grants P50GM082251 and 2RO1LM007994-05, and NSF grant CCF-1144281.
Conflict of Interest: none declared.
REFERENCES
- Ashburner M, et al. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, et al. NCBI GEO: archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011;39:D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liu M. Prediction of protein-protein interactions using random decision forest framework. Bioinformatics. 2005;21:4394–4400. doi: 10.1093/bioinformatics/bti721. [DOI] [PubMed] [Google Scholar]
- Chen KC, et al. Associations between HIV and human pathways revealed by protein-protein interactions and correlated gene expression profiles. PLoS One. 2012;7:e34240. doi: 10.1371/journal.pone.0034240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll T, et al. Pig–the pathogen interaction gateway. Nucleic Acids Res. 2009;37:D647–D650. doi: 10.1093/nar/gkn799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer M, et al. Computational prediction of host-pathogen protein-protein interactions. Bioinformatics. 2007;23:i159–i166. doi: 10.1093/bioinformatics/btm208. [DOI] [PubMed] [Google Scholar]
- Dyer M, et al. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008;4:e32. doi: 10.1371/journal.ppat.0040032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer M, et al. The human-bacterial pathogen protein interaction networks of Bacillus anthracis, Francisella tularensis, and Yersinia pestis. PLoS One. 2010;5:e12089. doi: 10.1371/journal.pone.0012089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evgeniou T, Pontil M. SIGKDD. New York: ACM; 2004. Regularized multi-task learning. [Google Scholar]
- Fan R, et al. Liblinear: a library for large linear classification. J. Mach. Learn. Res. 2008;9:1871–1874. [Google Scholar]
- Garcia J, et al. Biana: a software framework for compiling biological interactions and analyzing networks. BMC Bioinformatics. 2010;11:56. doi: 10.1186/1471-2105-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jubelin G, et al. Pathogenic bacteria target NEDD8-conjugated cullins to hijack host-cell signaling pathways. PLoS Pathog. 2010;6:e1001128. doi: 10.1371/journal.ppat.1001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kshirsagar M, et al. Techniques to cope with missing data in host-pathogen protein interaction prediction. Bioinformatics. 2012;28:i466–i472. doi: 10.1093/bioinformatics/bts375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Nanduri B. HPIDB–a unified resource for host-pathogen interactions. BMC Bioinformatics. 2010;11(Suppl. 6):S16. doi: 10.1186/1471-2105-11-S6-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews L, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37:D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar MS, et al. Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science. 2011;333:596–601. doi: 10.1126/science.1203659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, et al. Evaluation of different biological data and computational classification methods for use in protein interaction prediction. Proteins. 2006;63:490–500. doi: 10.1002/prot.20865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, et al. Systematic prediction of human membrane receptor interactions. Proteomics. 2009;23:5243–5255. doi: 10.1002/pmic.200900259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, et al. Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins. Bioinformatics. 2010;26:i645–i652. doi: 10.1093/bioinformatics/btq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer CF, et al. PID: the pathway interaction database. Nucleic Acids Res. 2009;37:D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleker S, et al. The current salmonella-host interactome. Proteomics Clin. Appl. 2012;6:117–133. doi: 10.1002/prca.201100083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AP, Gordon GJ. KDD. New York: ACM; 2008. Relational learning via collective matrix factorization. [Google Scholar]
- Singh R, et al. Struct2net: integrating structure into protein-protein interaction prediction. Pac. Symp. Biocomput. 2006:403–414. [PubMed] [Google Scholar]
- Tastan O, et al. Prediction of interactions between HIV-1 and human proteins by information integration. Pac. Symp. Biocomput. 2009:516–527. [PMC free article] [PubMed] [Google Scholar]
- Tekir SD, et al. Infection strategies of bacterial and viral pathogens through pathogen-host protein–protein interactions. Front. Microbiol. 2012;3:46. doi: 10.3389/fmicb.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Ongoing and future developments at the universal protein resource. Nucleic. Acids Res. 2011;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RS, et al. Analysis on multi-domain cooperation for predicting protein-protein interactions. BMC Bioinformatics. 2007;8:39. doi: 10.1186/1471-2105-8-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer C, et al. RECOMB. Berlin: Springer-Verilag; 2010. Leveraging sequence classification by taxonomy-based multitask learning. [Google Scholar]
- Winnenburg R, et al. Phi-base update: additions to the pathogen host interaction database. Nucleic Acids Res. 2008;36:D572–D576. doi: 10.1093/nar/gkm858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, et al. Prediction of yeast protein-protein interaction network: insights from the gene ontology and annotations. Nucleic Acids Res. 2006;34:2137–2150. doi: 10.1093/nar/gkl219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Yang Q. A survey of transfer and multitask learning in bioinformatics. J. Comput. Sci. Eng. 2011;5:257–268. [Google Scholar]
- Xu Q, et al. 2010 Protein-protein interaction prediction via collective matrix factorization. In: International Conference on Bioinformatics and Biomedicine. IEEE, Hong Kong. [Google Scholar]
- Yu J, Joachims T. International Conference on Machine Learning. New York: ACM; 2009. Learning structural SVMS with latent variables. [Google Scholar]
- Yuille A, Rangarajan A. The concave-convex procedure. Neural Comput. 2003;15:915–936. doi: 10.1162/08997660360581958. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.