

Abstract
The specificities of glycosaminoglycan (GAG) modification enzymes, particularly sulfotransferases, and the locations and concentrations of these enzymes in the Golgi apparatus give rise to the mature GAG polysaccharides that bind protein ligands. We studied the substrate specificities of sulfotransferases with a stable isotopically labeled donor substrate, 3′-phosphoadenosine-5′-phosphosulfate. The sulfate incorporated by in vitro sulfation using recombinant sulfotransferases was easily distinguished from those previously present on the GAG chains using mass spectrometry. The enrichment of the [M + 2] isotopic peak caused by 34S incorporation, and the [M + 2]/[M + 1] ratio, provided reliable and sensitive measures of the degree of in vitro sulfation. It was found that both CHST3 and CHST15 have higher activities at the non-reducing end (NRE) units of chondroitin sulfate, particularly those terminating with a GalNAc monosaccharide. In contrast, both NDST1 and HS6ST1 showed lower activities at the NRE of heparan sulfate (HS) chains than at the interior of the chain. Contrary to the traditional view of HS biosynthesis processes, NDST1 also showed activity on O-sulfated GlcNAc residues.
Keywords: chondroitin sulfate, glycosaminoglycan, heparan sulfate, mass spectrometry, sulfotransferase
Introduction
Proteoglycans and their associated glycosaminoglycan (GAG) chains play indispensable roles in animal physiology (Bishop et al. 2007). The biological functions of GAGs range from being major components of extracellular matrices, to creating gradients for storage and/or sequestration of chemokines, to binding with specific signal transduction molecules such as growth factors and growth factor receptors (Bernfield et al. 1999; Lander and Selleck 2000; Kreuger et al. 2001). In the latter case, the binding of GAGs are determined by both the bulk chain properties and domain structures that directly participate in protein binding. The mature structures of GAGs are highly heterogeneous and are dynamically regulated, both temporally and spatially (Turnbull et al. 2001; Shriver et al. 2002). A majority of past efforts to characterize GAG structure have focused primarily on the disaccharide distribution profiling and the compositional analysis of the oligosaccharides as depolymerized products of mature GAG chains isolated from tissues. Recently, efforts using tandem mass spectrometry (MS) of oligosaccharide have begun to shed light onto the positional details of the sulfates and C5 epimerization in GAG oligosaccharides (Zaia, Costello, et al. 2003; Zaia, Li, et al. 2003; Wolff, Amster, et al. 2007; Wolff, Chi, et al. 2007; Wolff et al. 2008a, b, 2010; Huang et al. 2011; Kailemia et al. 2012; Shi et al. 2012).
With the successful identification and cloning of GAG biosynthetic and modification enzymes, scientists have begun to deconvolute structure–function relationships in the context of the polydisperse structure of GAGs (Bulow and Hobert 2006). Thus, the availability of GAG biosynthetic enzymes makes it possible to determine substrate specificities and enzymatic efficiencies which are important because the number and positions of sulfate groups and iduronic acid residues on a GAG chain is directly related to its protein binding properties (Lindahl et al. 1998). There are over a dozen sulfotransferases involved in the multiple-step GAG synthesis and modification, and there is not likely to be a rigidly defined order of biosynthetic reactions. These reactions do not usually go to completion (Wu and Lech 2005) and it is conceivable that a GAG sample contains variant chains of intermediate sulfation degree that can serve as substrates for the sulfotransferases. Thus, we expected that substrates with a different sulfation site occupancy would be necessary in order to generate information about specificities and efficiencies of each of these enzymes. In addition, there is a particular need for isotopically labeled sulfur donor, 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to distinguish sulfates that have already been in the GAG chain substrates from those that are newly incorporated into the chains by the sulfotransferases.
Sulfur has four major natural stable isotopes: 32S (95.02%), 33S (0.75%), 34S (4.21%) and 36S (0.02%). 35S-Labeling facilitates quantification of a GAG sample as the radioactivity can be easily monitored. Unfortunately, 35S-labeling is not applicable to MS analysis for the concern of radioactivity contamination. In contrast, 34S-labeling is compatible with MS analysis, as it provides safe and discernible features (+2 Da shift relative to the most abundant naturally occurring isotope), which allowed us to further decipher the sequences surrounding the incorporation sites and the substrate specificity of a sulfotransferase (Wu et al. 2004).
MS assays have been used for analysis of sulfotransferase reactions. The Leary group has developed a liquid chromatography (LC)-MS strategy to determine enzyme activity and kinetics for a series of sulfotransferases, including tryosylprotein sulfotransferases, bacterial NodST and mycobacterial Stf0 using synthetic oligopeptides, chitobiose and trehalose as substrates (Yu et al. 2007; Danan et al. 2008, 2010; Hoffhines et al. 2009; Jen et al. 2009). These studies demonstrated that electrospray ionization (ESI)-MS or LC-ESI-MS is a tool effective for sensitive, fast and accurate kinetic measurements for sulfotransferase activity.
We investigated the modification of nascent and mature GAG chains by four recombinant sulfotransferases using PAP34S as the sulfur donor. These enzymes include chondroitin sulfate [chondroitin sulfate (CS)]-specific carbohydrate sulfotransferases 3 (CHST3) and 15 (CHST15), heparan sulfate (HS)-specific N-deacetylase/N-sulfotransferase 1 (NDST1) and HS 6-O-sulfotransferase 1 (HS6ST1). From LC-MS and LC-tandem mass spectrometry (MS/MS) analyses of depolymerized GAG chains, we confirmed the incorporation of 34S into the GAG substrates. This study revealed the substrate specificities of CHST3 and CHST15, which included the preference of transferring sulfate to the interior vs the non-reducing end (NRE) of CS chains. This study also revealed that NDST1 can sulfate previously O-sulfated GlcNAc residues of HS chains. Overall, this study proved that LC-MS/MS is a highly sensitive and informative tool to determine the number and position of sulfate incorporation on GAG chains.
Results and discussion
CS sulfotransferases
CHST3, also known as chondroitin 6-O-sulfotransferase, transfers sulfate to position 6 of GalNAc residues on chondroitin (Uchimura et al. 2002). CHST15, also known as N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase, transfers sulfate to the 6-O position of GalNAc4S on CSA, as well as dermatan sulfate, generating the CSE disaccharide unit GlcAβ13GalNAc4S,6S (Ohtake et al. 2001).
CS sulfotransferases act on chondroitin
Chondroitin, consisting of repeating units of (4GlcAβ1-3GalNAcβ1-)n, corresponds to nascent CS chains after initial chain elongation prior to modification by sulfotransferase and C5 epimerase enzymes. Although chondroitin proteoglycans have been identified in nematodes (Olson et al. 2006; Suzuki et al. 2006), unmodified chondroitin does not occur abundantly in mammalian tissue and can only be obtained by either complete chemical de-O-sulfation of CS or by defructosylation of K4 polysaccharides produced by Escherichia coli bacteria. We selected commercial chemically desulfated chondroitin as the substrate for CHST3. Following the sulfotransferase reaction, the sample was digested with chondroitinase ABC to profile the substrate chains. Figure 1A shows the extracted ion chromatograms (EICs) of the resultant disaccharides from these chains as a result of chondroitinase ABC digestion. There were four types of disaccharides observed, the unsulfated dp2 D0a0, its saturated counterpart U0a0, singly sulfated dp2 D0a4/6 and its saturated counterpart U0a4/6. The disaccharide code (Lawrence et al. 2008) is defined in the abbreviation list. It is apparent that the chemical desulfation was not complete, with the non-sulfated dp2 accounting for only 60% of the total disaccharides. The inset shows the mass spectra of D0a4/6 and U0a4/6 in their normal isotopic distribution. Although this commercially available chondroitin was not as pure and uniform as we had hoped (there were also methyl esters on the GlcA residues from the incomplete acid hydrolysis in the preparation of chondroitin, see Supplementary data, Figure S1), we proceeded to use it as the best substrate available for CHST3 assay.
Fig. 1.

EICs of chondroitin-saturated and Δ4,5-unsaturated disaccharides before (A) and after (B) CHST3 incubation and subsequent depolymerization. The insets show the mass spectra of non-sulfated (A) and singly sulfated (B) disaccharides. The Lawrence disaccharide nomenclature (Lawrence et al. 2008) was modified to indicate the ambiguous sulfation position, i.e. D0a4/6. The chromatographic peaks do not differentiate the contributions from 34S or 34S, these data are listed in Table I.
After the incubation of CHST3 with chondroitin, followed by chondroitinase ABC digestion, the relative abundances of the LC-MS profiles showed a pronounced increase in singly sulfated disaccharides that resulted from the sulfotransferase reaction. The mass spectra of the CHST3-acted upon, singly sulfated disaccharide D0a4/6 exhibited the enrichment of the peak at m/z 460 (inset of Figure 1B) relative to that observed before the sulfotransferase reaction (inset of Figure 1A), an indication of the incorporation of 34S into the chondroitin chains. In addition, the saturated disaccharides U0a0 and U0a4/6 were observed before and after CHST3 modification. The integration of the EIC peak areas of both the saturated and Δ4,5-unsaturated disaccharides generated the relative ratios of these species listed in Table I. From Table I (and visual inspection of Figure 1), the abundance ratio of unsulfated disaccharides U0a0/[D0a0 + U0a0], increased from 14.9 to 27.0% following CHST3 modification. In contrast, the ratio of monosulfated disaccharides U0a4/6/[U0a4/6 + D0a4/6] remained nearly constant, (9.1 vs 9.7% after CHST3 action). However, the ratio of 34S-U0a4/6/[34S-U0a4/6 + 34S-D0a4/6] after sulfotransferase treatment is only 1.8%, which suggests that the NRE structure GlcA-GalNAc is not a preferred substrate compared those in the interior of the chondroitin chains.
Table I.
Ratios of saturated and unsaturated disaccharides present in GAGs with and without sulfotransferase treatment
| Unsaturated | m/z | Saturated | m/z | Sat./total (%) |
|---|---|---|---|---|
| Chondroitin, control | ||||
| D0a0 | 378 | U0a0 | 396 | 14.9 |
| D0a4/6 | 458 | U0a4/6 | 476 | 9.1 |
| GalNac(S)* | 300 | 0.7 | ||
| Chondroitin, CHST3 treatment | ||||
| D0a0 | 378 | U0a0 | 396 | 27.0 |
| D0a4/6 | 458 | U0a4/6 | 476 | 9.7 |
| 34S-D0a4/6 | 460 | 34S-U0a4/6 | 478 | 1.8 |
| GalNAc(S/34S)* | 302 | 9.4 | ||
| CSA, control | ||||
| D0a4/6 | 458 | U0a4/6 | 476 | 0.35 |
| D0a10 | 538 | U0a10 | 556 | 29.3 |
| CSA, CHST15 treatment | ||||
| D0a4/6 | 458 | U0a4/6 | 476 | 0.0 |
| D0a10 | 538 | U0a10 | 556 | 28.9 |
| 34S-D0a10 | 540 | 34S-U0a10 | 558 | 0.38 |
| Heparosan, control | ||||
| D0A0 | 378 | U0A0 | 396 | 0.90 |
| Heparosan, NDST1 treatment | ||||
| D034S0 | 418 | U034S0 | 436 | 0.55 |
Since the NRE of CS chains can terminate at either a GlcA-GalNAc unit or a GalNAc-GlcA-GalNAc unit (Plaas et al. 1997), we investigated the possible addition of sulfates by CHST3 to the latter. Chondroitinase ABC digestion of chains terminating in a GalNAc-GlcA-GalNAc unit may generate a monosaccharide with the composition of GalNAc(±S). We did not observe GalNAc (m/z 220) before or after CHST3 treatment in our size-exclusion chromatography (SEC)-MS analysis. GalNAc does not carry an acid group and thus has a very small hydrodynamic volume, resulting in a very late SEC elution time, co-eluting with salts in the chromatogram. Thus, it was not detectable. However, we observed GalNAc4/6, m/z 300, before and after the action of CHST3, and GalNAc(34S), m/z 302, after CHST3 action. A ratio of the abundance of GalNAc4/6 to the sum of all NRE terminating units (U0a0, U0a4/6 and GalNAc4/6) represents the percentage of GalNAc-terminating NRE among total chains. From Table I, the percentage of GalNAc4/6-terminating NRE increased from 0.7 to 9.4% (note since GalNAc itself was not detected in this chromatography, the actual GalNAc4/6 terminating unit can be higher than 0.7 or 9.4%). Supplementary data, Figure S2 shows that the increased GalNAc4/6 abundance was indeed caused by the incorporation of 34S into GalNAc, as evidenced by the enrichment of the peak at m/z 302 in the isotopic pattern. To summarize, the enrichment of 34S-GalNAc4/6 and extremely less abundance of 34S-U0a4/6 as a result of CHST3 action suggest that this sulfotransferase has a preference of GalNAc (NRE) > GlcA-GalNAc (interior) > GlcA-GalNAc (NRE) in these chondroitin chains in vitro.
We performed tandem MS experiments in the collision-induced dissociation (CID) mode for m/z 460 (34S-D0a4/6), together with seven mock mixtures of different ratios of D0a4 and D0a6. The characteristic ion for D0a4 is an Y1 ion at m/z 300 and that for D0a6 is a Z1 ion at m/z 282. The abundance ratio of Y1/[Y1 + Z1] plotted against that for D0a4/[D0a4 + D0a6] showed a straight line for seven artificial mixtures (Supplementary data, Figure S3). The fragmentation of m/z 460 (34S-D0a4/6) produced a Z1 ion that contains a 34S (m/z 284) indicates the addition of 34S occurred exclusively at the 6-O position of GalNAc residues (Supplementary data, Figure S4).
For CHST15, we used a commercial CSA as the substrate. First, we digested CSA using chondroitinase ABC and confirmed that the vast majority of the disaccharide units are singly sulfated (Figure 2A). The tandem MS experiments also showed that the singly sulfated disaccharides are D0a4 (Supplementary data, Figure S5). After treatment with CHST15, the majority of the singly sulfated disaccharides (m/z 458) were converted to doubly sulfated disaccharides eluting at 92 min (Figure 2B). The inset shows the major doubly sulfated dp2 is the product of the incorporation of 34S, at m/z 540. The positional specificity of the CHST15 was determined by tandem mass spectra of 34S-D0a10 at m/z 540. A small isolation window was selected to exclude any possible m/z 538 from D0a10. The most abundant product ion was m/z 458. An ion at m/z 460 was observed at ∼10% abundance relative to m/z 458, suggesting that the 34S that had been added by CHST15 was more easily dissociated during CID. When gradually increasing the collisional energy in a time-of-flight mass spectrometer, the product ion m/z 458 continued to fragment. As shown in Supplementary data, Figure S6, the dissociation of m/z 458 generated predominantly m/z of 300, which is the characteristic ion of D0a4. The characteristic ions of D0a6, m/z 282, and D2a0, m/z 237, were observed only at very low abundances. Therefore, it can be concluded that CHST15 added sulfate only to G0a4 units in the CSA chains.
Fig. 2.

The EICs of CSA-saturated and Δ4,5-unsaturated disaccharides before (A) and after (B) CHST15 treatment and subsequent depolymerization using chondroitinase ABC. The inset shows the mass spectrum of doubly sulfated disaccharides.
The saturated forms of these disaccharides were also observed, and their relative distributions are listed in Table I. After CHST15 action, the saturated singly sulfated dp2s, U0a4/6, were depleted, indicating that the vast majority were converted to 34S-U0a10. The ratio 34S-U0a10/[34S-U0a10 + 34S-D0a10] (0.38%) showed no significant difference from U0a4/6/[U0a4/6 + D0a4/6] (0.35%) for control CSA. Therefore, CHST15 has almost the same preference at the NRE G0a4 units compared with those in the interior of the chains for the CSA we tested. For this particular CSA substrate, however, signals corresponding to odd-number oligosaccharides were insufficient to determine the activity of CHST15 on the GalNAc-terminating NRE.
In summary, the incorporation of 34S by sulfotransferases CHST3 and CHST15 to the nascent or homogeneous substrate polysaccharides was observed and the preference of the incorporation was analyzed at the disaccharide level by MS.
CS sulfotransferases acting on mature CS chains
To date, many of the details on how different biosynthetic enzymes, including those involved in chain initiation, elongation and modification (epimerization and sulfation), are expressed, localized and organized in the Golgi remain unclear; however, it is commonly accepted that these enzymes work in a competing manner with each other, and the chain modifications rarely go to completion (Silbert 1978; Sugumaran and Silbert 1989; Fransson et al. 2000; Tiedemann et al. 2001; Wu and Lech 2005). Although the analyses in the previous sections using nascent or homogeneous GAG chains as substrates offer a useful readout for activity assays of GAG sulfotransferases, they do not reflect the complexity of biosynthesis in vivo during which many enzymes appear to act on a chain simultaneously. Therefore, highly heterogeneous, mature GAG chains may be better substrates for us to understand the complex process of CS biosynthesis. Mature GAG chains contain domains with varying degrees of modification and may contain different accepting sites for individual sulfotransferases. These potential benefits are offset by the expectation that the maturity of the chains will likely limit the degree of sulfate incorporation, thus demanding a very sensitive detection method. Fortunately, 34S-labeling and MS allow unambiguous identification of the number and location of the newly incorporated sulfate. Given these rationales, we chose a commercial mature and heterogeneous CS from bovine cartilage to study patterns of CHST3 and CHST15 modification.
Disaccharide analysis
Figure 3A shows the distribution of disaccharides with zero, one or two sulfates after chondroitinase ABC digestion of CS with and without CHST3 and CHST15 treatments. From the profiling in Figure 3A, it is evident that after CHST15 treatment, the percentages of singly sulfated disaccharides D0a4/6 decreased slightly, while the doubly sulfated disaccharide D0a10 increased slightly, suggesting that CHST15 installed one more sulfate to singly sulfated disaccharides. However, the profile changes for CHST3 appeared to be minor and in some cases, inconclusive. This is partially because the CS sample was recalcitrant to chondroitinase ABC digestion. Even after multiple additions of chondroitinase ABC and prolonged incubation time, larger oligosaccharides, particularly disulfated tetrasaccharides, remained. Although a complete glycomics survey, including larger oligosaccharides, will be very informative, the lack of standards for the unusual oligosaccharides makes quantification of these species impractical. In this study, by tracking the changes in the isotopic pattern of disaccharides, we measured the degree of sulfate incorporation and identified the exact location of the newly incorporated sulfate on CS chains.
Fig. 3.

(A) The relative distributions of disaccharides for native and CHST3- and CHST15-treated CS samples. (B) [M + 2]/[M + 1] ratio of monosulfated disaccharides, D0a4/6 m/z 458. (C) The [M + 2]/[M + 1] ratio of disulfated disaccharides, D0a10 m/z 538. Error bars reflect the standard deviations of triplicate LC-MS runs.
The incorporation of 34S resulted in increased abundances of the [M + 2] isotopic peak of the product ion species but not the [M + 1] isotopic peak; therefore, the ratio of the integrated peak areas for EICs for [M + 2] and [M + 1] reflects the degree of 34S incorporation. The [M + 2]/[M + 1] ratio for monosulfated disaccharide D0a4/6 for non-treated as well as CHST3 and CHST15 treated CS are shown in Figure 3B. The apparent distribution of M, [M + 1] and [M + 2] in EIC peak areas, as well as in actual spectra, is shown in Supplementary data, Figure S7. The fact that the ratio of [M + 2]/[M + 1] in untreated samples agrees with the theoretical value suggests that this is a precise, accurate and sensitive indication of the degree of the addition of 34S into the GAG chains. CHST3 treatment increased the [M + 2]/[M + 1] ratio for D0a4/6, suggesting the incorporation of 34S to these disaccharides. However, CHST3 did not change the [M + 2]/[M + 1] ratio for the doubly sulfated D0a10, suggesting that CHST3 was not active on units of GlcA-GalNAc4S/6S in the polysaccharide. In contrast, CHST15 treatment changed the [M + 2]/[M + 1] ratio for D0a10 but not for D0a4/6, suggesting that CHST15 installed one 34SO3 to the GlcA-GalNAc4S/6S but not GlcA-GalNAc units in the polysaccharide. From the isotopic pattern changes, 2.6% of the D0A4/6 in the CHST3-treated sample contained incorporated 34S, and 54% of the D0a10 in the CHST15-treated sample contains incorporated 34S.
To identify the exact position of the newly incorporated 34S in the complex chain context, we performed tandem MS experiments in the CID mode for m/z 458–460 in the same manner described for Supplementary data, Figure S3. As shown in Supplementary data Figure S4, the intercept and slope of the response curve between Y1/[Y1 + Z1] and D0a4/[D0a4 + D0a6], the percentage of D0a4 in total singly sulfated disaccharide units was calculated. This method assumes that D2a0 is insignificant in abundance among the singly sulfated Δ4,5-disaccharides. Standing alone, U2a0 is usually rare because the 2-O-sulfation frequently occurs next to GalNAc with 4-O- or 6-O-sulfate. This was further verified by the fact that for D2a0, a B1 ion at m/z 237 was not observed in the untreated CS samples (Supplementary data, Figure S8). Therefore, the binary system of D0a4 and D0a6 is a valid approximation for the compositional analysis of singly sulfated dp2 for this particular CS sample.
Since we established that the Y1 (m/z 300) and Z1 (m/z 282) ions were characteristic of D0a4 and D0a6, respectively, the examination of the isotopic pattern of the product ions, specifically, the enrichment of the 2 Da peak as a result of incorporation of 34SO3, revealed the positional specificity of these two sulfotransferases. From Table II, it is clear that CHST3 produced elevated [M + 2] abundance for the Z1 ion, but not for the Y1 ion. In contrast, CHST15 did not affect this pattern for Y1 and Z1 ions of singly sulfated m/z 458. This suggests that only CHST3 added 34S specifically to the 6-O position of GlcA-GalNAc units in the polysaccharide. This conclusion was further supported by examining the percentage of D0a4 among all singly sulfated disaccharides, as shown in Table II. This percentage was 62.3% for untreated CS samples. After CHST3 treatment, this percentage decreased to 60.8%, as this enzyme converted some GlcA-GalNAc to GlcA-GalNAc6S, thus decreasing the proportion of D0a4 in the subsequent chondroitinase digested mixture. A total of 2.4% of singly sulfated dp2s contained 34SO3, in good agreement with the 2.6% obtained from the calculation of the degree of +2 Da enrichment.
Table II.
Percentage of 4-O-sulfated dp2 (D0a4) among all singly sulfated dp2s as determined by tandem MS
| D0a4/(D0a4 + D0a6) | [A + 2]/[A + 1]a (Z1, m/z 282) | [A + 2]/[A + 1]a (Y1, m/z 300) | |
|---|---|---|---|
| Native | 62.3% | 0.65 | 0.66 |
| CHST3 | 60.8% | 1.26 | 0.68 |
| CHST15 | 60.7% | 0.67 | 0.67 |
a[M + 2]/[M + 1] abundance ratios for product ions of singly sulfated dp2s.
As shown in Figure 3C, CHST15 treatment significantly enriched the +2 Da peak of D0a10, but not D0a4/6, indicating that CHST15 only added one extra sulfate to singly sulfated disaccharides. After CHST15 treatment, the percentage of 4-O-sulfation decreased to 60.7% (Table II). This was due to the conversion of some of the GlcA-GalNAc4S to GlcA-GalNAc(4S,634S), which resulted in the increase in disulfated disaccharides by more than 10-fold.
Oligosaccharide analysis
Unlike homogeneous CSA chains, even after extensive chondroitinase ABC digestion, mature and complex CS samples still contained significant amounts of oligosaccharides other than disaccharides. Similar to that used for HS oligosaccharides, we used a compositional coding with five numerals to represent the numbers of [ΔHexA, HexA, GalN, Ac and SO3] for CS oligosaccharides. For CS before and after CHST15 treatment, we identified and quantified oligosaccharide compositions of [1,1,2,2,X], [0,2,3,3,X], [0,1,2,2,X], [0,0,1,1,X] and [1,1,1,1,X], where X denotes varying SO3 numbers. Among them, [1,1,2,2,X] are tetrasaccharides originated from the interior of the chains. Subsequent digestion with chondroitinase results in a trisaccharide. A series of odd-numbered compositions [0,2,3,3,X], [0,1,2,2,X] and [0,0,1,1,X] derived either from GalNAc-terminating NRE of the original CS chain or from the NRE of a chain that has been undergone endoglucuronidase cleavage (Figure 4). These compositions, absent or very low in abundance in chondroitinase digestions of the more uniform chondroitin and CSA, provided a series of substrates from which we can examine the different specificities of sulfotransferases. It should be noted that the absolute quantities of all these oligosaccharides studied showed only slight changes from control to CHST3- and CHST15-treated samples. Moreover, disaccharide analyses showed that the incorporation of the sulfate into these mature CS chains was limited to only a few percent. In addition, the patterns of lyase digestion were not changed by modification by these sulfotransferases.
Fig. 4.

Suggested mechanisms whereby the observed odd-number oligosaccharides in CS were formed.
The relative abundances of the tetrasaccharide series, [1,1,2,2,X], with X equal to 0, 1, 2 and 3 sulfates, were normalized to the sum of the four and are shown in Supplementary data, Figure S9. Although the non-sulfated tetrasaccharide remains almost unchanged, the relative abundances of singly sulfated tetrasaccharide decreased and the doubly sulfated tetrasaccharide increased after CHST3 treatment. This implies that CHST3 preferred to act on a non-sulfated G0a0 adjacent to a sulfated dp2 unit over an unsulfated tetrasaccharide unit. In comparison, after CHST15 treatment, both [1,1,2,2,0] and [1,1,2,2,1] remained unchanged, whereas [1,1,2,2,2] decreased with a comparable increase in [1,1,2,2,3]. This implies that CHST15 preferred a G0a4 as a substrate adjacent to another sulfated dp2 unit. The absence of the tetrasaccharide with four sulfates also suggests that CHST15 did not add sulfate groups to triply sulfated tetrasaccharide to form repeating GlcA-GalNAc4S6S units. The mass spectra in Supplementary data, Figure S10, showed a pronounced enrichment in the [M + 2] isotope of the monoisotopic peak of triply sulfated [1,1,2,2,3], which confirms that the stoichiometric changes of this species was indeed caused by the incorporation of the heavy sulfate resulting from enzyme activity. More definitive understanding on the specificities of these enzymes on local structures will require structurally defined oligosaccharides standards.
The odd-number dp1, dp3 and dp5 were partially preserved after chondroitinase ABC digestion for control, CHST3 and CHST15 CS-treated samples. Their relative distributions from all contributing ionic species of different charge states within the same size are shown in Figure 5. For [0,2,3,3,X], a pentasaccharide with backbone of GalNAc-GlcA-GalNAc-GlcA-GalNAc and 2–4 sulfates, CHST3 treatment modestly increased the abundances of [0,2,3,3,3] and decreased the abundances of [0,2,3,3,2]. In comparison, more than 20% of [0,2,3,3,3] was converted to [0,2,3,3,4] by CHST15. This means that CHST15 was highly active on this pentasaccharide at the NRE structures that terminate with a GalNAc. This similar pattern was observed for trisaccharide series [0,1,2,2,X] (Figure 5), which suggests that the sulfate was incorporated to the first or the second GalNAc at the NRE. The observation of the monosaccharide [0,0,1,1,X] series, the decrease in [0,0,1,1,1] and the concomitant increase in [0,0,1,1,2] after CHST15 treatment continue to argue that CHST15 was able to the add a sulfate to the first GalNAc of the NREs of CS chains. In all cases, the stoichiometric changes caused by CHST3 were far less pronounced than CHST15. The mass spectra of the most heavily sulfated species in all series confirmed the incorporation of the 34S (Supplementary data, Figure S11–S13). In summary, both sulfotransferases, to a greater degree CHST15, had higher activities for GalNAc-terminating NRE sites than interior sites in CS chains (up to 20% at NRE sites vs ∼2.5% in the interior sites). In addition, the conservation of the activity of CHST15 on dp1, a single GalNAc unit, and the lack of more than one sulfate added to dp3 and dp5, imply that the most preferred site for CHST15 was at the NRE GalNAc residue.
Fig. 5.
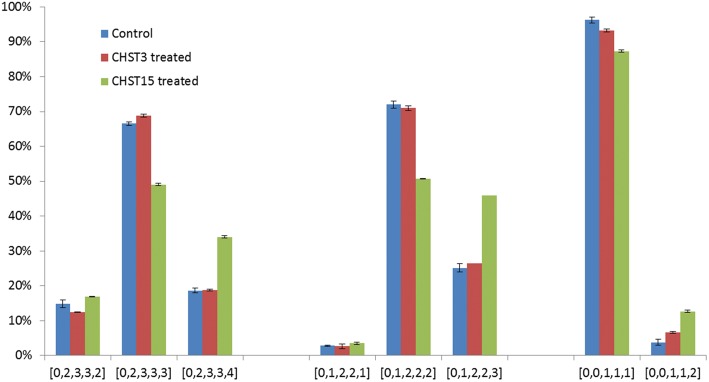
The distribution of dp5, dp3 and dp1 after chondrotinase ABC digestion for control and CHST3- and CHST15-treated CS samples. The percentages are normalized to the sum of abundances of saccharides of the same size. Error bars reflect the standard deviations of triplicate LC-MS runs. The oligosaccharide compositions are given as [ΔHexA, HexA, GalN, Ac, SO3].
A dp3, [1,1,1,1,1] (m/z: 634.2−), can be assigned to a digestion product of an endoglucuronidase and chondroitinase ABC. The digestion of CS chains by an endoglucuronidase produces saccharides with GlcA at the reducing end. Subsequent digestion with chondroitinase on such oligosaccharides will generate oligosaccharides with unsaturated uronic acid at NRE. The shortest oligosaccharides with NRE unsaturated uronic acid and a reducing end GlcA residue is a trisaccharide with composition [1,1,1,1,1] (ΔUA-GalNAc-GlcA plus sulfate). Trisaccharide with composition [1,1,1,1,1], m/z 634.2, was indeed observed. Tandem MS of this oligosaccharide showed the existence of both Y1 and Z1 ions of GalNAc(S). However, no addition of 34S to this oligosaccharide to form [1,1,1,1,2] (m/z 716.2) by CHST15 was observed. In addition, the ratio for abundances of [1,1,1,1,0]/[1,1,1,1,1] remained unchanged after CHST3 treatment, suggesting that CHST3 does not act on GlcA-GalNAc-GlcA.
The high activity of CHST15 toward NRE GalNAc residues on CS chains is of great interest. It has been shown that highly sulfated saccharides could be involved in the termination of the CS/DS chain synthesis (Midura et al. 1995). Ohtake et al. (2003) identified a structure of GalNAc4S-GlcA2S-GalNAc6S serves as a substrate for CHST15 to form GalNAc4S6S-GlcA2S-GalNAc6S. Therefore, the expression level of CHST15 plays a role in determining the type of the monosaccharide at the NRE (GlcA vs GalNAc) and the length of the chain. Moreover, Sugahara et al. have demonstrated that the E units in the CS/DS chain and/or the CHST15 level increases with malignant transformation in human lung carcinoma and murine osteosarcoma (Basappa et al. 2009). They suggested that mimics of CS/DS units have potential as anti-cancer agents. Tollefsen et al. have shown that 4,6-sulfated Dermatan sulfate (DS) have heparin co-factor II binding and anticoagulant activity (Halldorsdottir et al. 2006; He et al. 2008). DS treatment was found to inhibit thrombotic and inflammatory processes after arterial injury in an animal model (Godoy et al. 2011). Thus, our results, by increasing the understanding the specificities of these sulfotransferases, will inform the chemoenzymatic synthesis of such compounds for therapeutic uses.
HS sulfotransferases
We performed similar experiments for two HS sulfotransferases, NDST1 and HS6ST1. NDST1 is a dual-function enzyme that removes the N-acetyl group and replaces it with a sulfate group. HS6ST1 adds a sulfate to the 6-O position of GlcNS or GlcNAc residues. Both heparosan from E. coli K5 polysaccharide that is devoid of sulfation and HS from porcine intestinal mucosa were used as acceptor substrates to examine the activity of NDST1.
HS sulfotransferases act on their “natural” substrates
A sample of heparosan was first depolymerized by a combination of heparin lyase I, II and III and analyzed by MS. The digestion products contained 99.1% unsaturated disaccharide D0A0 and 0.9% saturated disaccharide U0A0 (Table I). This result suggests that an average K5 polysaccharide chain contained 110 disaccharides, which is longer than a typical mature HS chain of around 50 disaccharides. Another aliquot of heparosan was first modified with NDST1 using PAP34S as the sulfate donor. To keep the reaction under optimal conditions (pH 6.5, in the presence of 9 mM MnCl2), only 1/13 equivalent of PAP34S was used in the experiment. The modified sample was then depolymerized and analyzed the same way as for unmodified heparosan. Significant amounts of singly sulfated disaccharide were observed (Supplementary data, Figure S14). When modification was performed at pH 8.0, NDST1 activity substantially decreased (Supplementary data, Figure S15). The intermediate product D0H0 may be present in digested samples, but was not detected, as it is a zwitterion and elutes at the same time with the bulk buffer salts in SEC chromatogram. In summary, PAP34S and heparosan can be utilized for studying NDST1 activity in a way similar to that for studying CS-specific sulfotransferases.
HS sulfotransferases act on mature HS chains
Supplementary data, Figure S16 shows the disaccharide profiles for unmodified and NDST1 and HS6ST1 modified porcine intestinal HS samples. There are some general trends in the disaccharide glycomics profiling, e.g. the increased abundances of GlcNS-containing dp2s after NDST1 treatment. This approach alone, however, did not have an acceptable mass balance because the action of the NDST1 significantly changes the susceptibility of the chain to heparin lyase digestion. It also did not provide details of the position of the newly incorporated sulfate.
D034S0 and its saturated counterpart, U034S0, are products of NDST1 acting on GlcA-GlcNAc units. Figure 6 shows the abundance ratio of [M + 2]/[M + 1] in native and NDST1- and HS6ST1-treated samples. The increased [M + 2]/[M + 1] ratio for D0S0 after NDST1 treatment indicated the incorporation of 34S into the chain (∼6% of D0S0 contained 34S). In comparison, the enrichment of [M + 2] peak of the NRE U0S0 was not as pronounced as that of D0S0, suggesting that NDST1 by itself prefers interior sites of the chain over the NRE sites, and other NDSTs or cofactors may be required for efficient N-sulfation of NRE disaccharide units. Compared with theoretical values, the elevated [M + 2]/[M + 1] ratio of U0S0 for native samples is due to the inclusion of [M + 1] isotopic peak of the ammonia adduct of D0S0 (+17 D), which is indistinguishable from the U0S0 [M + 2] peak. It has been shown that U0S0 is highly expressed at the NRE of HS chains (Staples, Shi, et al. 2010). The HS6ST1-treated sample exhibited the same [M + 2]/[M + 1] ratio for D0S0 and U0S0 as the native sample, as expected.
Fig. 6.

The [M + 2]/[M + 1] ratio of D0S0 and U0S0 for control and NDST1- and HS6OST1-treated HS samples. Error bars reflect the standard deviations of triplicate LC-MS runs.
Figure 7 shows [M + 2]/[M + 1] for doubly sulfated dp2, including both D2S0 and D0S6 (m/z 496) and their saturated counterparts, U2S0 and U0S6 (m/z 514). After HS6ST1 treatment, there was a visible [M + 2] peak enrichment for m/z 496 but very little for m/z 514, indicating that HS6ST1 preferred interior sites rather than NRE sites. Tandem MS experiments were carried out for the binary mixture of m/z 496 which included D2S0 and D0S6. As shown in Supplementary data, Figures S17 and S18, the increased relative abundance of D0S6 after HS6ST1 treatment confirmed the addition of a sulfate to the 6-O position of D0S0. Interestingly, the NDST1-treated sample also showed a slight, but statistically significant, increase of m/z 496. This observation is consistent with the conclusion that NDST1 converted D0A6 to D0S6 and/or D2A0 to D2S0, the former being more likely as D0A6 was the dominant species (greater than 95%) relative to its isomer D2A0 in reported mammalian HS compositions (Shi and Zaia 2009). Although N-de-acetylation/N-sulfation is widely considered to be the first step in HS chain modification, the observation that NDST1 acted on D0A6/D2A0 in this in vitro experiment suggests that there is a possibility for NDST1 acting on O-sulfated intermediate chains.
Fig. 7.

The [M + 2]/[M + 1] ratio of doubly sulfated disaccharides, D2S0/D0S6 and U2S0/U0S6, for control, and NDST1- and HS6OST1-treated HS samples. Error bars reflect the standard deviations of triplicate LC-MS runs.
In addition to acting on D0S0, HS6ST1 added one sulfate to the 6-O position of D2S0 to form D2S6. This activity is shown in Figure 8. The [M + 2]/[M + 1] ratio of D2S6, m/z 576, showed a significant increase after HS6ST1 treatment, whereas native and NDST1 treatment samples remained unchanged and were similar to theoretical values. The enzyme activity on NRE U2S0 was not conclusive, due to poor quantitation at m/z 594 because of (i) the less abundances of U2S6 and (ii) the complicating of a more significant ammonia-adducted form of D2S6 (+17 D).
Fig. 8.

The [M + 2]/[M + 1] ratio of triply sulfated disaccharides, D2S6, for control and NDST1- and HS6OST-treated HS samples. Error bars reflect the standard deviations of triplicate LC-MS runs.
With the treatment of the combination of heparin lyases I, II and III, these HS samples did not produce abundant signals from oligosaccharides. Therefore, it was not clear from these experiments how these enzymes acted on larger oligosaccharides of HS. We also digested HS samples with lyase III only and collected the oligosaccharide fractions by size-exclusion chromatography. These fractions were then subjected to hydrophilic interaction chromatography (HILIC)-MS profiling (Staples et al. 2009; Staples, Naimy, et al. 2010). The enrichment of the [M + 2] peaks of the oligosaccharides ions was much less distinct than those of disaccharides, primarily because oligosaccharides resulting from the addition of one or more sulfate groups were very low in abundance relative to pre-existing oligosaccharides with the same number of sulfate groups. In addition, lyase III seems to be more sensitive to the structural changes within the HS chain, making comparison between sulfotransferase treated and untreated HS samples difficult. We therefore expect that informative and accurate studies of the specificity of HS sulfotransferases will require structurally defined oligosaccharide substrates.
Summary
We used LC-MS methods to quantify the incorporation of 34S into nascent and/or uniform GAG chains by recombinant CHST3, CHST5, NDST1 and HS6ST1 and elucidate their substrate preferences at the disaccharide level. We used the stable isotope ratio of [M + 2]/[M + 1] as a sensitive index of 34S incorporation. By analyzing the mass spectral data on disaccharide and oligosaccharide profiling, we showed that these sulfotransferases have different preferences for interior sites and NRE sites on GAG chains. We also showed that the exceptional capability of tandem MS to unambiguously identify the locations of the newly incorporated 34S in very complex GAG chains.
Our results showed that both CHST3 and CHST15 had high activity for GalNAc at the NRE of CS chains. These results are consistent with CHST15 participating in formation of 4,6-sulfated GalNAc at the NRE of CS chains. Such NRE saccharides are thought to be involved in chain termination during biosynthesis (Plaas et al. 1998).
The mature structures of HS chains result from the interplay of biosynthetic enzyme activities, resulting in regulated diversity (Lindahl et al. 1998). The knowledge of the activities and specificities of these enzymes, in different isoforms, on different substrates is necessary for us to understand how specific protein binding epitopes and bulk chain properties are generated. Finally, we reported that NDST1 added sulfate to singly O-sulfated disaccharide units on substrate HS chains, suggesting that O-sulfation can precede N-sulfation on the HS synthesis in some circumstances. We plan to apply the same strategy to oligosaccharides to elucidate with greater specificity the activities and specifies of GAG sulfotransferases. We foresee that this method will greatly aid efforts to synthesize defined GAG saccharides using recombinant enzymes.
Materials and methods
Nomenclature for GAG disaccharides
The CS and HS disaccharide coding used is according to a method established by Lawrence et al. (2008). Detailed codes are given in the abbreviations. Oligosaccharide compositions for both CS and HS oligosaccharides are given as [ΔHexA, HexA, HeXNAc, Acetate, sulfate].
Materials
Sulfotransferases, including CHST3, CHST15, NDST1, HS6ST1, PAPS and PAP34S, were from R&D Systems (Minneapolis, MN). HS from porcine intestinal mucosa was from Celsus Labs (Cincinnati, OH). CS and chondroitinase ABC were from Sigma-Aldrich (St Luis, MO). Chondroitin and CSA were from Seikagaku (Tokyo, Japan). CS from bovine cartilage was purchased from Sigma-Aldrich. Heparosan was from Iduron (Manchester, UK). Heparan lyases I, II and III were from IBEX Technologies (Montreal, Canada).
Sulfotransferase reactions
Chondroitin (4.3 µg) was mixed in Tris–HCl buffer (50 mM, pH 7.45) in the presence of MgCl2 (6.7 mM), PAP34S (4.0 µg) and CST-3 (0.5 µg). The reaction mixture (total volume of 30 µL) was incubated at 37°C for overnight. After that, 3 µL of 100 mM ammonia acetate and 10 mU chondroitinase ABC in 5 µL were added to the mixture and digestions were continued for 2 more hours. For a negative control, an untreated chondroitin sample was also subjected to s chondroitinase ABC digestion under same condition.
CSA (4.0 µg) was mixed in 2-(N-morpholino)ethanesulfonic acid buffer (44 mM, pH 7.0) in the presence of MgCl2 (3.0 mM), MnCl2 (3.0 mM), 3 mM Ca(OAc)2, PAP34S (4.0 µg) and CHST15 (0.5 µg). The reaction mixture (total volume of 35 µL) was incubated at 37°C overnight. A volume of 3 µL of 100 mM ammonia acetate and 10 mU chondroitinase ABC in a volume of 5 µL were added to the mixture and incubations were continued for 2 more hours. CSA untreated with sulfotransferases was incubated with chondroitinase ABC digestion under the same conditions.
Heparosan (10 µg) was dissolved in (3-(N-morpholino)propanesulfonic acid) (33 mM, pH 6.5), NDST1 (0.5 µg), PAP34S (1 µg) and MnCl2 (9 mM). The reaction mixture (total volume of 35 µL) was incubated at 37°C overnight and then desalted using a PD-10 cartridge (GE Bioscience, Piscataway, NJ). The dried, modified heparosan chain was depolymerized using heparin lyases I, II and III (2 mU each) in Tris–HCl (50 mM, pH 7.45, 2 mM Ca(OAc)2) for 3 h. The untreated heparosan was also subjected to heparin lyase digestion under the same conditions.
For modifying mature GAG chains, 100 µg of GAG was mixed with 1 µg of a specific sulfotransferase and 10 µg of PAP34S in 50 mM Tris–HCl (pH 8.0) in a total volume of 50 µL. The reaction was then incubated at 37°C for 3 h. Untreated CS, CHST3- and CHST15-treated CS samples were further incubated with chondroitinase ABC (10 mU) in 50 mM Tris–HCl buffer (pH 8.0) in the presence of 5 mM NH4OAc at 37°C for 3 h. Untreated HS, NDST1 and HS6OST1 (10 µg each) samples were incubated with heparin lyase I (2 mU), II (2 mU) and III (2 mU) in 40 µL of Tris–HCl buffer (pH 7.45) in the presence of 3 mM Ca(OAc)2 at 37°C for overnight.
SEC-MS and MS/MS
Lyase-treated GAG samples were subjected to SEC-MS and MS/MS analyses for oligosaccharide and disaccharide profiling. The instrumental conditions were similar to those described previously (Shi and Zaia 2009). Briefly, a 3.2 mm × 300 mm Superdex Peptide column (GE Healthcare, Piscataway, NJ) was used to separate oligosaccharides and disaccharides with a 16-µL/min mobile phase (12.5 mM formic acid in water containing 10% (V/V) acetonitrile, titrated by ammonia to pH 4.4), delivered by a Waters Acquity UPLC system (Milford, MA). After an in-line UV detector, the eluents were sprayed into an Applied Biosystem/Sciex QSTAR time-of-flight mass spectrometer (currently ABSciex, Framingham, MA) via a TurboIonSpray interface. Typically, 2–5 µg of GAG samples prior to digestion was injected in a volume of 5–10 µL, and triplicates of the samples were run to obtain the standard deviations of MS analyses. Error bars reflect the standard deviations of triplicate LC-MS runs. For tandem MS experiments, the collision gas was set at 3 and collision energy at 30. A series of artificial mixtures of D0S6 and D2S0 for HS dp2s and ΔDi-4S and ΔDi-6S for CS dp2 to produce response curves of respective diagnostic ions for these two pair of isomers.
HS samples (2 µg each) were also digested with heparin lyase III (2 mU) in Tris–HCl, pH 7.45, and 3 mM Ca(OAc)2 at 37°C overnight. The oligosaccharides generated were fractionated by SEC, collected, desalted, dried and profiled using HILIC-MS according to previously reported procedures.
Supplementary data
Supplementary data for this article is available online at http://glycob.oxfordjournals.org/.
Funding
This work was supported by National Institutes of Health (P41GM104603 and R01HL098950).
Conflict of interest
None declared.
Abbreviations
CS disaccharide codes and masses: D0a0: ΔHexA-GalNAc, 379.1115; D0a4/6: ΔHexA-GalNAc-4/6-sulfate, 459.0683; D0a10: ΔHexA-GalNAc-4,6-sulfate, 539.0251; U0a0: HexA-GalNAc, 397.1220; U0a4/6: HexA-GalNAc-4/6-sulfate, 477.0788; U0a10: HexA-GalNAc-4,6-sulfate, 557.0357. HS disaccharide codes: D0A0: ΔHexA-GlcNAc, 379.1115; D2A0: ΔHexA-2-O-sulfate-GlcNAc, 459.0683; D0A6: ΔHexA-GlcNAc-6-O-sulfate, 459.0683; D2A6: ΔHexA-2-O-sulfate-GlcNAc-6-O-sulfate, 539.0251; D0S0: ΔHexA-GlcN-N-sulfate, 417.0577; D0S6: ΔHexAGlcNAc-6-O-sulfate, 497.0145; D2S6: ΔHexA-2-O-sulfate-GlcN-N-sulfate, 6-O-sulfate, 576.9713; D0H6: ΔHexA-GlcN-6-O-sulfate, 417.0577; D2H0: ΔHexA-2-O-sulfate-GlcN, 417.0577; D2H6: ΔHexA-2-O-sulfate-GlcN-6-O-sulfate, 497.0145; U0A0: HexA-GlcNAc, 397.1220; U0S0: HexA-GlcN-N-sulfate, 435.0683; U2S0: HexA-2-O-sulfate-GlcN-N-sulfate, 515.0251; U0S6: HexA-GlcN-N-sulfate, 6-O-sulfate, 515.0251; CHST, carbohydrate sulfotransferases; CID, collision-induced dissociation; CS, chondroitin sulfate; dp, degree of polymerization; EIC, extracted ion chromatogram; GAG, glycosaminoglycan; HILIC, hydrophilic interaction chromatography; HS, heparan sulfate; HS6ST1, HS 6-O-sulfotransferase 1; LC, liquid chromatography; MS, mass spectrometry; NDST1, N-deacetylase/N-sulfotransferase; NRE, non-reducing end; PAPS, 3′-phosphoadenosine-5′-phosphosulfate; SEC, size-exclusion chromatography.
Supplementary Material
References
- Basappa Murugan S, et al. Involvement of chondroitin sulfate E in the liver tumor focal formation of murine osteosarcoma cells. Glycobiology. 2009;19:735–742. doi: 10.1093/glycob/cwp041. [DOI] [PubMed] [Google Scholar]
- Bernfield M, Gotte M, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, et al. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Bulow HE, Hobert O. The molecular diversity of glycosaminoglycans shapes animal development. Annu Rev Cell Dev Biol. 2006;22:375–407. doi: 10.1146/annurev.cellbio.22.010605.093433. [DOI] [PubMed] [Google Scholar]
- Danan LM, Yu Z, et al. Mass spectrometric kinetic analysis of human tyrosylprotein sulfotransferase-1 and -2. J Am Soc Mass Spectrom. 2008;19:1459–1466. doi: 10.1016/j.jasms.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danan LM, Yu Z, et al. Catalytic mechanism of Golgi-resident human tyrosylprotein sulfotransferase-2: A mass spectrometry approach. J Am Soc Mass Spectrom. 2010;21:1633–1642. doi: 10.1016/j.jasms.2010.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson LA, Belting M, et al. Biosynthesis of decorin and glypican. Matrix Biol. 2000;19:367–376. doi: 10.1016/s0945-053x(00)00083-4. [DOI] [PubMed] [Google Scholar]
- Godoy JA, Block DB, et al. Dermatan sulfate and bone marrow mononuclear cells used as a new therapeutic strategy after arterial injury in mice. Cytotherapy. 2011;13:695–704. doi: 10.3109/14653249.2010.548378. [DOI] [PubMed] [Google Scholar]
- Halldorsdottir AM, Zhang L, et al. N-Acetylgalactosamine 4,6-O-sulfate residues mediate binding and activation of heparin cofactor II by porcine mucosal dermatan sulfate. Glycobiology. 2006;16:693–701. doi: 10.1093/glycob/cwj117. [DOI] [PubMed] [Google Scholar]
- He L, Giri TK, et al. Vascular dermatan sulfate regulates the antithrombotic activity of heparin cofactor II. Blood. 2008;111:4118–4125. doi: 10.1182/blood-2007-12-127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffhines AJ, Jen CH, et al. Tyrosylprotein sulfotransferase-2 expression is required for sulfation of RNase 9 and Mfge8 in vivo. J Biol Chem. 2009;284:3096–3105. doi: 10.1074/jbc.M808434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Shi X, et al. Improved LC-MS/MS of heparan sulfate oligosaccharides via chip-based pulsed make-up flow. Anal Chem. 2011;83:8222–8229. doi: 10.1021/ac201964n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen CH, Moore KL, et al. Pattern and temporal sequence of sulfation of CCR5 N-terminal peptides by tyrosylprotein sulfotransferase-2: An assessment of the effects of N-terminal residues. Biochemistry. 2009;48:5332–5338. doi: 10.1021/bi900285c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailemia MJ, Li L, et al. Complete mass spectral characterization of a synthetic ultralow-molecular-weight heparin using collision-induced dissociation. Anal Chem. 2012;84:5475–5478. doi: 10.1021/ac3015824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuger J, Salmivirta M, et al. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001;276:30744–30752. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- Lander AD, Selleck SB. The elusive functions of proteoglycans: In vivo veritas. J Cell Biol. 2000;148:227–232. doi: 10.1083/jcb.148.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence R, Lu H, et al. Disaccharide structure code for the easy representation of constituent oligosaccharides from glycosaminoglycans. Nat Methods. 2008;5:291–292. doi: 10.1038/nmeth0408-291. [DOI] [PubMed] [Google Scholar]
- Lindahl U, Kusche-Gullberg M, et al. Regulated diversity of heparan sulfate. J Biol Chem. 1998;273:24979–24982. doi: 10.1074/jbc.273.39.24979. [DOI] [PubMed] [Google Scholar]
- Midura RJ, Calabro A, et al. Nonreducing end structures of chondroitin sulfate chains on aggrecan isolated from Swarm rat chondrosarcoma cultures. J Biol Chem. 1995;270:8009–8015. doi: 10.1074/jbc.270.14.8009. [DOI] [PubMed] [Google Scholar]
- Ohtake S, Ito Y, et al. Human N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase cDNA is related to human B cell recombination activating gene-associated gene. J Biol Chem. 2001;276:43894–43900. doi: 10.1074/jbc.M104922200. [DOI] [PubMed] [Google Scholar]
- Ohtake S, Kimata K, et al. A unique nonreducing terminal modification of chondroitin sulfate by N-acetylgalactosamine 4-sulfate 6-O-sulfotransferase. J Biol Chem. 2003;278:38443–38452. doi: 10.1074/jbc.M306132200. [DOI] [PubMed] [Google Scholar]
- Olson SK, Bishop JR, et al. Identification of novel chondroitin proteoglycans in Caenorhabditis elegans: Embryonic cell division depends on CPG-1 and CPG-2. J Cell Biol. 2006;173:985–994. doi: 10.1083/jcb.200603003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaas AH, West LA, et al. Glycosaminoglycan sulfation in human osteoarthritis. Disease-related alterations at the non-reducing termini of chondroitin and dermatan sulfate. J Biol Chem. 1998;273:12642–12649. doi: 10.1074/jbc.273.20.12642. [DOI] [PubMed] [Google Scholar]
- Plaas AH, Wong-Palms S, et al. Chemical and immunological assay of the nonreducing terminal residues of chondroitin sulfate from human aggrecan. J Biol Chem. 1997;272:20603–20610. doi: 10.1074/jbc.272.33.20603. [DOI] [PubMed] [Google Scholar]
- Shi X, Huang Y, et al. Tandem mass spectrometry of heparan sulfate negative ions: Sulfate loss patterns and chemical modification methods for improvement of product ion profiles. J Am Soc Mass Spectrom. 2012;23:1498–1511. doi: 10.1007/s13361-012-0429-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Zaia J. Organ-specific heparan sulfate structural phenotypes. J Biol Chem. 2009;284:11806–11814. doi: 10.1074/jbc.M809637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver Z, Liu D, et al. Emerging views of heparan sulfate glycosaminoglycan structure/activity relationships modulating dynamic biological functions. Trends Cardiovasc Med. 2002;12:71–77. doi: 10.1016/s1050-1738(01)00150-5. [DOI] [PubMed] [Google Scholar]
- Silbert JE. Biosynthesis of chondroitin sulfate. Chain termination. J Biol Chem. 1978;253:6888–6892. [PubMed] [Google Scholar]
- Staples G, Bowman M, et al. A chip-based amide-HILIC LC/MS platform for glycosaminoglycan glycomics. Proteomics. 2009;9:686–695. doi: 10.1002/pmic.200701008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples GO, Naimy H, et al. Improved hydrophilic interaction chromatography LC/MS of heparinoids using a chip with postcolumn makeup flow. Anal Chem. 2010;82:516–522. doi: 10.1021/ac901706f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples GO, Shi X, et al. Extended NS domains reside at the non-reducing end of heparan sulfate chains. J Biol Chem. 2010;285:18336–18343. doi: 10.1074/jbc.M110.101592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugumaran G, Silbert JE. Biosynthesis of chondroitin sulfate. Organization of sulfation. J Biol Chem. 1989;264:3864–3868. [PubMed] [Google Scholar]
- Suzuki N, Toyoda H, et al. Chondroitin acts in the guidance of gonadal distal tip cells in C. elegans. Dev Biol. 2006;300:635–646. doi: 10.1016/j.ydbio.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Tiedemann K, Larsson T, et al. The glucuronyl C5-epimerase activity is the limiting factor in the dermatan sulfate biosynthesis. Arch Biochem Biophys. 2001;391:65–71. doi: 10.1006/abbi.2001.2376. [DOI] [PubMed] [Google Scholar]
- Turnbull J, Powell A, et al. Heparan sulfate: Decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/s0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]
- Uchimura K, Kadomatsu K, et al. Functional analysis of the chondroitin 6-sulfotransferase gene in relation to lymphocyte subpopulations, brain development, and oversulfated chondroitin sulfates. J Biol Chem. 2002;277:1443–1450. doi: 10.1074/jbc.M104719200. [DOI] [PubMed] [Google Scholar]
- Wolff JJ, Amster IJ, et al. Electron detachment dissociation of glycosaminoglycan tetrasaccharides. J Am Soc Mass Spectrom. 2007;18:234–244. doi: 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JJ, Chi L, et al. Distinguishing glucuronic from iduronic acid in glycosaminoglycan tetrasaccharides by using electron detachment dissociation. Anal Chem. 2007;79:2015–2022. doi: 10.1021/ac061636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JJ, Laremore TN, et al. Electron detachment dissociation of dermatan sulfate oligosaccharides. J Am Soc Mass Spectrom. 2008a;19:294–304. doi: 10.1016/j.jasms.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JJ, Laremore TN, et al. Influence of charge state and sodium cationization on the electron detachment dissociation and infrared multiphoton dissociation of glycosaminoglycan oligosaccharides. J Am Soc Mass Spectrom. 2008b;19:790–798. doi: 10.1016/j.jasms.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JJ, Leach FE, et al. Negative electron transfer dissociation of glycosaminoglycans. Anal Chem. 2010;82:3460–3466. doi: 10.1021/ac100554a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZL, Lech M. Modification degrees at specific sites on heparan sulphate: An approach to measure chemical modifications on biological molecules with stable isotope labelling. Biochem J. 2005;389:383–388. doi: 10.1042/BJ20041827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZL, Lech M, et al. Determining heparan sulfate structure in the vicinity of specific sulfotransferase recognition sites by mass spectrometry. J Biol Chem. 2004;279:1861–1866. doi: 10.1074/jbc.M311398200. [DOI] [PubMed] [Google Scholar]
- Yu Y, Hoffhines AJ, et al. Determination of the sites of tyrosine O-sulfation in peptides and proteins. Nat Methods. 2007;4:583–588. doi: 10.1038/nmeth1056. [DOI] [PubMed] [Google Scholar]
- Zaia J, Costello CE. Tandem mass spectrometry of sulfated heparin-like glycosaminoglycan oligosaccharides. Anal Chem. 2003;75:2445–2455. doi: 10.1021/ac0263418. [DOI] [PubMed] [Google Scholar]
- Zaia J, Li X-Q, et al. Tandem mass spectrometric strategies for determination of sulfation positions and uronic acid epimerization in chondroitin sulfate oligosaccharides. J Am Soc Mass Spectrom. 2003;14:1270–1281. doi: 10.1016/S1044-0305(03)00541-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.