

Abstract
Tetrad analysis has been a gold standard genetic technique for several decades. Unfortunately, the manual nature of the process has relegated its application to small-scale studies and limited its integration with rapidly evolving DNA sequencing technologies. We have developed a rapid, high-throughput method, called Barcode Enabled Sequencing of Tetrads (BEST), that replaces the manual processes of isolating, disrupting and spacing tetrads. BEST uses a meiosis-specific GFP fusion protein to isolate tetrads by fluorescence-activated cell sorting and molecular barcodes that are read during genotyping to identify spores derived from the same tetrad. Maintaining tetrad information allows accurate inference of missing genetic markers and full genotypes of missing (and presumably nonviable) individuals. By removing the bottleneck of manual dissection, hundreds or even thousands of tetrads can be isolated in minutes. We demonstrate the approach in Saccharomyces cerevisiae, but BEST is readily transferable to microorganisms in which meiotic mapping is significantly more laborious.
INTRODUCTION
Meiotic mapping is a linkage-based method for analyzing the recombinant progeny of a cross that has long been a cornerstone of genetics. The method is possible in a wide range of eukaryotes, including genetically facile yeasts and less tractable microorganisms, such as the filamentous fungus Neurospora crassa and the unicellular green alga Chlamydomonas reinhardtii. The approach is enabled by tetrad dissection, a technique for isolating and cultivating “with complete certainty all of the spores [meiotic progeny] derived from individual asci [tetrads]“ that was first developed in S. cerevisiae1. In the 75 years since that publication, the method has catalyzed yeast genetic research. However, the manual process of dissecting tetrads severely limits its throughput, even for experienced researchers with access to specialized equipment. One prominent yeast geneticist, Cora Styles, documented 12,157 yeast crosses over a career that spanned 30 years (Gerald Fink, personal communication), a number few could hope to replicate.
Many approaches have tried to circumvent this bottleneck. While details differ between methods and organisms, they generally employ one of three strategies. The first strategy, “random spore analysis”, enriches for tetrads based on properties of the ascus, which protects spores from a variety of insults that kill vegetative cells2. Spores are then randomly dispersed on solid media to recover the recombinant progeny. A second strategy avoids much of the high variability and low specificity of random spore analysis by using a selectable reporter gene (HIS3) under the control of a mating-type-specific transcriptional promoter3. This approach has been applied with great success to generate specific classes of recombinant progeny needed to test synthetic growth defects3-5 and linkage between traits and gene deletions6. The third strategy, “bulk segregant analysis”7 or more recently “extreme QTL (X-QTL) mapping”8, has been used in organisms ranging from yeast to plants. Bulk segregant methods use a pooled genotyping strategy to identify genomic regions common to the majority of progeny selected under a specific condition, e.g. high drug concentrations. While the three strategies have been applied effectively to specific problems, they each have limitations that fall short of the broad applicability of conventional tetrad analysis. Chief amongst these is the inability to recover all viable meiotic progeny (either due to the progeny generation method or the phenotypic selection imposed for bulk comparisons) and the loss of the tetrad relationships (i.e. knowledge of which sister spores were members of the same original tetrad).
The limited throughput of manual tetrad dissection constrains two research areas in which meiotic mapping can be powerfully applied. The first area is the mapping of complex traits resulting from combinations of naturally occurring polymorphisms. The ability to detect only a small fraction of the genetic loci that underlie complex traits of medical and agricultural relevance by meiotic mapping is at least partially due to the lack of sufficiently large numbers of individuals9. The second area is the study of the molecular mechanisms of recombination. The study of some recombination and segregation processes depends on capturing all the events from an individual meiosis. A striking example is the study of gene conversion, for which one seminal paper utilized over 19,000 tetrads10. In the absence of high-throughput alternatives, such fields are unable to effectively leverage the current revolution in DNA sequencing technology, the costs of which have decreased 100,000-fold over the past decade and continue to outpace Moore's Law11.
Here, we describe a new high-throughput method, which we call Barcode Enabled Sequencing of Tetrads (BEST). BEST expands upon current methods in high-throughput genetics by enabling the generation and genotyping of large numbers of progeny that are isolated, genotyped, and maintained as individuals in a manner that allows the sister-spore relationships of all four meiotic products to be recovered. The approach is an example of how techniques that marry the power of conventional genetics with ultra-high-throughput DNA sequencing can enable the study of problems that were previously intractable.
RESULTS
Tetrad dissection has two critical steps that are difficult to automate because they are performed manually with a micromanipulator mounted to a microscope. The first is the isolation of tetrads away from unsporulated cells in the culture, which out-number tetrads 99 to 1 in the commonly used FY strain background (Swain-Lenz and Fay, unpublished result). The second is physically separating the spores of a tetrad and arranging them in a grid. In S. cerevisiae, spores are held together by both an outer ascus, the remnant of the cell wall of the original diploid cell, and a set of interspore bridges12. In conventional tetrad dissection, enzymatic digestion removes the ascus, and a researcher uses a micromanipulator to identify tetrads, break the interspore bridges and array the spores in a gridded pattern. The grid separates spores to prevent interspore mating and also preserves the knowledge of which spores came from the same tetrad. BEST (Fig. 1) overcomes both of these bottlenecks with a process in which tetrads are isolated and disrupted to give single spores, whose tetrad relationships are reconstructed using molecular barcodes.
Figure 1. BEST method.

(a) A pool of barcoded plasmids is transformed into the diploid strain from a cross. (b) Transformants are then grown on selection and ~10,000 transformed colonies are pooled and sporulated. During meiosis each spore of a tetrad inherits a copy of the barcoded plasmid. (c) Tetrads are separated from unsporulated cells by FACS and collected on agar plates, where they are digested and disrupted to allow each spore to form a colony. (e) Colonies are then picked into 96 well plates, phenotyped and genotyped. During genotyping the plasmid barcode is read and used to identify the four members of each tetrad.
Tetrad isolation and disruption
Florescence-activated cell sorting (FACS) permits an easy, rapid separation of 4-spore tetrads from a mixed population that includes vegetative cells, dead cells, clumped cells, and 2-spore dyads (Fig. 2). Several reporter genes have been used to fluorescently label tetrads or individual spores13-15. We chose the SPS2-GFP fusion, because it has been successfully used to quantitate sporulation in a number of genetically diverse, non-laboratory strains. This construct was introduced onto a 2-micron plasmid, which when transformed into yeast is expressed in meiosis and fluorescently labels tetrads. We then established a series of FACS gating parameters (Online Methods, Supplementary Fig. 1) that reproducibly yield 95% 4-spore tetrads (Fig. 2), even from strains with sporulation efficiencies less than 1% (Scott, Sirr, and Dudley, unpublished results). The inclusion of this FACS sorting step is where BEST achieves its largest gain in throughput. Because FACS sorters are able to query thousands of events per second, the identification and isolation of 10 tetrads can be accomplished by FACS in less than a second, while the equivalent manual process takes an experienced researcher several minutes. In crosses with poor sporulation efficiency, the burden of manual dissection increases, as the researcher must hunt through a large excess of unsporulated cells to find tetrads. Consequently, cell sorting provides an even greater advantage in these strain backgrounds.
Figure 2. Isolation of tetrads.
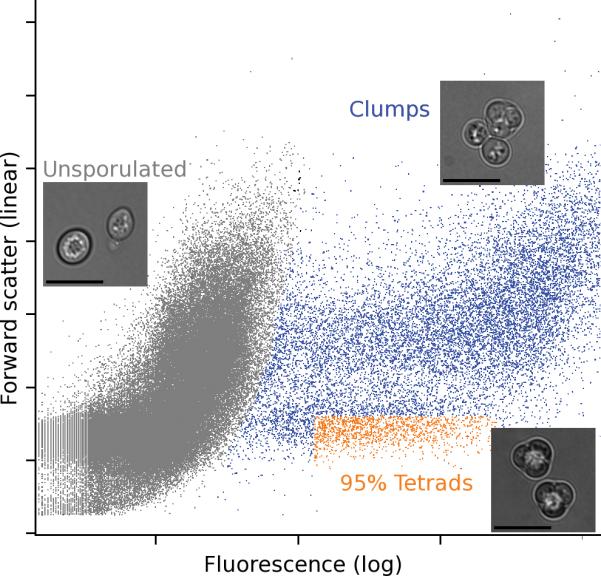
The sporulation-specific fluorescent reporter on the plasmid allows the use of FACS to quickly identify and collect tetrads from the sporulation culture. The gating parameters (orange region) separate tetrads from vegetative, dead, and clumped cells as well as 2-spore dyads (gray and blue regions). Inset images show representative examples of cells present in each region of the FACS plot. Images were acquired with an Applied Precision DeltaVision microscope with a 60x oil-immersion objective and Photometrics CoolSnap HQ2 camera. The scale bar is 5 μm.
Following tetrad isolation, the ascus must be digested and the spores disrupted and physically separated to grow as individual clones (colonies). To avoid significant spore loss during liquid transfer steps, we developed a procedure for digesting, disrupting, and distributing the spores on an agar plate (Online Methods). We sort GFP-positive tetrads directly into a pool of zymolyase solution on the plate, where the ascus is enzymatically digested. Spores are then separated by agitation with glass beads, which provides the mechanical force necessary to break the interspore bridges connecting sister spores. This process also physically disperses spores randomly across the plate, far enough apart that most colonies are pure clonal isolates of each recombinant spore. Once cells have grown to single colonies, they are individually picked into the 96-well liquid plate format in which they are stored as frozen stocks for subsequent genotype and phenotype analysis. In this way, a rapid, high-throughput method replaces the manual processes of isolating, disrupting and spacing 4-spore tetrads.
Tetrad reconstruction
The ease of random plating strategies and their ability to be automated come at the expense of losing information about which progeny originated from the same tetrad. To facilitate the reconstruction of sister spore relationships, we devised a molecular barcoding strategy that satisfied four main criteria. First, the pool of barcodes must be complex enough to ensure that most individuals recovered share a common barcode because they were members of the same tetrad. Second, the barcode must be reliably transmitted to all four tetrad spores. Third, the presence of the barcode should be phenotypically neutral. Finally, the barcode should be compatible with the method used to determine the progeny genotypes, allowing the barcode to be read as part of the genotyping workflow. No existing barcoding resource satisfies all of these criteria. For example, strategies that integrate barcodes at a neutral genomic location16 will be heterozygous in a diploid genome and thus only present in half of the tetrad's spores.
To satisfy these requirements, we chose a highly complex, barcoded 2-micron plasmid. A random barcode sequence, flanked by restriction sites that ensure its representation in the sequencing reads of the chosen genotyping method, can be inserted into the plasmid. 2-micron plasmids are maintained in high copy (10-40 copies per cell) and stably segregate during cell division17, increasing the likelihood of plasmid transmission to all four spores. Like the native 2-micron circle17, the presence of engineered 2-micron plasmids should have relatively neutral impacts on most traits. However, because the plasmid is no longer required after the strain is genotyped, direct counter selection or simple failure to maintain selection could facilitate plasmid loss.
We constructed a 2-micron-based plasmid library that contains the SPS2-GFP sporulation-specific fluorescent reporter, a complex DNA barcode flanked by restriction sites compatible with our genotyping protocol and a selectable drug resistance marker for plasmid maintenance (Online Methods, Supplementary Fig. 2). This plasmid library is transformed into the heterozygous diploid cells of the cross (Fig. 1). The complexity of the library (Supplementary Information) is conferred by the presence of a randomized 15 nucleotide sequence, which permits a theoretical 109 unique sequences. By pooling thousands of yeast transformants, we create a mixed population of barcoded diploid cells that fluoresce only when cells have undergone sporulation and that pass on a tetrad-specific barcode to each spore of the tetrad (Fig. 1). Because the pool of barcodes (Supplementary Fig. 3), determined by the number of initial yeast transformants, is much larger than the number of tetrads that will be mixed together on the same agar plate (Online Methods), the probability of isolating the same barcode in multiple tetrads is relatively low.
Sequencing-based genotyping and reconstruction of tetrads
While BEST is compatible with numerous genotyping platforms, we favor sequencing-based approaches that permit the simultaneous determination of the strain's genotype and plasmid-borne tetrad barcode (Fig. 3a) in a manner that yields individual, rather than pooled, genotypes. Because recombination in yeast generates relatively few crossover events per chromosome per meiosis18, most of a recombinant genome's sequence can be imputed from a relatively sparse set of genetic markers (Fig. 3b). We, therefore, chose a highly multiplexed genome reduction strategy known as restriction-site associated DNA tag sequencing (RAD-seq)19, which directs genome sequencing to positions with a specific restriction site pattern (Fig. 3, Online Methods). Our choice of restriction enzymes and 40 base Illumina single-end reads (Online Methods), allow us to sequence the same 2-3% of every yeast strain using a highly multiplexed sequencing strategy (Online Methods). For crosses between strains with a sufficient number of DNA polymorphisms, this provides a high-density set of genetic markers and drastically reduces costs relative to whole genome sequencing each progeny strain. If the complete genome sequences of both of the parental strains are known, RAD-seq markers permit the imputation of essentially the entire genome sequence of each recombinant individual. However, the rapid decrease of sequencing costs will ultimately enable cost effective whole genome sequencing of large numbers of progeny.
Figure 3. Sequence-based tetrad reconstruction and genotyping.

The progeny strains from a cross are genotyped by sequencing, and during this process the plasmid barcode of each strain is read. (a) The members of a single tetrad can be established by identifying four strains that share a common plasmid barcode. (b) Once strains have been assigned to tetrads, missing markers can be inferred and a full genome sequence with recombination events can be deduced for each strain.
Because the plasmid-borne tetrad barcode is flanked by the same restriction sites used in our RAD-seq method, it is captured by the genotyping reads. After RAD-seq, strains arising from the same FACS-sorted plate that share a common plasmid barcode sequence are assigned to the same tetrad (Fig. 3a), a hypothesis that is confirmed by a series of computational quality control metrics (Online Methods). The small proportion of strains (<5%) that lack a tetrad barcode in their sequence reads can then be assigned to tetrads based on the expectation of 2:2 allele segregation of markers within tetrads (Online Methods). Thus, while the presence of a usable tetrad barcode in the sequencing data simplifies tetrad assignment, strains lacking this sequence still have the potential to be assigned to tetrads.
Missing marker and complete genotype inference
Missing data arising from stochastic lack of sequence coverage is a common problem in large-scale genetic analyses, even in samples with otherwise high sequence representation. In these cases, the expected 2:2 segregation of each allele in a tetrad can be leveraged to infer the values of markers that are not confidently assigned (Fig. 3b). Except in the case of rare gene conversion events, it should always be possible to correctly infer a missing marker from a complete (4-spore) tetrad, if the status of that marker in the other three members of the tetrad is known. Similarly, if a marker is missing in two members of a tetrad, it is possible to infer their values from the other two spores 50% of the time (Online Methods). Missing markers can also be inferred probabilistically based on the genetic distances between markers, i.e. an untyped marker that is close to a typed marker has a high probability of being derived from the same parent as the typed marker. The use of both genetic distance and the known haplotypes of all spores in the tetrad (Online Methods) can improve the accuracy of inference, sometimes greatly (Supplementary Fig. 4), by incorporating the probability of all possible recombination patterns at the tetrad level. In the pilot crosses below, 5-10% of the final set of allele calls were made using these inference methods.
Tetrad-based genotype inference can be further extended to infer the full genome sequence of non-viable spores. In the crosses below, removing all sequence information for one member of a 4-spore tetrad and reconstructing its genotype by inference (i.e. simulating the inference of a dead spore) recovered ~98% of the original allele calls with ~98% accuracy. Such analysis could enable the discovery of synthetic interactions, like those seen in synthetic lethal screens, resulting from combinations of naturally occurring polymorphisms and in a way that is less limited by strain background and the number of interacting genes than current methods. For example, it should be possible to uncover a synthetic interaction between four genes in two previously uncharacterized wild strains.
Pilot yeast crosses
We piloted BEST by generating two crosses comparable in size to those commonly published for complex trait studies in yeast (~100 tetrads). The first cross between two well characterized and commonly used laboratory strains, FY420 and Σ1278b21, showed high (98%) spore viability by manual dissection. The second cross between the laboratory strain S288c22 and the wild oak isolate YPS16323 showed a spore viability (86%) more typical of crosses between genetically distant strains. In the FY4 × Σ1278b (pilot) cross, 77% of the progeny that passed our quality control filters could be assembled into 3 or 4 spore tetrads. The lower viability cross, S288c × YPS163 (pilot), had 70% of quality-filtered progeny assembled into 3- or 4-spore tetrads. Using this “percent in tetrads” metric as a measure of efficiency, these results demonstrate that BEST can be successfully applied to crosses between strains at a range of genetic distances and beyond laboratory strain backgrounds (Table 1). For both crosses, we were also able to generate a dense set of genetic markers, where the larger number of markers in S288c × YPS163 reflects the greater sequence divergence of the two parental strains.
Table 1.
BEST results for three crossesa.
| Cross | Colonies recovered | Strains Passing QC | Strains in 3- or 4-spore tetrads | Number of Markers | Mean Marker Separation |
|---|---|---|---|---|---|
| FY4 × Σ1278b (pilot) | 385 | 325 | 77% | 481 | 21 kb |
| S288c × YPS163 (pilot) | 378 | 347 | 70% | 831 | 14 kb |
| FY4 × Σ1278b (full scale) | 4,354 | 3,652 | 63% | 579 | 18 kb |
Parental SNP tables, progeny genotypes and barcode sequences are presented (Supplementary Tables 1-8).
To assess the performance of the method in a large-scale implementation, we repeated the high viability cross, FY4 × Σ1278b (full scale). In this experiment, a single researcher performed BEST from the start of FACS sorting through the completion of platin ~3,725 tetrads in three hours, a scale that would require almost a month of manual dissection. To assess efficiency, we sequenced strains from a subset of the agar plates generated in the experiment. The resulting 4,354 strains produced 1.7 billion high quality sequencing reads that mapped to the genome sequence or to the tetrad barcode. Of the 3,652 progeny strains that passed our quality control filters, 63% could be assembled into 4- or 3-spore tetrads (Table 1). Taken together, these results demonstrate that BEST can be applied on a scale beyond that commonly performed for conventional studies.
Thus, BEST removes the major bottleneck of tetrad analysis, the generation of the progeny strains, with a relatively modest reduction in efficiency compared to manual dissection. Factors reducing efficiency could include loss of spores caused by adhesion to the glass beads during spreading, plasmid loss, increased spore death due to the mechanical stresses of the process or failure to separate sister spores. These decreases in efficiency are easily overcome by the large advance in throughput that BEST affords. The rapid fluorescence-based identification of tetrads allows large numbers of tetrads to be collected quickly, and the glass-bead based separation method allows those tetrads to be “dissected” in parallel. As a result, an individual researcher can prepare separated spores for several thousand tetrads in a few hours, a vast improvement over manual dissection.
DISCUSSION
The ability to isolate large numbers of meiotically-derived recombinant progeny in a manner that retains their sister spore relationships advances a technique that has remained essentially unchanged for 75 years. BEST combines progeny isolation and genotyping using three main strategies. First is the introduction of a reporter construct that GFP-labels cells that have undergone meiosis so that tetrads can be isolated by FACS. Second is use of a complex pool of DNA barcodes in a form that transmits the same, unique sequence to all four spores of a tetrad. Third is the genotyping step, which reads a consistent 2-3% of the genome, including the tetrad-specific barcode. The recovery of tetrad relationships along with the empirically-derived genotyping data from the cross allows the accurate inference of missing genotype information, from markers with low sequence coverage to the complete genotypes of inviable (and therefore unrecoverable) individuals. We have applied the method in S. cerevisiae, the most commonly used microorganism for meiotic mapping. However, minor substitutions of organism-specific reagents, e.g. different sporulation-specific proteins fused to GFP, make the method readily transferrable to other microorganisms, including organisms in which meiotic mapping is significantly more labor intensive or currently intractable.
ONLINE METHODS
Yeast strains, media, and manipulation
Unless noted, standard media and methods were used for growth and genetic manipulation of yeast24. S. cerevisiae strains used in this study are listed in Supplementary Table 9.
pCL2_BC barcode library construction
The plasmid-based barcode library (pCL2_BC) was constructed in two steps.
First, the pCL2 plasmid backbone was constructed by gap repair in yeast as follows: the yeast 2-micron ADE2 plasmid, pRS42225, was cut with BglII. The ADE2-containing fragment was discarded and the remaining plasmid backbone was treated with Antarctic Phosphatase (New England Biolabs, NEB) to prevent re-ligation and gel-purified. An SPS2::EGFP::kanMX4 cassette14 was amplified from BC257 (gift of Barak Cohen) using primers Gap1.1_F and Gap1.1_R (Supplementary Table 10) that bear homology to both the SPS2 genomic and plasmid DNA sequences. The resulting PCR product was co-transformed with the plasmid fragment into yeast. Transformants were selected on YPD plates containing 200 μg/ml G418. G418 resistant clones were scraped and pooled; DNA was prepared and transformed into OneShot TOP10 chemically competent bacteria (Life Technologies). Bacterial transformants were selected on LB-carbenicillin plates and analyzed by restriction digest to identify the repaired plasmid.
Next, a complex library of random barcodes was inserted as follows: 20 nmoles of a 200-mer oligo (Supplementary Table 10), including a high complexity 15-base degenerate region, was amplified by 20 rounds of PCR using Phusion High-Fidelity DNA Polymerase (Thermo Fisher) with BC_F and BC_R primers (Supplementary Table 10) at a final concentration of 20 pM each. The DNA from 24 separate reactions was pooled and ligated to the linearized pCL2 at its unique SmaI site using the In-Fusion HD Cloning System (Clontech). To maintain complexity, five ligation reactions were carried out and used for 18 independent bacterial transformations with LB-carbenicillin selection. Each transformation produced an average of 3.5 × 104 colonies. A pilot ligation, transformed and screened by X-gal blue-white, showed a low plasmid re-ligation background of ~5%. The transformants were scraped from the plates, re-suspended and divided into 5 separate pools. Plasmid DNA from each pool was extracted and purified using a Qiagen Plasmid Maxi Purification kit (Qiagen).
pCL2 BC barcode complexity determination
The barcode complexity of the pCL2_BC library was assessed by Illumina DNA sequence analysis (Supplementary Table 11). Briefly, 1.5 μg of the plasmid library was fragmented by digestion with MfeI and Sau3A1 (a DAM-methylation insensitive isoschizomer of MboI). Digests were incubated for 2 hrs. at 37°C in a 20 μl reaction with 2 units of Sau3A1 and 10 units MfeI (NEB), followed by heat inactivation at 65°C for 20 min. The annealed P2 adaptors and four sets of annealed barcoded P1 adaptors (Supplementary Table 10) were then ligated onto the plasmid fragments at room temperature for 20 min in a single 25 μl reaction containing 1 μg of digested plasmid, 400 units T4 DNA ligase (NEB), 2.5 μl 10x T4 ligase buffer and 6 μl of a combined P1 (25 nM) and P2 (1 μM) adaptor mix (Supplementary Table 10). T4 ligase was heat inactivated for 20 min. at 65°C, and ligated plasmid DNA was concentrated to 10 μl using a MinElute PCR Purification Kit. The DNA was size selected and extracted, as below. Approximately 10 ng of the purified plasmid DNA library was enriched with a PCR reaction and sequenced in a single flow cell lane of a Genome Analyzer IIx (Illumina).
Generation of barcoded yeast tetrads
Heterozygous diploids resulting from crosses between two parental strains were grown to ~2 × 107 cells/ml and transformed with ~2 μg of the pCL2_BC barcoded plasmid library using a standard protocol26 modified to include 8% DMSO in the transformation mix. After the 30 min. 42°C heat shock step, the transformed cells were gently washed with 1 ml of YPD, resuspended in 1 ml of YPD, and allowed to recover by sitting at room temperature for 3 hrs. Transformants were then selected by plating 200 μl of the recovered culture per YPD + 200 μg/ml G418 plate, a total of five plates per transformation. This protocol yielded a library of ~104 single colonies. Transformants were pooled by scraping the plates. A portion of the pool was saved as a frozen glycerol stock to set up sporulation cultures at a later date. All crosses described in this work were performed with frozen stocks revived by an overnight growth in liquid YPD + 200 μg/ml G418, washed, and transferred to liquid sporulation medium3 containing 200 μg/ml G418. Sporulation was performed at room temperature with agitation and monitored daily. Cultures were deemed ready for sorting when sporulation had reached the point of completing well-formed tetrads, without significant numbers of dyads. In the crosses described here, spore separation was improved by allowing spores to sit at room temperature (without agitation) for an additional 7-10 days (Supplementary Information).
Tetrad Isolation by FACS
Tetrads were isolated from the sporulation culture by FACS with a FACSAria II equipped with an Automated Cell Deposition Unit (BD Biosciences). GFP fluorescence was detected using the 488nm laser and 530/30 filter. To achieve a reproducibly high proportion of tetrads we implemented a series of gating steps. Selecting a narrow width of the FSC and SSC signals, while permitting a large range of FSC and SSC heights filtered out events containing cell or media debris as well as those containing multiple cells per droplet (Supplementary Fig. 1a,b). A GFP vs. FSC area gate was used to identify fluorescent (and therefore sporulated) cells (Supplementary Fig. 1c). The population selected by these steps consisted of two subpopulations: one subpopulation was composed of clumps of tetrads and tetrads with a small bud attached, while the other subpopulation was primarily composed of isolated tetrads. These subpopulations were distinguished from each other on the basis of their FSC signal. The clumps and budded tetrads had a higher FSC than the isolated tetrads, though the distribution of FSC in these two subpopulations did overlap as indicated by the overlapping peaks in (Supplementary Fig. 1d). To enrich for isolated tetrads, we set a final gate to include events with a low FSC (Supplementary Fig. 1d). During gate assignment, tetrad recovery was assessed by sorting 1,000 events onto a microscope slide and manually counting tetrads.
Spore separation by on-plate digestion and glass bead spreading
To prevent spore loss during liquid handling, tetrads were sorted directly onto YPD + 200 μg/ml G418 agar plates with a 25 μl drop of 1mg/ml zymolyase in 0.7 M sorbitol on top of the agar. Tetrads were sorted into the drop by positioning the plate on top of the 96-well plate adaptor and directly under the sorting stream. To reduce the chance of recovering two tetrads with the same plasmid barcode on the same plate and to ensure the development of single, isolated colonies, only 25 tetrads were sorted per plate. Each plate was incubated at 37°C for 90 min. to digest the asci and 15-25 glass beads (Sigma-Aldrich, 425-600 μM) were added per plate. Plates were shaken vigorously for 4 min. in stacks of 5 plates and then incubated face up (with glass beads in place) at 30°C for 2 days. After colonies appeared, plates were carefully inverted to remove the glass beads without disturbing the colonies and the number of single colonies on each plate was counted to assess the success of the spore separation treatment. In our hands, different sporulation conditions have different, strain-specific effects on the ability to disrupt tetrads (Supplementary Information). Each colony was picked into a well of a 96-well plate containing liquid YPD + 200 μg/ml G418. Information about which colonies came from each agar plate was recorded. These colonies were cultured for genotyping and preserved as frozen glycerol stocks.
RAD-seq progeny genotyping
Yeast genomic DNA was isolated for RAD-seq as follows. 96-well format plates were used to seed 0.5 ml cultures in 2 ml deep-well plates of YPD with 200 μg/ml of G418 and grown overnight at 30°C on a VibraTranslator electromagnetic shaker (Union Scientific Corp). Cells were pelleted at 1,000xg for 5 min. and resuspended with 400 μl of lysis buffer (10 mM Tris (pH 8.0), 1 mM EDTA, 2% Triton x-100, 1% SDS, 100 mM NaCl), and the suspension was transferred to a lysis rack (Costar 4413, 1.2 ml tubes) containing 0.5 mm glass beads. The racks were processed at 1,300 rpm for 2 min. in a bead beater (Geno/Grinder 2010, SPEX Sample Prep). After centrifugation, 250 μl of supernatant was transferred to a 96 deep-well block and 250 μl 6M Guanidine HCl DNA binding buffer was added. The mixture was then transferred to the 96-well DNA binding plates (Pall Acroprep #8032). Centrifugation, washing and elution procedures followed the manufacturer's protocol.
RAD-seq was carried out as previously described27. Briefly, yeast genomic DNA was fragmented by restriction enzyme digestion with MfeI and MboI. P1 and P2 Adaptors were then ligated onto the fragments. The P1 adaptor contains the Illumina PCR Forward sequencing primer sequence followed by one of 48 unique 4 nucleotide barcodes and finally the MfeI overhang sequence. The two pilot crosses, FY4 × Σ1278B (pilot) and S288c × YPS163 (pilot), were sequenced in-house on a Genome Analyzer IIx (Illumina) and used the P2 adaptor sequences (Supplementary Table 10) allowing a multiplexing of 48 strains per lane. The larger cross, FY4 × Σ1278B (full scale), required a higher level of multiplexing on a HiSeq 2000 (Illumina) and therefore used the P2-BC adaptors (Supplementary Table 10), which contain the Illumina PCR Reverse primer sequence, a second barcode (a 6 nucleotide sequence based on Ilumina Truseq indexes 1-12), and the MboI restriction enzyme compatible overhang. After ligation, the barcoded ligation products were pooled, concentrated, and size selected on agarose gels. Fragments between 150 to 500 base pairs were extracted from the gel. Gel-extracted DNA was further pooled to multiplex 48 uniquely barcoded samples in one sequencing library that was then enriched with a PCR reaction using Illumina PCR Forward and Reverse primers (Supplementary Table 10). For the full-scale cross, 200 ng of a set of 12 libraries, each with a different P2 barcode, were combined and concentrated by QIAquick PCR Purification column (Qiagen). This combined, dual-indexed pool of 576 yeast strains was then sequenced in a single Illumina flow-cell lane. Dual-indexed sequencing runs were performed on the HiSeq 2000 (Northwest Genomics Center, University of Washington) utilizing 50 base pair single-end reads and the second index read only. Sequencing runs performed on the Genome Analyzer IIx for the pilot studies generated 40 base pair single-end reads.
Strain genotype and tetrad determination
For each lane of sequencing, raw read sequences were split into pools based on their P1 barcode sequences, and also their P2 barcodes for the larger cross. Reads with unexpected strain barcodes or with barcodes having Phred (-10 log10 Perror) quality scores less than 20 or ambiguous (“N”) calls at any barcode base were discarded. Reads with more than 2 “N” calls in the body of the sequencing read were also discarded. The P1 barcodes were then removed from the read sequences. In each of the resulting strain-specific pools of reads, the sequences were searched for reads carrying the plasmid (tetrad) barcode. Tetrad barcodes were identified using the pattern <read-start>NNNNNTGCCGACCC<barcode>GCAGG, with the barcode restricted to a length of 11-19 nucleotides. A single mismatch or nucleotide deletion was allowed in the pattern match outside the barcode. The most frequent barcode sequence was then identified from the set of all plasmid barcode reads coming from each strain.
The strain-specific read pools were then used to infer the genotypes of the progeny strains. From each strain pool, the sequence reads were aligned to one parental genome. In both crosses, one parent was S288c/FY4, therefore the S288c genome sequence was used as a reference in both crosses. Read alignment was carried out using BWA (v5.8)28 allowing 6 mismatches and using quality trimming (threshold of Phred=20). The SAMtools (v0.1.18)29 mpileup (-u -C 50) and bcftools view (-c) commands were then used to generate a variant call format (vcf) file for each strain. A table of SNPs (Supplementary Tables 1 and 2) between the two parental genomes was generated by aligning the parental genomes using Mummer (v3.22)30. The bases called in the vcf file were then compared to the expected parent 1 (P1) and parent 2 (P2) alleles from this SNP file to convert the base calls into P1 or P2 allele calls. When the count of one parental allele was at least 5-fold higher than the count of the other parental allele, a P1 or P2 allele assignment was made. Otherwise, the allele was defined as mixed/heterozygous.
Marker quality filtering was then carried out, markers were removed unless they were called at least 30% as frequently as the most called marker and unless the ratio P1/(P1+P2) lay between 0.3 and 0.7.
The strains were then grouped into tetrads based on common tetrad barcode sequences and strain quality control filters were applied. Strains derived from each plate of 25 sorted tetrads were analyzed independently to reduce the risk of encountering more than 1 tetrad with the same plasmid barcode. Strains with too few reads (<50,000) or too many mixed/heterozygous sites (>20%) were flagged and removed from further processing and analysis. Duplicate strains were identified (>90% identical allele calls across >100 markers) and the lower coverage strain removed. Strains with unique barcodes, where the total number of tetrad barcode reads was <30 or where the most common barcode sequence comprised less than 80% of all observed barcode sequences had their tetrad barcode removed and were relabeled as un-barcoded. Among the remaining tetrads, any “tetrads” with more than 5 spores, any 4-spore tetrads with >20% 3:1 or 4:0 segregation patterns and any 3-spore tetrads with >10% 3:0 segregation patterns were dissolved. All strains in dissolved tetrads were relabelled as unbarcoded. An attempt was then made to create 3-spore tetrads out of the group of unbarcoded strains. This was done by examining the frequency of allele misegregation (3:0) among each subset of 3 un-barcoded strains. Any set showing <10% 3:0 segregation over at least 100 sites was identified as a tetrad. After this process, an attempt was made to add the remaining unbarcoded strains to each 2 or 3 spore tetrad. Again, this was done by examining the frequency of abnormal allele segregation in each existing tetrad when each single un-barcoded strain was added and requiring <20% misegregation over at least 100 informative sites. Finally, for each 2 and 3 spore tetrad, the genotypes of the missing/dead spores were initialized to 0 (missing data) at all marker positions.
Missing data inference
Having generated an initial haplotype for each strain and assigned strains to tetrads, the tetrad organization was used to infer missing allele calls (including mixed calls). This included inferring alleles for any missing/dead spores. Based on the expected 2:2 allele segregation pattern in a tetrad, any marker called as P1 or P2 in 2 of the strains allows assignment of missing marker alleles in either of the other strains with almost complete confidence.
After the tetrad inference step, the data were then used to calculate a genetic map for each cross using R/qtl (version 1.21-2). This map was then used to carry out linkage-based inference of remaining missing values on a tetrad-basis. Specifically, missing alleles were inferred based on the relative probability of all possible local crossover patterns within the tetrad, anchored at flanking positions with allele calls in all 4 spores. Allele calls with probabilities greater than 0.99 were then accepted.
Linkage-based inference methods can suffer from two sources of error, gene conversion and markers with abnormal linkage patterns. To address the latter, haplotypes were first generated without linkage-based inference and then analyzed using R/qtl (version 1.21-2). Markers with abnormal linkage patterns (e.g. unlinked to any marker, linked to different chromosomes or linked to a distant region of the same chromosome) were identified and flagged. These flagged markers were then removed and the final haplotypes and genetic map were regenerated.
Supplementary Material
ACKNOWLEDGEMENTS
We thank B. Cohen, A. Sherman, and members of the Dudley lab for helpful discussions. We thank D. Nickerson and B. Paeper (Northwest Genomics Center, University of Washington) for assistance with Illumina HiSeq 2000 sequencing and S. Bloom for assistance with Illumina GA IIx sequencing. This work was supported by an NIH/NHGRI Genome Scholar/ Faculty Transition Award (K22 HG002908) to A.M.D. and a strategic partnership between the ISB and the University of Luxembourg. We note with sadness the passing of our friend and co-author Teresa Gilbert.
Footnotes
AUTHOR CONTRIBUTIONS
C.L.L., A.C.S., A.S., E.W.J., T.L.G. and A.M.D. contributed experimental design; A.C.S. and C.L.L. contributed strains and plasmids; A.C.S., A.S., M.H., C.L.L., E.W.J. and T.L.G. contributed experiments; G.A.C., A.C.S., P.M. and J.L. contributed data analysis and scripts; A.C.S., C.L.L., G.A.C. and A.M.D. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Winge O, Laustsen O. On two types of spore germination, and on genetic segregations in Saccharomyces demonstrated through single spore cultures. C.R. Trav. Lab. Carlsberg Ser. Physiol. 1937;24:263–315. [Google Scholar]
- 2.Amberg DC, Burke DJ, Strathern JN. Random spore analysis in yeast. CSH Protoc. 2006;2006 doi: 10.1101/pdb.prot4162. doi:10.1101/pdb.prot4162. [DOI] [PubMed] [Google Scholar]
- 3.Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. doi:10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 4.Pan X, et al. A robust toolkit for functional profiling of the yeast genome. Mol Cell. 2004;16:487–496. doi: 10.1016/j.molcel.2004.09.035. doi:10.1016/j.molcel.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 5.Schuldiner M, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. doi:S0092-8674(05)00868-8 [pii] 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 6.Snitkin ES, et al. Model-driven analysis of experimentally determined growth phenotypes for 465 yeast gene deletion mutants under 16 different conditions. Genome Biol. 2008;9:R140. doi: 10.1186/gb-2008-9-9-r140. doi:gb-2008-9-9-r140 [pii] 10.1186/gb-2008-9-9-r140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michelmore RW, Paran I, Kesseli RV. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:9828–9832. doi: 10.1073/pnas.88.21.9828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrenreich IM, et al. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature. 2010;464:1039–1042. doi: 10.1038/nature08923. doi:10.1038/nature08923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackay TF, Stone EA, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10:565–577. doi: 10.1038/nrg2612. doi:10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- 10.Fogel S, Mortimer R, Lusnak K, Tavares F. Meiotic gene conversion: a signal of the basic recombination event in yeast. Cold Spring Harb Symp Quant Biol. 1979;43(Pt 2):1325–1341. doi: 10.1101/sqb.1979.043.01.152. [DOI] [PubMed] [Google Scholar]
- 11.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. doi:10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 12.Coluccio A, Neiman AM. Interspore bridges: a new feature of the Saccharomyces cerevisiae spore wall. Microbiology. 2004;150:3189–3196. doi: 10.1099/mic.0.27253-0. doi:10.1099/mic.0.27253-0. [DOI] [PubMed] [Google Scholar]
- 13.Chin BL, Frizzell MA, Timberlake WE, Fink GR. FASTER MT: Isolation of Pure Populations of a and alpha Ascospores from Saccharomycescerevisiae. G3 (Bethesda) 2012;2:449–452. doi: 10.1534/g3.111.001826. doi:10.1534/g3.111.001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerke JP, Chen CT, Cohen BA. Natural isolates of Saccharomyces cerevisiae display complex genetic variation in sporulation efficiency. Genetics. 2006;174:985–997. doi: 10.1534/genetics.106.058453. doi:genetics.106.058453 [pii] 10.1534/genetics.106.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thacker D, Lam I, Knop M, Keeney S. Exploiting spore-autonomous fluorescent protein expression to quantify meiotic chromosome behaviors in Saccharomyces cerevisiae. Genetics. 2011;189:423–439. doi: 10.1534/genetics.111.131326. doi:genetics.111.131326 [pii] 10.1534/genetics.111.131326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan Z, et al. Yeast Barcoders: a chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nat Methods. 2008;5:719–725. doi: 10.1038/nmeth.1231. doi:10.1038/nmeth.1231. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong KA, Som T, Volkert FC, Rose A, Broach JR. Propagation and expression of genes in yeast using 2-micron circle vectors. Biotechnology. 1989;13:165–192. [PubMed] [Google Scholar]
- 18.Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature. 2008;454:479–485. doi: 10.1038/nature07135. doi:10.1038/nature07135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baird NA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. doi:10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winston F, Dollard C, Ricupero-Hovasse SL. Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast. 1995;11:53–55. doi: 10.1002/yea.320110107. doi:10.1002/yea.320110107. [DOI] [PubMed] [Google Scholar]
- 21.Dowell RD, et al. Genotype to phenotype: a complex problem. Science. 2010;328:469. doi: 10.1126/science.1189015. doi:10.1126/science.1189015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mortimer RK, Johnston JR. Genealogy of principal strains of the yeast genetic stock center. Genetics. 1986;113:35–43. doi: 10.1093/genetics/113.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sniegowski PD, Dombrowski PG, Fingerman E. Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Res. 2002;1:299–306. doi: 10.1111/j.1567-1364.2002.tb00048.x. [DOI] [PubMed] [Google Scholar]
- 24.Rose M, Winston F, Hieter P. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- 25.Brachmann CB, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. doi:10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 26.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 27.Lorenz K, Cohen BA. Small- and large-effect quantitative trait locus interactions underlie variation in yeast sporulation efficiency. Genetics. 2012;192:1123–1132. doi: 10.1534/genetics.112.143107. doi:10.1534/genetics.112.143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. doi:10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. doi:10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome biology. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. doi:10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.