

Abstract
The skin interstitium sequesters excess Na+ and Cl– in salt-sensitive hypertension. Mononuclear phagocyte system (MPS) cells are recruited to the skin, sense the hypertonic electrolyte accumulation in skin, and activate the tonicity-responsive enhancer-binding protein (TONEBP, also known as NFAT5) to initiate expression and secretion of VEGFC, which enhances electrolyte clearance via cutaneous lymph vessels and increases eNOS expression in blood vessels. It is unclear whether this local MPS response to osmotic stress is important to systemic blood pressure control. Herein, we show that deletion of TonEBP in mouse MPS cells prevents the VEGFC response to a high-salt diet (HSD) and increases blood pressure. Additionally, an antibody that blocks the lymph-endothelial VEGFC receptor, VEGFR3, selectively inhibited MPS-driven increases in cutaneous lymphatic capillary density, led to skin Cl– accumulation, and induced salt-sensitive hypertension. Mice overexpressing soluble VEGFR3 in epidermal keratinocytes exhibited hypoplastic cutaneous lymph capillaries and increased Na+, Cl–, and water retention in skin and salt-sensitive hypertension. Further, we found that HSD elevated skin osmolality above plasma levels. These results suggest that the skin contains a hypertonic interstitial fluid compartment in which MPS cells exert homeostatic and blood pressure–regulatory control by local organization of interstitial electrolyte clearance via TONEBP and VEGFC/VEGFR3–mediated modification of cutaneous lymphatic capillary function.
Introduction
Mechanisms causing salt-sensitive hypertension are imperfectly defined (1). Guyton et al. attributed long-term blood pressure regulation to the kidney, arguing that blood flow, auto-regulation, and pressure natriuresis control blood pressure (2, 3). This model suggests a close relationship among total body Na+, total body volume, and blood pressure. It assumes isosmolarity of body fluids among the bodily compartments (2). Along with others (4–7), we (8–14) showed earlier that electrolytes are distributed in a more complex 3-compartment model, in which intravascular and the interstitial fluids do not equilibrate as readily as believed (15, 16). We underscored the importance of Na+ binding to negatively charged proteoglycans in the skin, the largest organ with the most extracellular space (8, 11). We suggested that, in addition to renal control, local extrarenal regulatory mechanisms for electrolyte clearance of interstitial fluid are operative to maintain extracellular electrolyte clearance and blood pressure. We postulated that electrolyte accumulation in the skin occurs in excess of water and causes local hypertonicity. Mononuclear phagocyte system (MPS) cells respond to osmotic stress via the transcription factor tonicity-responsive enhancer-binding protein (TONEBP) that provokes a tissue-specific, MPS-driven, regulatory response (15, 16). MPS cells infiltrate the salt-overloaded interstitium, initiate TONEBP-driven VEGFC expression, and restructure the interstitial lymphatic capillary network, while increasing eNOS expression in blood vessels. Blocking this MPS-driven regulatory process leads to reduced cutaneous lymphatic capillary density, skin electrolyte accumulation, reduced eNOS expression in blood vessels, and increased blood pressure. The findings suggest that immune cells are regulators of internal environment and blood pressure homeostasis (15, 16).
Our model implies that the local skin microenvironment is hypertonic to plasma, that MPS cells dictate regulatory events via TONEBP, and that skin VEGFC is important for systemic blood pressure control. It was unclear whether MPS cells influence blood pressure via VEGFC/VEGFR3–driven lymphatic electrolyte clearance or perhaps by VEGFC/VEGFR2–driven modulation of eNOS expression. Furthermore, the relationship between Na+ and Cl– disposition in the microenvironment was also ill defined. Here, we show that selective depletion of TONEBP in MPS cells, blockade of VEGFR3 with antibody leaving VEGFR2 intact, and deletion of VEGFC signaling in skin all disrupt cutaneous lymphatic capillary architecture and result in predominantly Cl– accumulation in the skin, which is paralleled by salt-sensitive hypertension. Finally, we document with several independent methods the hypertonic electrolyte concentrations of the interstitial microenvironment in the skin. These findings strengthen our proposal of a third, locally regulated, skin fluid compartment relevant to systemic blood pressure regulation.
Results
Eliminating TONEBP in MPS cells reduces skin Cl– clearance and causes salt-sensitive hypertension.
To understand the role of TONEBP in MPS cells in modulating lymphatic density and skin electrolyte storage, we investigated the TONEBP/VEGFC regulatory axis in mice with MPS cell–specific conditional TonEBP gene deletion (LysMcreTonEBPfl/fl mice). We first harvested macrophages from LysMWTTonEBPfl/fl mice (without TONEBP deficiency) and from LysMcreTonEBPfl/fl mice (with TONEBP deficiency). We exposed the cells to standard cell culture medium, to NaCl-mediated osmotic stress, or urea-mediated hyperosmolality (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI60113DS1). Increasing NaCl, which is an effective osmolyte, caused increased TONEBP expression in macrophages without TONEBP deficiency. In contrast, primary bone marrow macrophages from LysMcreTonEBPfl/fl mice showed reduced TONEBP protein expression under normal cell culture medium conditions and with NaCl-mediated osmotic stress. We also tested TonEBP mRNA expression in vivo in the mice at various organs. The mice all received high-salt diet (HSD) to induce electrolyte storage. The only tissue in which we found a genotype-dependent decrease in TonEBP mRNA expression was the skin (Supplemental Figure 1). These findings suggest that MPS/TONEBP–driven responses to HSD are particularly manifest in the skin.
To further substantiate this hypothesis, we next tested whether or not MPS-specific TONEBP depletion abolishes TONEBP/VEGFC–driven increases in cutaneous lymphatic capillary density in response to skin electrolyte storage. Similar to FVB strain mice (genetic background control), LysMWTTonEBPfl/fl mice fed HSD showed increased lymphatic capillary density (Figure 1A). However, no increase occurred in LysMcreTonEBPfl/fl mice with MPS-specific deletion of TonEBP gene. Western blotting (Figure 1B) confirmed that VEGFC protein expression in LysMcreTonEBPfl/fl mice was reduced compared with that in the two controls. Statistical analysis (Figure 1C) showed significant reduction in TonEBP and Vegfc mRNA and protein levels in LysMcreTonEBPfl/fl mouse skin, while CD68 expression reflecting macrophage infiltration was unaffected. The resulting decrease in cutaneous lymphatic capillary density was paralleled by a salt-sensitive increase in mean arterial blood pressure (MAP) in LysMcreTonEBPfl/fl mice.
Figure 1. TONEBP in MPS cells is essential for VEGFC-driven lymphatic capillary hyperplasia and clearance of Cl– in the skin and buffers systemic blood pressure.

(A) Representative whole-mount staining of lymphatic capillary density (anti–Lyve-1 antibody, green) in ears of FVB mice (WT control group) fed LSD and HSD and of LysMWTTonEBPfl/fl mice (without MPS-specific TONEBP deletion) and LysMcreTonEBPfl/fl mice (with MPS-specific TONEBP deletion), both after HSD. Scale bar: 500 μm. (B) Representative VEGFC protein (85 kDa) expression in FVB mice and LysMWTTonEBPfl/fl controls compared with that in LysMcreTonEBPfl/fl mice. β-Actin (42 kDa) expression was used as a loading control. The mice were fed HSD. (C) TonEBP and Vegfc mRNA expression and VEGFC and CD68 protein expression in skin as well as cutaneous lymphatic capillary density (LCD; arbitrary units) and MAP (mmHg) in LysMWTTonEBPfl/fl mice (n = 16) and in LysMcreTonEBPfl/fl mice (n = 12) fed a HSD. (D) Na+ and Cl– content and concentrations in skin compared with plasma concentrations in the same mice. (E) GAG charge densities in the same mice. *P (genotype) < 0.05.
In skin (Figure 1D), Cl– content and concentration were both increased in LysMcreTonEBPfl/fl mice, while no differences were observed in plasma. We found no significant differences in skin Na+ content and skin water content between LysMWTTonEBPfl/fl and LysMcreTonEBPfl/fl mice. Disaccharide analysis of skin glycosaminoglycans (GAGs) showed no differences in GAG charge density between LysMcreTonEBPfl/fl and LysMWTTonEBPfl/fl mice (Figure 1E). These data show that MPS cell–specific TONEBP depletion blocks VEGFC-driven increases in cutaneous lymphatic capillary density after HSD. Absence of increased lymphatic capillary density resulted in Cl– retention in the skin and increased MAP. We conclude that MPS cells locally regulate skin Cl– content by a TONEBP-dependent mechanism.
Anti-VEGFR3 treatment causes skin-Cl– retention and salt-sensitive hypertension.
We next performed experiments with anti-VEGFR3 antibodies to specifically block MPS/TONEBP/VEGFC–driven lymphatic capillary hyperplasia distal to TONEBP. As shown previously, HSD increased lymphatic capillary density in mice (Figure 2A), which was paralleled by increased CD68 and VEGFC expression (Figure 2B) and increased phosphorylated eNOS (p-eNOS) expression (Figure 2B). Treatment with the mF4-31c1 anti-VEGFR3 antibody blocked the skin lymphatic capillary response to HSD completely (Figure 2A and Table 1) but not the MPS cell count, CD68 expression, or p-eNOS expression. The mF4-31c1 treatment in mice fed HSD resulted in increased infiltration of VEGFC-positive cells into the cutaneous interstitium. These data suggest that MPS cells responded to VEGFR3 blockade in an effort to restructure the lymphatic capillary network when challenged with HSD. While treatment with the anti-VEGFR3 antibody abolished the increased lymphatic capillary density after HSD, mF4-31c1 treatment did not change eNOS and p-eNOS expression. We conclude that mF4-31c1 did not interfere with the MPS/VEGFC/VEGFR2/eNOS signaling pathway. Because blockade of VEGFC/VEGFR3–driven lymphatic capillary hyperplasia was paralleled by a 17-mmHg increase in MAP (Figure 2C), the data suggest that the unopposed MPS/VEGFC/VEGFR2/eNOS signaling pathway, which had a MPS-driven increase in eNOS and p-eNOS expression (Figure 2B and Table 1) in the same mice, was not able to buffer blood pressure in mice in which the VEGFC/VEGFR3 interaction was blocked. Without HSD, mF4-31c1 did not increase blood pressure (Figure 2C). We determined skin lymphatic capillary density with whole-mount staining in each mouse (Supplemental Figure 2) and assessed the relationship between lymphatic capillary density and MAP (Figure 2D). We found that MAP increases were inversely related to lymphatic capillary density in the skin. Complete blockade of the increased skin lymphatic capillary density resulted in the highest MAP values, while less complete blockade resulted in lower MAP responses to HSD. The finding that blood pressure and lymphatic capillary density are inversely correlated after HSD suggests that skin electrolyte clearance through cutaneous lymph capillaries could be coupled with systemic blood pressure.
Figure 2. Blocking the VEGFC/VEGFR3 interaction with mF4-31c1 eliminates the MPS/VEGFC–driven lymphatic capillary hyperplasia after HSD and leads to salt-sensitive blood pressure increase despite unaltered VEGFC/VEGFR2–mediated increased eNOS expression.

(A) Quantification of lymphatic capillary density by whole-mount staining (anti–Lyve-1 antibody) in whole ears of FVB mice fed LSD or HSD, with and without mF4-31c1 treatment. Red squares are the computerized quantitated area; numerical values are lymphatic capillary density (arbitrary units). (B) Representative Western blots for CD68 (100 kDa), VEGFC (85 kDa), eNOS (132 kDa), and p-eNOS (140 kDa) in mouse skin. β-Actin (42 kDa) expression was used as a loading control. (C) Lymphatic capillary density in ear and MAP in the mice. In mice fed HSD, blockade of lymphatic capillary hyperplasia was paralleled by a 17-mmHg increase in MAP with mF4-31c1. (D–F) Relationship among lymphatic capillary density and MAP, skin Cl– content, and skin Cl– concentration in the same mice. SKW, skin water content; SKCl–, skin Cl– content; rSKCl–, skin Cl– content relative to DW. *P < 0.05 versus LSD WT; †P < 0.05 versus LSD plus mF4-31c1; ‡P < 0.05 versus HSD WT; #P (diet*mF4-31c1) < 0.05.
Table 1.
The effect of dietary salt loading in WT mice with or without VEGFR3 blockade on MPS infiltration, MPS TONEBP/VEGFC expression, lymphatic capillary density, and eNOS expression in the skin
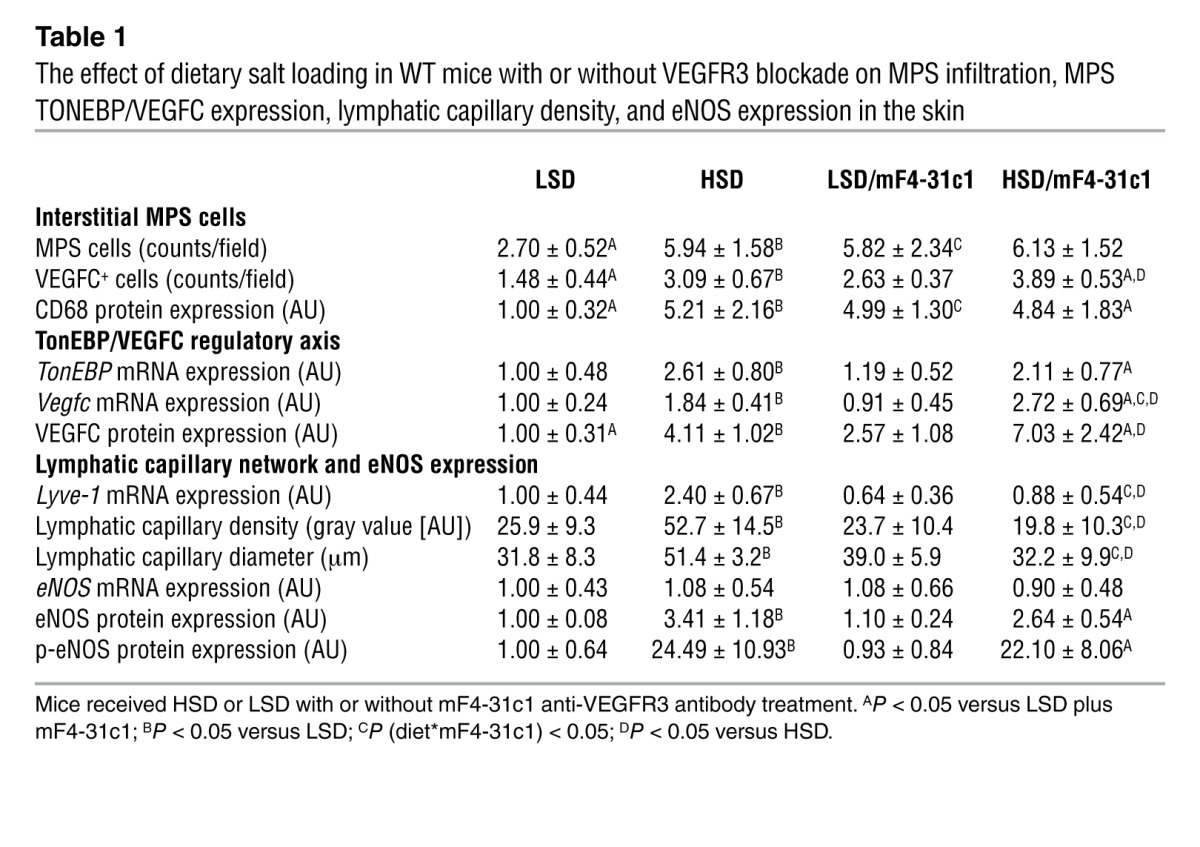
We therefore next tested whether or not salt-sensitive blood pressure increases after VEGFR3 blockade were paralleled by electrolyte and water retention in the skin. Blocking VEGFC/VEGFR3 interaction resulted in higher skin Cl– content (Figure 2E and Table 2) and increased skin Cl– concentrations (Figure 2F and Table 2). Both were inversely correlated with lymphatic capillary density. In contrast, mF4-31c1 treatment neither increased skin Na+ nor skin water content in the same mice (Table 2 and Supplemental Figure 3). These findings suggest that MPS/VEGFC–driven hyperplasia of cutaneous lymph capillaries specifically promotes clearance of Cl– from skin tissue and that this clearance process might be coupled with blood pressure.
Table 2.
The effect of dietary salt loading on blood pressure and on internal environment composition in WT mice, in WT mice with mF4-31c1 anti-VEGFR3 treatment, and in K14-FLT4 mice
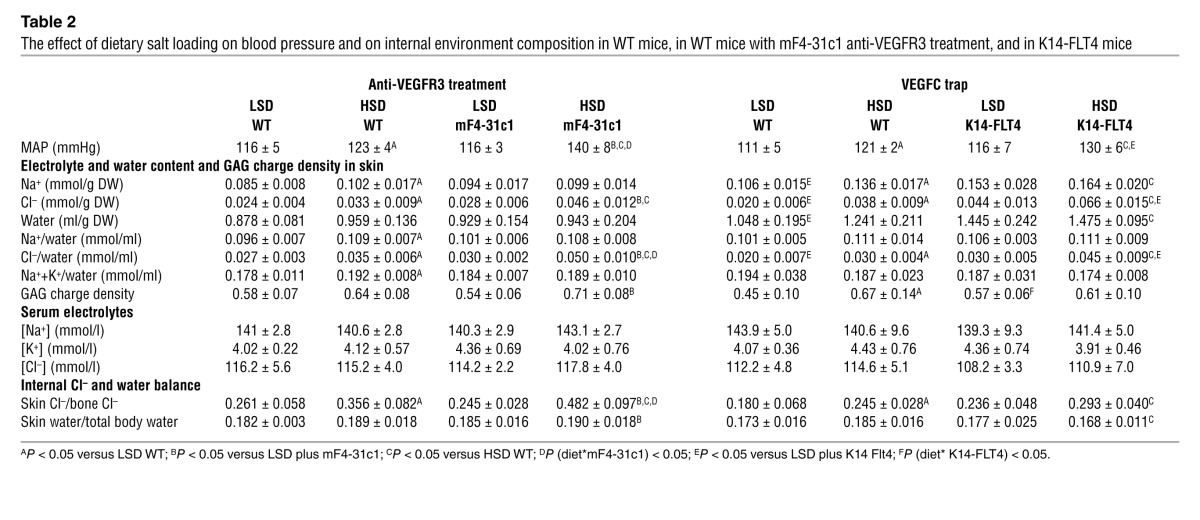
To test this hypothesis further, we next investigated the relationship between skin Na+ and Cl– content and blood pressure in each individual mouse fed HSD. The mF4-31c1 treatment increased MAP and skin Cl– content (Figure 3A) but not skin Na+ content (Figure 3B). Compared with low-salt diet (LSD), HSD increased skin Na+ and Cl– content (Figure 3C). Additional mF4-31c1 treatment increased Cl– further but not Na+. Expressing the data as skin Cl–/Na+ (Figure 3C) underscores the finding that absence of MPS/VEGFC–driven hyperplasia of cutaneous lymph capillaries resulted in selective Cl– retention in skin tissue. The salt-sensitive blood pressure increase with mF4-31c1 treatment was associated neither with increases in skin Na+ content (Figure 3B) nor with changes in skin water content (Supplemental Figure 4). In contrast, increased MAP in the mF4-31c1–treated mice was paralleled by increased skin Cl– content (Figure 3C) and with increased skin Cl–-to-water ratio (Supplemental Figure 4). These findings suggest that the salt-sensitive blood pressure increase was neither Na+ nor volume related, but rather Cl– dependent. We therefore inspected the relationship between skin Na+, Cl–, and water in more detail. In HSD control mice, increased Na+ and Cl– content resulted in increased skin water content, underscoring the role of these effective osmolytes to control body fluid content (Figure 3D). However, skin Cl– content remained more than 2-fold lower than skin Na+ content, indicating an important role of unmeasured anionic osmolytes for maintenance of fluid tonicity and electroneutrality. Additional blockade of VEGFC/VEGFR3–driven lymphatic capillary hyperplasia changed this relationship substantially (Figure 3D). In HSD mice given mF4-31c1 treatment, Cl– was accumulated in the skin without commensurate water retention. The resulting rightward shift of the skin Cl–-to-water ratio (Figure 3D) further increased skin Cl– concentration (Figure 3D and Table 2). Because these changes were Cl–-specific, while skin Na+ content did not change, we conclude that when the mF4-31c1 antibody prevents the increase in lymphatic capillary density, the amount of unmeasured anionic osmolytes in skin fluid is reduced. These data suggest that MPS/TONEBP/VEGFC–driven increases in lymphatic capillary density facilitate Cl– clearance from skin tissue and that extrarenal Cl– removal, rather than skin Na+ and water clearance, is tightly linked to blood pressure regulation (Supplemental Video 1).
Figure 3. Relationship among Na+ and Cl– accumulation, water retention, MAP, and unmeasured anions in mice without and with mF4-31c1 treatment.

Relationship between (A) Cl– accumulation and (B) Na+ accumulation in the skin and MAP in control and in mF4-31c1–treated mice fed a HSD. Elevated blood pressure with anti-VEGFR3 treatment was paralleled by increased skin Cl– content but not with increased skin Na+ content. (C) Skin Na+ content, skin Cl– content, and Cl–-to-Na+ ratio in the mice. With HSD, blockade of cutaneous lymphatic capillary density by mF4-31c1 treatment selectively increased skin Cl– content. (D) Relationship between skin Na+ (orange) and Cl– (blue) accumulation and skin water content in control mice and in mF4-31c1–treated mice fed HSD. Increasing skin Na+ or Cl– content increased skin water. However, the skin Cl–-to-water ratio was shifted to the right with mF4-31c1 treatment (0.035 ± 0.006 mmol/ml [control HSD] versus 0.050 ± 0.010 mmol/ml [HSD plus mF4-31c1]; P < 0.05), indicating a reduction in the gap between skin Na+ and Cl– content, which represents unmeasured anionic osmolytes. rSKNa+, skin Na+ content relative to DW; rSKW, skin water content relative to DW. *P < 0.05 versus LSD WT; †P < 0.05 versus LSD plus mF4-31c1; ‡P < 0.05 versus HSD WT.
Skin VEGFC depletion reduces electrolyte clearance and increases blood pressure.
To test the hypothesis that skin VEGFC is relevant for skin electrolyte clearance and blood pressure, we performed additional experiments in K14-FLT4 mice designed to block the effect of VEGFC in the skin (17). These transgenic mice overexpress soluble VEGFR3 (sVEGFR3, also known as FLT4) in skin keratinocytes, which increases sVEGFR3 levels, resulting in a VEGFC trap that results in hypoplasia of skin lymph capillaries (17) but not of renal lymph vessels (Supplemental Figure 5). While WT mice showed increased MPS cell count only with HSD, we found higher MPS cell counts in K14-FLT4 mice even with LSD (Figure 4A and Table 3). Moreover, K14-FLT4 mice exhibited cutaneous lymphatic capillary hypoplasia (Figure 4B and Table 3) and had higher eNOS and phosphorylated blood capillary eNOS expression after both LSD and HSD (Figure 4, C and D). Unlike that in WT mice (Figure 4, D and E, and Table 3), HSD did not increase lymphatic capillary density and p-eNOS expression in K14-FLT4 mice. This situation was paralleled by a 1.5-fold increase in skin Na+ content and more than a 2-fold increase in Cl– content in the skin of K14-FLT4 mice fed HSD (Table 2). Furthermore, salt-sensitive hypertension resulted (Figure 4E). The data suggest that selective trapping of skin VEGFC by sVEGFR3 blocks the VEGFC-dependent electrolyte clearance from skin tissue in K14-FLT4 mice and induces salt-sensitive hypertension (Supplemental Video 1), illustrating a further link between skin electrolyte homeostasis and blood pressure regulation.
Figure 4. A skin-specific VEGFC trap in mice with overexpression of sVEGFR3 in keratinocytes (K14-FLT4 mice) leads to cutaneous lymphatic capillary hypoplasia, skin electrolyte retention, and salt-sensitive blood pressure increase.

(A) Representative staining of MPS cells in ears (anti-CD68 staining; red) of WT and K14-FLT4 mice given LSD or HSD. Scale bar: 50 μm. (B) Representative whole-mount staining of lymphatic capillaries (anti–Lyve-1 staining; green) in the same group of mice. Scale bar: 200 μm. (C) Representative Western blots for CD68 (100 kDa) and VEGFC (85 kDa) expression in mouse skin from the same groups. β-Actin (42 kDa) expression was used as a loading control. (D) Representative Western blots for eNOS (132 kDa) and p-eNOS (140 kDa), with β-actin (42 kDa) as a loading control. (E) Lymphatic capillary density and MAP in WT and in K14-FLT4 mice fed LSD or HSD. *P < 0.05 versus LSD WT; †P < 0.05 versus LSD K14-FLT4; ‡P < 0.05 versus HSD WT; #P (diet*K14-FLT4) < 0.05.
Table 3.
The effect of dietary salt loading on MPS infiltration, MPS TonEBP/VEGFC expression, lymphatic capillary density, and eNOS expression in WT and K14-FLT4 mice with overexpression of sVEGFR3 in the skin

Verifying interstitial hypertonicity in salt-sensitive hypertension in rats.
Our experiments in mice with deletion of TONEBP in MPS cells suggest that the skin microenvironment is characterized by osmotic stress, suggesting osmotic gradients between blood plasma and the interstitial fluid matrix. To further support this hypothesis, we aimed to document the hypothesis that interstitial osmolality can be higher than that in plasma and investigated rats for this purpose. In line with our experiments in mice, HSD in rats showed no effect on plasma Na+ and Cl– concentrations or osmolality but increased skin Na+ and Cl– content and concentration, as determined by chemical analysis (Figure 5A). In parallel with skin Na+ storage, TonEBP and Vegfc mRNA expressions, as well as MPS cell count, increased in the skin (Figure 5B). The rats developed a salt-sensitive increase in MAP and in interstitial fluid pressure, the latter representing a driving force for lymph flow (18). To probe for electrolyte composition in prenodal lymph capillaries, we additionally induced pronounced skin Na+ storage in rats by deoxycorticosterone acetate treatment. We then measured electrolyte concentrations using energy-dispersive x-ray electron microprobe analysis of primary lymph vessels, which drain interstitial fluid (Figure 5C). We found substantially higher Na+ concentrations in lymph capillaries than in blood plasma from the carotid artery, while differences in Cl– concentrations did not reach statistical significance under these conditions with this technique. We also tested whether or not we could find higher electrolyte concentrations in microdialysate from skin. We found that Na+ concentrations and osmolality were higher in microdialysate from skin than in blood plasma (Figure 5D); however, we identified no statistically significant differences in dialysate electrolyte concentrations between rats fed LSD or HSD (Supplemental Table 1). Finally, we measured osmolality in snap-frozen skin samples compared with that in plasma. HSD increased skin osmolality by approximately 20 mOsmol/kg H2O, while plasma values were unchanged (Figure 5E). Despite some inter-methodological differences, these data, derived from four distinct methodological approaches, suggest that HSD leads to Na+ and Cl– storage in the skin, resulting in increased interstitial osmolality and osmotic stress in the skin microenvironment. The osmotic stress activates TONEBP in MPS cells and drives VEGFC gene expression in the immune cells.
Figure 5. HSD leads to skin Na+ and Cl– storage and osmotic stress that is not reflected in plasma.

(A) Na+, Cl–, and osmolality in plasma in rats remained unchanged with HSD, while Na+ and Cl– content and concentrations increased in skin tissue. (B) HSD increased TonEBP and Vegfc mRNA expression and MPS cell count as well as MAP and interstitial fluid pressure (IFP) in the same rats. (C) Electron-dispersion x-ray scanning electron microprobe analysis of Na+ and Cl– concentrations in skin lymph capillaries in DOCA-HSD rats. The arrow denotes lymphatic capillary site from which the spectra were obtained. Scale bar: 20 mm. Lymphatic capillary Na+ was higher than that in plasma. The Cl– values were not significantly different. (D) Na+ concentration and osmolality in skin microdialysate and in plasma in rats. (E) Direct plasma and skin-tissue vapor pressure osmolality measurements in rats after 2 weeks of LSD and HSD. DOCA, deoxycorticosterone acetate. *P (diet) < 0.05; †P (fluid composition) < 0.05.
Discussion
To verify the role of MPS cells in regulating interstitial electrolyte concentrations, we genetically induced TONEBP deficiency in MPS cells. TONEBP binds to the VEGFC promoter and increases VEGFC expression in MPS cells in vitro (16). In vivo, reduced skin TONEBP expression in LysMcreTonEBPfl/fl mice fed HSD was directly coupled with reduced VEGFC expression. VEGFC binds to VEGFR3 on lymph endothelial cells and increases lymphatic capillary density in mice with Na+- and Cl–-overloaded skin (15, 16). We found that deletion of MPS cell TONEBP blocked the expected increase in lymphatic capillary density in response to skin electrolyte storage-induced osmotic stress. Skin electrolyte composition analysis showed increased skin Cl– content in LysMcreTonEBPfl/fl mice. In contrast, MPS-specific TONEBP deletion did not change skin Na+ and water content in the mice. We conclude that TONEBP is necessary to enable MPS cells to signal a VEGFC-dependent increase in cutaneous lymphatic capillary density and induces local Cl– clearance. Our data indicate that TONEBP is an essential component by which MPS cells regulate extracellular fluid composition.
Our additional experiments with an antibody directed against VEGFR3 (mF4-31c1), to selectively block the VEGFC/VEGFR3–mediated increase in lymphatic capillary density, allowed us to investigate homeostatic regulation of local electrolyte clearance distal to TONEBP-mediated VEGFC expression. These experiments were designed to test the hypothesis that lymph capillaries are relevant for systemic blood pressure regulation. We found that the antibody eliminated the lymphatic capillary density response but not the increased eNOS and p-eNOS expression in response to HSD. Thus, we were able to eliminate diminished eNOS production as the responsible mechanism for increased blood pressure in response to HSD. Similar to our findings with specific TONEBP deletion in MPS cells, the most robust inverse correlations between lymphatic capillary density and skin electrolyte content were with skin Cl–, rather than Na+ concentrations. Our comparisons between skin Na+ and Cl– content suggest that this Cl– accumulation is paralleled by a commensurate reduction of unmeasured anionic osmolytes. We speculate that a hyperplastic cutaneous lymphatic capillary network secretes increased amounts of negatively charged organic molecules (such as GAGs) into the skin interstitium. It is unclear to us why skin Cl– content, and not Na+ and water content, was so tightly associated with salt-sensitive blood pressure increases in our mice. Recently, evidence from our group suggests that skin Na+ and Cl– storage in rats is associated with increased in vitro contractility of skin resistance vessels (19). We speculate that changes in interstitial Cl– ion concentration, or unidentified organic components secreted by the lymphatic capillary network or immune cells, may modify vascular resistance in the skin and increase blood pressure. The steep inverse relationship between blood pressure and skin lymphatic capillary density in our anti-VEGFR3 experiments supports the idea that the lymphatic capillary endothelium could secrete factors relevant for systemic blood pressure regulation.
To address our hypothesis that lymphatic regulation of electrolyte composition in the skin is relevant for systemic blood pressure control, we also tested skin electrolyte homeostasis and blood pressure in a transgenic model that functionally eliminates skin VEGFC and thereby disrupts cutaneous lymph capillaries (17). HSD in K14-FLT4 mice led to pronounced Na+, Cl–, and water retention in the skin, which was paralleled by increased blood pressure. The blood pressure results of these experiments are in accord with those obtained using the antibody directed against VEGFR3 and confirmed our initial hypothesis. Na+, Cl–, and water retention in the skin accompanied the increased blood pressure in K14-FLT4 mice. In contrast, blockade of MPS/VEGFC/VEGFR3–driven hyperplasia of skin lymph capillaries in WT mice resulted in selective tissue Cl– retention, which was paralleled by hypertension, even in the absence of parallel Na+ and water accumulation in skin. We interpret these findings to indicate that preexisting hypoplasia in cutaneous lymph capillaries in K14-FLT4 mice results in expected parallel electrolyte and volume retention with HSD, while HSD-induced hyperplasia of intact cutaneous lymph capillaries seems to specifically enhance clearance of Cl– from tissue. The underlying mechanisms responsibly for this Cl– specificity are unknown.
Finally, we show that HSD results in Na+ and Cl– storage in the skin in rats and in mice. This electrolyte storage is not associated with commensurate water retention, and skin electrolyte storage is not entirely osmotically inactive but instead increases osmolality. We used 5 separate approaches to substantiate our hypotheses. First, chemical analysis showed increases in skin Na+ and Cl– content, without commensurate increases in tissue water. This finding suggests that electrolytes are stored in tissue, either by binding to negatively charged matrix components (GAGs) or by exerting hyperosmolality (4–6, 11, 13). Second, electron microprobe analysis provided the first measurements of electrolyte concentrations in prenodal skin lymph vessels, demonstrating that Na+ concentrations in pericapillary interstitial skin fluid are substantially higher than those in plasma (20). Third, skin microdialysis experiments showed increased Na+ concentrations and higher osmolality in microdialysate compared with that in plasma. Fourth, direct vapor pressure measurements of plasma and skin showed that HSD increased tissue osmolality. Fifth, TONEBP, which induces expression of VEGFC from MPS cells in response to osmotic stress, was concomitantly expressed with skin electrolyte storage, indicating a hypertonic microenvironment. These 5 independent observations, in line with findings of other investigators (5, 7), lead us to conclude that salt storage in the skin is characterized by a local hypertonic microenvironment. The resulting osmotic stress triggers homeostatic macrophages to exert their regulatory activity and modulate interstitial electrolyte composition by TONEBP/VEGFC–driven modulation of cutaneous lymphatic capillary function, which may include enhanced pumping activity of the lymph vessels (21).
This series of events suggests that MPS cells regulate skin interstitial electrolyte homeostasis. MPS cells constitute a subgroup of monocytes, macrophages, and dendritic cells that respond to infection or tissue damage to restore the integrity of the body (22). Macrophages exhibit a phenotypic continuum, with overlapping functions ranging from classically activated macrophages to alternatively activated, wound healing, and regulatory macrophages (23). While classically activated macrophages protect against fungi, bacteria, and viruses, they contribute to extracellular matrix destruction and tissue damage. However, the cells of the MPS not only combat invading pathogens, but also orchestrate an antiinflammatory milieu, in which proinflammatory activation is inhibited while expression profiles promoting cell proliferation, tissue repair, extracellular matrix construction, and angiogenesis are expanded (24–26). In addition to these classical immunological roles, our evidence suggests that MPS cells are empowered to play an important homeostatic role to maintain the steady-state electrolyte composition of the tissue. Our findings support the notion of “homeostatic immune function” dating back to the work of Metchnikoff, demonstrating that macrophages play a central role in tissue development (27).
The idea that lymph capillaries are relevant for blood pressure control (15, 16, 28) might shed new light on the current discussion on how VEGF kinase inhibitors may elevate blood pressure (29, 30). Blood capillary rarefaction (31–34) or reduced eNOS expression (35–43) as well as lymphatic capillary rarefaction may lead to arterial hypertension in patients treated with angiogenesis inhibitors. In contrast to selective VEGFR2 blockade (44), however, the hypertension induced by VEGFR3 blockade in our mice was strictly salt sensitive. Dietary salt restriction therefore may serve to lower blood pressure in patients treated with angiogenesis inhibitors.
Current concepts on lymphatic function rely on the idea that changes in lymphatic morphology may modulate lymphatic pumping and thereby interstitial flow (18, 21, 45–51). Our data suggest that MPS cells may modulate lymphatic function by not only changing the quantity of lymph flow, but also by altering the relationship among electrolytes, water, and tonicity of interstitial fluids. The resulting change in the relationship between tissue electrolyte and water contents in the skin resembles the interstitial changes in the renal medulla (20), in which specialized tubular segments provide active transport mechanisms. As a result, segmental impermeability for water and countercurrents creates a local hypertonic interstitial microenvironment. In the kidney, this hypertonic interstitial microenvironment modifies urinary concentration to maintain a constant internal composition of the blood. While the physiological processes and molecular mechanisms involved are elusive, the presence of electrolyte transporters in skin lymph endothelial cells (52), combined with endothelial tight junctions in lymph collecting vessels (53–55), suggests that blood vascular countercurrents and lymph vessels in the skin may form a functional network for local regulation of internal environmental composition and blood pressure control.
Despite substantial MPS/VEGFC–driven changes in the dermal lymphatic capillary network, we found no activation of the MPS/TONEBP/VEGFC regulatory axis in the kidney after HSD. We also observed no changes in renal VEGFC expression in LysMcreTonEBPfl/fl mice. Furthermore, TonEBP gene expression in LysMcreTonEBPfl/fl mice was reduced in skin but not in kidney. We also observed no abnormalities in renal lymph vessels in K14-FLT4 mice. The renal inner medulla lacks lymphatic vessels that evidently are not necessary for fluid transport from the medulla to the cortex (56). Finally, in experiments with genetic depletion of the TONEBP/VEGFC regulatory axis in MPS cells and in experiments antagonizing lymph vessel formation with VEGFR3 blockade during HSD, we found selective Cl– accumulation in the skin, a direct correlation between skin Cl– content and blood pressure increases, and no relationship between Na+ and water content and blood pressure but a massive increase in skin Cl– concentration. In contrast, plasma Cl– concentration, which is undoubtedly under exclusive renal control, was not perturbed in the same mice. The findings suggest that MPS cells exert homeostatic immune function by extrarenal control of interstitial electrolyte composition via skin lymph capillaries.
In summary, we have elucidated an interstitial environment that is hypertonic to the plasma compartment. MPS cells outfitted with TONEBP can detect this local osmotic stress when aggravated by HSD. The local signaling regulates lymphatic capillary density. We hypothesize that an additional biological clearance system exists in the skin interstitium, which is necessary to protect tissues from electrolyte overload. In contrast to the view that renal electrolyte elimination is sufficient for the control of the internal environment (57–59), MPS-driven lymphatic capillary hyperplasia provides an additional extrarenal regulatory component. Na+ retention with other accompanying anions than Cl– does not increase blood pressure (14, 60–65). We are intrigued by the observation that tissue Cl– content was directly related to blood pressure in that regard. We are aware that these studies do not explain why peripheral vascular resistance in skin vessels is increased in salt-consuming animals (19). Although we believe that our findings regarding lymphatic capillary density and its importance to skin electrolyte clearance are relevant to this process, future research must pursue this issue further. Finally, we have the impression that our findings are directly clinically relevant. Our long-term observations on Na+ balance in humans (66), showing infradian rhythms of Na+ storage, independent of body weight or blood pressure, are consistent with the interstitial electrolyte storage that we observed in our animal studies. We have since implemented 23Na magnetic resonance imaging techniques to make Na+ storage visible in animals and patients (67, 68) and have shown that essential hypertension in humans is paralleled by tissue Na+ storage (69).
Methods
Experiments in mice.
For the investigation of TONEBP deficiency in MPS cells, we had TonEBPfl/fl mice generated by inGenious Targeting Laboratory Inc. using standard gene-targeting techniques (Supplemental Figure 6). The targeting construct was designed such that the short homology arm extends about 2.45 kb 3′ to exon 4, whereas the long homology arm extends about 5.25 kb 5′ to exon 4. A single LoxP site, containing an engineered StuI site for Southern blot analysis, was inserted 166 bp upstream of exon 4, and a LoxP/FRT flanked Neo cassette was inserted 202 bp downstream of exon 4. Thus, the target region is 561 bp long and includes exon 4. BA1 (C57BL/6 × 129/SvEv) hybrid embryonic stem cells were electroporated with 10 μg of linearized targeting vector and selected with G418 antibiotic. Four PCR-positive clones were further confirmed by Southern blot analysis. Targeted iTL BA1 (C57BL/6N × 129/SvEv) hybrid embryonic stem cells were microinjected into C57BL/6 blastocysts. Resulting chimeras, with a high percentage agouti coat color, were mated to C57BL/6 homozygous FLP mice to remove the Neo cassette. For standard genotyping of the offspring, we used primers 5′-GTTGTGCTTTGGATTCTCAGAAC-3′ and 5′-CTTCTACCCTTCTATTTCAGGAAGC-3′, which amplified a 675-bp fragment in case successful Neo deletion occurs. Finally, we crossbred LysMcreTonEBPfl/fl mice with LysMWTTonEBPfl/fl mice to generate male LysMcreTonEBPfl/fl mice (n = 16) and LysMWTTonEBPfl/fl mice (n = 12), which were fed a HSD (4% NaCl chow plus 0.9% saline to drink) for 2 weeks to investigate differences in expression of their TONEBP/VEGFC regulatory axis.
For the selective blockade of VEGFC/VEGFR3–driven hyperplasia of lymph vessels, we randomly assigned 44 male FVB mice (Charles River) aged 10–12 weeks to 4 groups. Groups 1 and 2 received a LSD (<0.1% NaCl chow) and tap water. Groups 3 and 4 received a HSD (4% NaCl chow plus 0.9% saline to drink). Groups 2 and 4 were additionally treated with 45 μg mF4-31c1 per g body weight i.p. every 48 hours (provided by ImClone Systems).
For the skin-specific genetic depletion of VEGFC, we used 14 male WT and 12 male mice with heterozygous overexpression of sVEGFR3 under the control of the keratinocyte promoter K14 (17). K14-FLT4 mice overexpress sVEGFR3 in their skin, resulting in hypoplasia of the subcutaneous lymphatic capillary system (17, 70). WT and K14-FLT4 mice were randomly assigned to 4 groups. Groups 1 and 2 received a LSD. Groups 3 and 4 received a HSD. In all experiments with mice, we kept the mice on their specified diets for 2 weeks. Mouse diets were from Altrominy. At the end of the experiment, we anesthetized the mice with 1.5% to 2% isoflurane anesthesia and catheterized the common carotid artery. We connected arterial lines to BP Transducers and a PowerLab 8/30 Channel Recorder (ADInstruments) and measured MAP in conscious animals 1 hour after the operation. Thereafter, we took blood samples and sacrificed the animals. We analyzed arterial blood gases with a clinical blood gas analyzer (Radiometer Copenhagen), including Na+, K+, and Cl– measurements by ion-selective electrodes.
Experiments in rats.
We obtained 20 male Sprague Dawley rats (12 weeks of age) that were assigned to a LSD (<0.1% NaCl and tap water) and a HSD (8% NaCl and 0.9% saline water to drink) for 2 weeks. Chow was obtained from Altromin. We then characterized skin electrolyte accumulation by chemical analysis, MAP, skin interstitial fluid pressure, and gene expression of the TONEBP/VEGFC regulatory axis in the same animals. For the electron microprobe electrolyte measurements in primary lymph vessels, we used 3 deoxycorticosterone acetate–treated rats (50 mg/pellet) that received 1% saline water to drink for 3 consecutive weeks. For the microdialysis of skin interstitial fluid determinations, we assigned 20 male rats to 2 groups and fed them LSD or HSD for 2 consecutive weeks. To directly measure skin tissue osmolality, we assigned 12 male rats to 2 groups and fed them LSD or HSD for 2 consecutive weeks.
Cell culture experiments.
We isolated bone marrow–derived macrophages from LysMWTTonEBPfl/fl and LysMcreTonEBPfl/fl mice, as reported previously (71). Briefly, total bone marrow cells were cultured in hydrophobic Teflon bags (DuPont, purchased via Cadillac Plastic) with 10% CO2/ 90% humidified air in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 0.05 mmol/l 2-ME, 1% nonessential amino acids, 10% fetal calf serum (PAA), 5% horse serum (Cell Concepts), and 15% L929 cell-conditioned medium. After 8 days of culture, the cell population regularly contained more than 90% macrophages (CD11bhiF4/80hi). We then stimulated the primary macrophages by increasing the culture medium osmolality by adding 40 mM NaCl or 80 mM urea for 24 hours.
Chemical analysis of tissue electrolyte and water content.
We completely skinned the carcasses. We separately weighed the completely skinned carcasses and skin (wet weight) and then desiccated both at 90°C for 72 hours (dry weight [DW]). Because weights were unchanged with further drying, the difference between wet weight and DW was considered as tissue water content. We then ashed the tissues at 190°C and 450°C for 24 hours at each temperature level and sieved the bones from the carcass ashes. We further ashed the separated tissues at 600°C for 48 hours and then dissolved them in 5% or 10% HNO3. We measured Na+ and K+ concentration by atomic adsorption spectrometry (Model 3100, Perkin Elmer) and Cl– concentration by titration with 0.1 N silver nitrate (Model Titrando, German Metrohm).
Electron microprobe analysis.
Electron microprobe analysis was performed by energy-dispersive x-ray spectroscopy, as described previously (20). Tissue from the rat ear was shock frozen in an isopentane/propane mixture (1:4) at –196°C, small pieces were subsequently mounted in a precooled ultracryomicrotome (modified Ultrotome V, LKB), and 1-μm-thick cryosections were cut at –80°C. The sections were sandwiched between 2 films (formvar and collodium) and subsequently freeze-dried at –80°C and 10-4 Pa. X-ray microanalysis was performed in a scanning transmission electron microscope (S 150, Cambridge Instruments) equipped with an energy-dispersive x-ray–detecting system (Link Systems). Small areas (1 μm2) in the vessels devoid of erythrocytes (presumably lymphatic vessels) were analyzed using an acceleration voltage of 20 kV and a probe current of 0.3 nA. The preparation and electron microprobe analysis of freeze-dried cryosections and the quantification of the x-ray spectra have been described previously (72).
Measurement of tissue and plasma osmolality.
Osmolality in skin samples was measured using a method slightly modified from that described by Tornheim (73) and Knepper (74). Immediately after terminating the experiment, the rat back skin was closely clipped, removed, and snap frozen in liquid nitrogen. Frozen samples were wrapped in aluminum foil and transferred to double plastic bags to prevent evaporation and stored at –20°C until analysis. At the day of analysis, samples were transferred to dry ice. Skin discs were prepared with a cork bore, 7.5 mm, and chilled on dry ice, and discs were again wrapped in plastic foil and stored on dry ice. From these, 0.5-mm-thick discs were cut using a chilled tissue slicer (Thomas Stadie-Riggs Tissue Slicer, Thomas Scientific); immediately after cutting, they were transferred to the sample chamber of the osmometer (5500 Wescor vapor pressure osmometer, Wescor Inc.) that had been calibrated with standards 50–850 mOsmol/l. Reported values are the average of 3 to 5 individual samples, and the calibration was checked after each rat. Similarly, we determined osmolality in plasma from blood samples from the same rats by vapor pressure osmometry.
Measurement of interstitial fluid pressure.
During experiment 4, we measured interstitial fluid pressure in the skin and intraarterial blood pressure simultaneously in anesthetized rats (1.5%–2% isoflurane anesthesia; n = 4 per salt diet). We measured interstitial fluid pressure by micropuncture technique, as described in detail previously (75). A sharpened glass capillary (diameter; 4–7 μm) connected to an automatic counterpressure system was inserted through intact back skin. We simultaneously monitored intraarterial blood pressure after cannulation of the common carotid artery as described above in the mouse experiments.
Microdialysis.
We anesthetized rats with 1.5% to 2% isoflurane anesthesia and placed intracutaneous microdialysis probes (20MD Elite Probe, CMA Microdialysis) in neck skin and cannulated the common carotid artery. We then perfused probes with Ringer’s solution at flow rate of 2.1 μl/min for 1 hour for equilibration. We then reduced flow rate to 0.3 μl/min and collected the microdialysate for 4 hours.
Real-time PCR.
We extracted total RNA from animal tissue with RNeasy minicolumns (Qiagen), homogenizing skin slices (~10–20 mg) in 500 μl RLT buffer reagent with an Ultra-Turrax for 30 seconds. After homogenization, we added 950 μl water and 16 μl proteinase K (25 U/μl) and incubated the sample at 55°C for 10 minutes and then centrifuged it at 13,500 g for 3 minutes. After addition of 1 ml 96%–100% ethanol, we transferred the solvent to the mini-columns and eluted according to the standard protocol. First-strand cDNA was synthesized with TaqMan RT reagents (Applied Biosystems), with random hexamers used as primers. We performed real-time PCR with an ABI PRISM 7000 sequence detector and SYBR green reagents (Applied Biosystems) according to the manufacturer’s instructions. Primers used for amplification are shown in Supplemental Figure 1. All samples were run in duplicate. We normalized the relative amount of the specific mRNA of interest with respect to 18S rRNA content in the sample. Dissociation curves confirmed the specificity of the PCR.
Quantification of protein expression.
For protein purification, cells were washed 3 times with PBS and incubated in 8 M urea, including protease inhibitors, for 10 minutes on ice. We then removed the cells from plates and centrifuged cells for 10 minutes at 16,000 g. The supernatant was used for immunoblot analysis. We homogenized cells and matrix proteins from shock-frozen skin in 500 μl lysis buffer (50 mM Tris HCl, pH 7.2, 150 mM NaCl, 1% Triton X-100, 1 mM sodium orthovanadate, 50 μg/ml sodium pyrophosphate, 100 mM sodium fluoride, 0.01% aprotinin, 4 μg/ml pepstatin A, 10 μg/ml leupeptin, and 1 mM phenylmethylsulfonyl fluoride) with an Ultra-Turrax for 30 seconds.
Immunoblot.
For immunoblotting, we separated equal amounts of total protein on 8% or 12% SDS-polyacrylamide gel under reducing conditions and electroblotted onto a PVDF membrane. We blocked the blots with 5% nonfat milk in PBS and 0.1% Tween 20, pH 7.5, for 1 hour at room temperature and then incubated them overnight at 4°C with anti-TONEBP (ABR) diluted 1:1,000; anti-CD68 (Serotec) diluted 1:1,000; anti-VEGFC (Abcam) diluted 1:1,000; eNOS (Anaspec) diluted 1:500; p-eNOS (Ser1177; Cell Signaling) diluted 1:500; or β-actin (Sigma-Aldrich) diluted 1:5,000. After 3 washes in PBS with 0.1% Tween 20, pH 7.5, we incubated the blots for 1 hour with horseradish peroxidase–conjugated secondary antibody (Pierce) diluted 1:1,000 in blocking solution at room temperature. We visualized antibody binding using an enhanced chemiluminescence system (Pierce). We scanned specific bands and quantified them by densitometry.
Immunhistochemistry.
We fixed animal tissues in 5% formalin and embedded them in paraffin. We performed all staining using the Avidin/Biotin Blocking Kit (Vector Laboratories) and HRP Super Staining Kit (ID Labs) according to manufacturer’s instructions. Briefly, we deparaffinized slides and boiled them twice for about 5 minutes at 600 W in a microwave in 0.1 M citrate buffer (pH 6.0). We cooled down the slides to room temperature and incubated them in 3% H2O2 for 10 minutes. We then blocked the slides with the Avidin/Biotin Blocking Kit (Vector Laboratories) according to manufacturer’s protocol and with SuperBlock for about 7 minutes. After washing 3 times with PBS, we incubated slides with anti-CD68 (Serotec) diluted 1:1,000 in 1% BSA for about 1 hour, followed by incubation with the polyvalent antibody and HRP for 10 minutes. We washed the slides 3 times with PBS between every step. We detected specific staining using AEC Chromogen/Substrate (3-amino-9-ethylcarbazole; ID Labs). CD68-positive cells were counted throughout the whole diameter of a crosswise cut mouse ear at 500-fold amplification under the microscope. Starting from the same edge of the ear, we counted MPS cells in 10 consecutive sections and normalized MPS cell counts per field.
Whole-mount staining of lymph capillaries.
We fixed ears in 4 % PFA overnight at 4°C. We removed cartilage under the microscope and attached ears on Sylgaard plates with insect pins, washed with 0.3% PBS-TritonX (4 hours, at least 10 times), and blocked with Immunomix (0.2% BSA, 5% goat serum in 0.3% PBS-TritonX) for 2 hours. We then incubated the samples with the primary antibody LYVE-1 (1:100 in Immunomix; Reliatech) overnight with shaking. They were washed in 0.3% PBS-TritonX for 6 hours (at least 5 times). Afterward we incubated samples with the secondary antibody (anti-rabbit Alexa Fluor A488; 1:500 in 0.3% PBS-TritonX) overnight at room temperature with shaking, and we washed samples for 6 hours (at least 5 times) after that. We performed post fixing by incubating samples in 4% PFA for 10 minutes at room temperature. After washing 5 times with PBS for 30 minutes at room temperature, the ears were mounted with Vectashield (Vector Laboratories). They were analyzed with fluorescent microscopy. Quantification of fluorescence was performed using Image J.
GAG disaccharide analysis.
We cut approximately 0.2 g of abdominal skin from mice into small slices and digested with 7.0 mg proteinase K in 5 ml 20 mM Tris-buffer (pH 7.0) at 55°C for 16 hours (then boiled for 5 minutes). After centrifugation, we added 50 ml acetone to the supernatant and stored the tubes at –20°C for 16 hours. After precipitation, the tubes were centrifuged at 1,500 g for 5 minutes, and the precipitate was dried at 90°C overnight. We dissolved the dried material in 10 ml distilled water and introduced it into glass columns containing 9 ml of the anion-exchange resin (DEAE, Sigma-Aldrich). After washing the resin twice with 10 ml 100 mM NaCl, pH 4, we eluted the GAGs with 15 ml 2.5 M NaCl, pH 4, and transferred the eluate to centrifugation tubes and again precipitated with 150 ml acetone at –20°C for 16 hours. After precipitation, the tubes were centrifuged at 1,500 g for 5 minutes, and the precipitate was dried further at 90°C for 16 hours. We dissolved the dried material with the purified GAGs in 10 ml water, transferred it to a dialysis bag, and dialyzed it against distilled water for 24 hours. After lyophilization, we dissolved the purified skin GAGs in 0.5 ml deionized water for HPLC analysis.
HPLC disaccharide analysis.
We treated 10 μl of samples with 5 mU chondroitin ABC lyase in 50 μl 100 mM Tris/150 mM sodium acetate buffer, pH 8.0, at 37°C for 16 hours. Boiling the solution for 1 minute blocked the reaction. We analyzed the unsaturated disaccharides generated from hyaluronan (ΔdiHA) and dermatan/chondroitin sulphate (Δdi4s and Δdi6s) after enzymatic treatment of the purified GAGs by strong anion exchange HPLC (column: Sphere-Image 80-5 SAX, Knauer) separation and detected at 232 nm. Isocratic separation was from 0 to 15 minutes with 10 mM NaHPO, pH 4.0, and linear gradient separation was from 15 to 35 minutes with 10 mM NaH2PO, pH 4.0, to 33% 750 mM NaH2PO, pH 4.0. Flow rate was 1.2 ml/min. HPLC equipment was from Shimadzu.
Statistics.
Comparison of means of data from animal experiments with more than one effector (salt and genotype; salt and mF4-31c1 treatment) was calculated by multivariate or univariate analysis using the general linear measurements procedure. We tested for the effect of diet, genotype, and mF4-31c1 treatment. Differences between plasma and interstitial electrolyte concentrations and osmolality were investigated by paired t test (dependent samples) or 1-way ANOVA (independent samples). All data in the manuscript are presented as average ± SD. The terms “increased” or “decreased” are used only if the results were significant at P < 0.05. Statistical analysis was performed with SPSS software (version 15.0).
Study approval.
We performed all animal experiments in accordance with the guidelines of the American Physiological Society, and the animal care and use committee of local government authorities approved the studies (Regierung von Mittelfranken, Ansbach, Germany).
Supplementary Material
Acknowledgments
This study was supported by grants from the Interdisziplinäres Zentrum für klinische Forschung (IZKF), Erlangen; the Bundesministerium für Wirtschaft und Technologie – Forschung unter Weltraumbedingungen (DLR/BMBF; grant 50WB0620 to J. Titze); and the Deutsche Forschungsgemeinschaft (DFG; grant Ti345/2 to J. Titze; grant SFB643/TP B16 to J. Jantsch and J. Titze) as well as from the ELAN-Fonds Erlangen. The Grieg Foundation and the Research Council of Norway supported H. Wiig. NIH grants R01HL039006, P01HL058000, P01HL095070, P01GM015431, and R01HL105294 supported D.G. Harrison. We are indebted to Elke Prell and Sabrina Cabric for their technical assistance and to Bronek Ptykowski (ImClone) for providing us with the mF4-31c1 antibody.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exist.
Citation for this article:J Clin Invest. 2013;123(7):2803–2815. doi:10.1172/JCI60113.
References
- 1.Marvar PJ, Gordon FJ, Harrison DG. Blood pressure control: salt gets under your skin. Nat Med. 2009;15(5):487–488. doi: 10.1038/nm0509-487. [DOI] [PubMed] [Google Scholar]
- 2.Guyton AC, Coleman TC, Cowley AW, Jr, Manning RD, Jr, Norman RA, Jr, Ferguson JD. A systems analysis approach to understanding long-range arterial blood pressure control and hypertension. Circ Res. 1974;35:159–176. doi: 10.1161/01.RES.35.2.159. [DOI] [Google Scholar]
- 3.Guyton AC, Coleman TG. Quantitative analysis of the pathophysiology of hypertension. Circ Res. 1969;24(5 suppl):1–19. doi: 10.1161/01.RES.24.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Haljamae H. Anatomy of the interstitial tissue. Lymphology. 1978;11(4):128–132. [PubMed] [Google Scholar]
- 5.Haljamae H, Linde A, Amundson B. Comparative analyses of capsular fluid and interstitial fluid. Am J Physiol. 1974;227(5):1199–1205. doi: 10.1152/ajplegacy.1974.227.5.1199. [DOI] [PubMed] [Google Scholar]
- 6.Ivanova LN, Archibasova VK, Shterental’ ISh. [Sodium-depositing function of the skin in white rats]. Fiziol Zh SSSR Im I M Sechenova. 1978;64(3):358–363. [PubMed] [Google Scholar]
- 7.Szabo G, Magyar Z. Electrolyte concentrations in subcutaneous tissue fluid and lymph. Lymphology. 1982;15(4):174–177. [PubMed] [Google Scholar]
- 8.Schafflhuber M, et al. Mobilization of osmotically inactive Na+ by growth and by dietary salt restriction in rats. Am J Physiol Renal Physiol. 2007;292(5):F1490–F1500. doi: 10.1152/ajprenal.00300.2006. [DOI] [PubMed] [Google Scholar]
- 9.Titze J. Water-free sodium accumulation. Semin Dial. 2009;22(3):253–255. doi: 10.1111/j.1525-139X.2009.00569.x. [DOI] [PubMed] [Google Scholar]
- 10.Titze J, et al. Internal sodium balance in DOCA-salt rats: a body composition study. Am J Physiol Renal Physiol. 2005;289(4):F793–F802. doi: 10.1152/ajprenal.00096.2005. [DOI] [PubMed] [Google Scholar]
- 11.Titze J, et al. Osmotically inactive skin Na+ storage in rats. Am J Physiol Renal Physiol. 2003;285(6):F1108–F1117. doi: 10.1152/ajprenal.00200.2003. [DOI] [PubMed] [Google Scholar]
- 12.Titze J, et al. Extrarenal Na+ balance, volume, and blood pressure homeostasis in intact and ovariectomized deoxycorticosterone-acetate salt rats. Hypertension. 2006;47(6):1101–1107. doi: 10.1161/01.HYP.0000221039.17735.1a. [DOI] [PubMed] [Google Scholar]
- 13.Titze J, et al. Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am J Physiol Heart Circ Physiol. 2004;287(1):H203–H208. doi: 10.1152/ajpheart.01237.2003. [DOI] [PubMed] [Google Scholar]
- 14.Ziomber A, et al. Sodium-, potassium-, chloride-, and bicarbonate-related effects on blood pressure and electrolyte homeostasis in deoxycorticosterone acetate-treated rats. Am J Physiol Renal Physiol. 2008;295(6):F1752–F1763. doi: 10.1152/ajprenal.00531.2007. [DOI] [PubMed] [Google Scholar]
- 15.Machnik A, et al. Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension. 2010;55(3):755–761. doi: 10.1161/HYPERTENSIONAHA.109.143339. [DOI] [PubMed] [Google Scholar]
- 16.Machnik A, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15(5):545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 17.Makinen T, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. 2001;7(2):199–205. doi: 10.1038/84651. [DOI] [PubMed] [Google Scholar]
- 18.Wiig H, Swartz MA. Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiol Rev. 2012;92(3):1005–1060. doi: 10.1152/physrev.00037.2011. [DOI] [PubMed] [Google Scholar]
- 19.Helle F, Karlsen TV, Tenstad O, Titze J, Wiig H. High-salt diet increases hormonal sensitivity in skin pre-capillary resistance vessels. Acta Physiol (Oxf). 2013;207(3):577–581. doi: 10.1111/apha.12049. [DOI] [PubMed] [Google Scholar]
- 20.Beck FX, Schmolke M, Guder WG, Dorge A, Thurau K. Osmolytes in renal medulla during rapid changes in papillary tonicity. Am J Physiol. 1992;262(5 pt 2):F849–F856. doi: 10.1152/ajprenal.1992.262.5.F849. [DOI] [PubMed] [Google Scholar]
- 21.Kwon S, Agollah GD, Chan W, Sevick-Muraca EM. Altered lymphatic function and architecture in salt-induced hypertension assessed by near-infrared fluorescence imaging. J Biomed Opt. 2012;17(8):080504-1. doi: 10.1117/1.JBO.17.8.080504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2011;327(5966):656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 25.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 26.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118(11):3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mekarski JE. Essential hypertension is lymphatic: a working hypothesis. Med Hypotheses. 1998;51(2):101–103. doi: 10.1016/S0306-9877(98)90101-3. [DOI] [PubMed] [Google Scholar]
- 29.Bhargava P. VEGF kinase inhibitors: how do they cause hypertension? Am J Physiol Regul Integr Comp Physiol. 2009;297(1):R1–R5. doi: 10.1152/ajpregu.90502.2008. [DOI] [PubMed] [Google Scholar]
- 30.Stortecky S, Suter TM. Insights into cardiovascular side-effects of modern anticancer therapeutics. Curr Opin Oncol. 2010;22(4):312–317. doi: 10.1097/CCO.0b013e32833ab6f1. [DOI] [PubMed] [Google Scholar]
- 31.Harper RN, Moore MA, Marr MC, Watts LE, Hutchins PM. Arteriolar rarefaction in the conjunctiva of human essential hypertensives. Microvasc Res. 1978;16(3):369–372. doi: 10.1016/0026-2862(78)90070-5. [DOI] [PubMed] [Google Scholar]
- 32.Prasad A, Dunnill GS, Mortimer PS, MacGregor GA. Capillary rarefaction in the forearm skin in essential hypertension. J Hypertens. 1995;13(2):265–268. [PubMed] [Google Scholar]
- 33.Hansen-Smith FM, Morris LW, Greene AS, Lombard JH. Rapid microvessel rarefaction with elevated salt intake and reduced renal mass hypertension in rats. Circ Res. 1996;79(2):324–330. doi: 10.1161/01.RES.79.2.324. [DOI] [PubMed] [Google Scholar]
- 34.Mourad JJ, des Guetz G, Debbabi H, Levy BI. Blood pressure rise following angiogenesis inhibition by bevacizumab. A crucial role for microcirculation. Ann Oncol. 2008;19(5):927–934. doi: 10.1093/annonc/mdm550. [DOI] [PubMed] [Google Scholar]
- 35.Gu JW, Manning RD, Jr, Young E, Shparago M, Sartin B, Bailey AP. Vascular endothelial growth factor receptor inhibitor enhances dietary salt-induced hypertension in Sprague-Dawley rats. Am J Physiol Regul Integr Comp Physiol. 2009;297(1):R142–R148. doi: 10.1152/ajpregu.90972.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am J Physiol. 1998;274(3 pt 2):H1054–H1058. doi: 10.1152/ajpheart.1998.274.3.H1054. [DOI] [PubMed] [Google Scholar]
- 37.Matsuura A, Yamochi W, Hirata K, Kawashima S, Yokoyama M. Stimulatory interaction between vascular endothelial growth factor and endothelin-1 on each gene expression. Hypertension. 1998;32(1):89–95. doi: 10.1161/01.HYP.32.1.89. [DOI] [PubMed] [Google Scholar]
- 38.Bouloumie A, Schini-Kerth VB, Busse R. Vascular endothelial growth factor up-regulates nitric oxide synthase expression in endothelial cells. Cardiovasc Res. 1999;41(3):773–780. doi: 10.1016/S0008-6363(98)00228-4. [DOI] [PubMed] [Google Scholar]
- 39.Shen BQ, Lee DY, Zioncheck TF. Vascular endothelial growth factor governs endothelial nitric-oxide synthase expression via a KDR/Flk-1 receptor and a protein kinase C signaling pathway. J Biol Chem. 1999;274(46):33057–33063. doi: 10.1074/jbc.274.46.33057. [DOI] [PubMed] [Google Scholar]
- 40.Li B, et al. KDR (VEGF receptor 2) is the major mediator for the hypotensive effect of VEGF. Hypertension. 2002;39(6):1095–1100. doi: 10.1161/01.HYP.0000018588.56950.7A. [DOI] [PubMed] [Google Scholar]
- 41.Veronese ML, et al. Mechanisms of hypertension associated with BAY 43-9006. J Clin Oncol. 2006;24(9):1363–1369. doi: 10.1200/JCO.2005.02.0503. [DOI] [PubMed] [Google Scholar]
- 42.Curwen JO, Musgrove HL, Kendrew J, Richmond GH, Ogilvie DJ, Wedge SR. Inhibition of vascular endothelial growth factor-A signaling induces hypertension: examining the effect of cediranib (recentin; AZD2171) treatment on blood pressure in rat and the use of concomitant antihypertensive therapy. Clin Cancer Res. 2008;14(10):3124–3131. doi: 10.1158/1078-0432.CCR-07-4783. [DOI] [PubMed] [Google Scholar]
- 43.Sandrim VC, Palei AC, Metzger IF, Gomes VA, Cavalli RC, Tanus-Santos JE. Nitric oxide formation is inversely related to serum levels of antiangiogenic factors soluble fms-like tyrosine kinase-1 and soluble endogline in preeclampsia. Hypertension. 2008;52(2):402–407. doi: 10.1161/HYPERTENSIONAHA.108.115006. [DOI] [PubMed] [Google Scholar]
- 44.Facemire CS, Nixon AB, Griffiths R, Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54(3):652–658. doi: 10.1161/HYPERTENSIONAHA.109.129973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gashev AA, Zawieja DC. Hydrodynamic regulation of lymphatic transport and the impact of aging. Pathophysiology. 2010;17(4):277–287. doi: 10.1016/j.pathophys.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zawieja DC. Contractile physiology of lymphatics. Lymphat Res Biol. 2009;7(2):87–96. doi: 10.1089/lrb.2009.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gasheva OY, Zawieja DC, Gashev AA. Contraction-initiated NO-dependent lymphatic relaxation: a self-regulatory mechanism in rat thoracic duct. J Physiol. 2006;575(pt 3):821–832. doi: 10.1113/jphysiol.2006.115212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zawieja D. Lymphatic biology and the microcirculation: past, present and future. Microcirculation. 2005;12(1):141–150. doi: 10.1080/10739680590900003. [DOI] [PubMed] [Google Scholar]
- 49.Boardman KC, Swartz MA. Interstitial flow as a guide for lymphangiogenesis. Circ Res. 2003;92(7):801–808. doi: 10.1161/01.RES.0000065621.69843.49. [DOI] [PubMed] [Google Scholar]
- 50.Mkonyi LE, Bletsa A, Fristad I, Wiig H, Berggreen E. Importance of lymph vessels in the transcapillary fluid balance in the gingiva studied in a transgenic mouse model. Am J Physiol Heart Circ Physiol. 2010;299(2):H275–H283. doi: 10.1152/ajpheart.01199.2009. [DOI] [PubMed] [Google Scholar]
- 51.Wiig H, Keskin D, Kalluri R. Interaction between the extracellular matrix and lymphatics: consequences for lymphangiogenesis and lymphatic function. Matrix Biol. 2010;29(8):645–656. doi: 10.1016/j.matbio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wick N, et al. Transcriptomal comparison of human dermal lymphatic endothelial cells ex vivo and in vitro. Physiol Genomics. 2007;28(2):179–192. doi: 10.1006/geno.1995.1129. [DOI] [PubMed] [Google Scholar]
- 53.Baluk P, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204(10):2349–2362. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corada M, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96(17):9815–9820. doi: 10.1073/pnas.96.17.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009;335(1):17–25. doi: 10.1007/s00441-008-0694-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tenstad O, Heyeraas KJ, Wiig H, Aukland K. Drainage of plasma proteins from the renal medullary interstitium in rats. J Physiol. 2001;536(pt 2):533–539. doi: 10.1111/j.1469-7793.2001.0533c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edelman IS, Leibman J, O’Meara MP, Birkenfeld LW. Interrelations between serum sodium concentration, serum osmolarity and total exchangeable sodium, total exchangeable potassium and total body water. J Clin Invest. 1958;37(9):1236–1256. doi: 10.1172/JCI103712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pitts RF. Physiology Of The Kidney And Body Fluids. 1974. Volume and composition of the body fluids. In: Pitts RF, ed. pp. 11–34. Chicago, Illinois, USA: Year Book Medical Publishers; [Google Scholar]
- 59.Rose BD. Regulation Of Water And Electrolyte Balance. New York, New York, USA: McGraw-Hill, Inc.; 1994. [Google Scholar]
- 60.Kurtz TW, Al-Bander HA, Morris RC., Jr “Salt-sensitive” essential hypertension in men. Is the sodium ion alone important? N Engl J Med. 1987;317(17):1043–1048. doi: 10.1056/NEJM198710223171702. [DOI] [PubMed] [Google Scholar]
- 61.Kurtz TW, Morris RC. Dietary chloride as a determinant of “sodium-dependent” hypertension. Science. 1983;222(4628):1139–1141. doi: 10.1126/science.6648527. [DOI] [PubMed] [Google Scholar]
- 62.Kotchen TA, Galla JH, Luke RG. Contribution of chloride to the inhibition of plasma renin by sodium chloride in the rat. Kidney Int. 1978;13(3):201–207. doi: 10.1038/ki.1978.30. [DOI] [PubMed] [Google Scholar]
- 63.Kotchen TA, Kotchen JM. Dietary sodium and blood pressure: interactions with other nutrients. Am J Clin Nutr. 1997;65(2 suppl):708S–711S. doi: 10.1093/ajcn/65.2.708S. [DOI] [PubMed] [Google Scholar]
- 64.Whitescarver SA, Ott CE, Jackson BA, Guthrie GP, Jr, Kotchen TA. Salt-sensitive hypertension: contribution of chloride. Science. 1984;223(4643):1430–1432. doi: 10.1126/science.6322303. [DOI] [PubMed] [Google Scholar]
- 65.Zicha J, Kunes J. Haemodynamic changes induced by short- and long-term sodium chloride or sodium bicarbonate intake in deoxycorticosterone-treated rats. Acta Physiol Scand. 1994;151(2):217–223. doi: 10.1111/j.1748-1716.1994.tb09740.x. [DOI] [PubMed] [Google Scholar]
- 66.Rakova N, et al. Long-term space flight simulation reveals infradian rhythmicity in human Na(+) balance. Cell Metab. 2013;17(1):125–131. doi: 10.1016/j.cmet.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 67.Kopp C, et al. Seeing the sodium in a patient with hypernatremia. Kidney Int. 2012;82(12):1343–1344. doi: 10.1038/ki.2012.314. [DOI] [PubMed] [Google Scholar]
- 68.Kopp C, et al. (23)Na magnetic resonance imaging of tissue sodium. Hypertension. 2012;59(1):167–172. doi: 10.1161/HYPERTENSIONAHA.111.183517. [DOI] [PubMed] [Google Scholar]
- 69.Kopp C, et al. 23Na magnetic resonance imaging-determined tissue sodium in healthy subjects and hypertensive patients. Hypertension. 2013;61(3):635–640. doi: 10.1161/HYPERTENSIONAHA.111.00566. [DOI] [PubMed] [Google Scholar]
- 70.Tammela T, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454(7204):656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 71.Wiese M, Castiglione K, Hensel M, Schleicher U, Bogdan C, Jantsch J. Small interfering RNA (siRNA) delivery into murine bone marrow-derived macrophages by electroporation. J Immunol Methods. 2010;353(1–2):102–110. doi: 10.1016/j.jim.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 72.Sone M, Albrecht GJ, Dorge A, Thurau K, Beck FX. Osmotic adaptation of renal medullary cells during transition from chronic diuresis to antidiuresis. Am J Physiol. 1993;264(4 pt 2):F722–F729. doi: 10.1152/ajprenal.1993.264.4.F722. [DOI] [PubMed] [Google Scholar]
- 73.Tornheim PA. Use of a vapor pressure osmometer to measure brain osmolality. J Neurosci Methods. 1980;3(1):21–35. doi: 10.1016/0165-0270(80)90031-X. [DOI] [PubMed] [Google Scholar]
- 74.Knepper MA. Measurement of osmolality in kidney slices using vapor pressure osmometry. Kidney Int. 1982;21(4):653–655. doi: 10.1038/ki.1982.73. [DOI] [PubMed] [Google Scholar]
- 75.Wiig H, Reed RK, Aukland K. Micropuncture measurement of interstitial fluid pressure in rat subcutis and skeletal muscle: comparison to wick-in-needle technique. Microvasc Res. 1981;21(3):308–319. doi: 10.1016/0026-2862(81)90014-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.