

Background: Scorpion α-toxins affect voltage-gated sodium channels in both mammals and insects.
Results: We perform thorough computational analyses of α-toxin molecular architecture and structure-function relationship.
Conclusion: Taxon specificity of “orphan” toxins can be predicted from a structural perspective.
Significance: The proposed surface mapping technique is a new tool to analyze protein-protein complexes.
Keywords: Ion Channels, Membrane Proteins, Molecular Dynamics, Neurotoxin, Protein Domains, Protein Dynamics, Protein Structure, Protein-Protein Interactions, Sodium Channels
Abstract
To gain success in the evolutionary “arms race,” venomous animals such as scorpions produce diverse neurotoxins selected to hit targets in the nervous system of prey. Scorpion α-toxins affect insect and/or mammalian voltage-gated sodium channels (Navs) and thereby modify the excitability of muscle and nerve cells. Although more than 100 α-toxins are known and a number of them have been studied into detail, the molecular mechanism of their interaction with Navs is still poorly understood. Here, we employ extensive molecular dynamics simulations and spatial mapping of hydrophobic/hydrophilic properties distributed over the molecular surface of α-toxins. It is revealed that despite the small size and relatively rigid structure, these toxins possess modular organization from structural, functional, and evolutionary perspectives. The more conserved and rigid “core module” is supplemented with the “specificity module” (SM) that is comparatively flexible and variable and determines the taxon (mammal versus insect) specificity of α-toxin activity. We further show that SMs in mammal toxins are more flexible and hydrophilic than in insect toxins. Concomitant sequence-based analysis of the extracellular loops of Navs suggests that α-toxins recognize the channels using both modules. We propose that the core module binds to the voltage-sensing domain IV, whereas the more versatile SM interacts with the pore domain in repeat I of Navs. These findings corroborate and expand the hypothesis on different functional epitopes of toxins that has been reported previously. In effect, we propose that the modular structure in toxins evolved to match the domain architecture of Navs.
Introduction
Voltage-gated sodium channels (Navs)6 are vital components of the nervous system and muscles, playing a central role in the excitability of these tissues (1, 2). Dysfunctions of Navs cause a number of channelopathies (3), and approaches for treatment of these diseases are eagerly awaited.
The pore-forming α-subunit of Navs (∼260 kDa) is organized in four non-identical but homologous repeats (pseudosubunits) (I–IV), each consisting of six transmembrane segments (S1–S6). Segments S1–S4 form a voltage-sensing domain (VSD) in each of the repeats (VSDs I-IV), whereas segments S5 and S6 from all four repeats contribute to the sole pore domain (PD) of Navs (PDs I–IV will denote S5-S6 from corresponding repeats). We should note here that the homologous repeats of Navs are more often referred to as “domains” in the literature. Although imperfect, we shall follow this conventional nomenclature.
Nav pharmacology is mostly determined by the α-subunit. Note that at present nine isoforms of α-subunit have been well characterized in humans (Nav1.1–1.9), and just one appears to function in insects (e.g. the Para protein in Drosophila) (4, 5). To date, a host of substances has been described affecting Navs through binding to different parts of the channels, so-called receptor sites (6). One of the most prominent examples is scorpion α-toxins, classical and potent modulators of Navs, which slow or inhibit channel inactivation, leading to prolongation of the action potential (7). Scorpion α-toxins bind to receptor site 3 of Navs, whereas the first two sites are targeted by tetrodotoxin and batrachotoxin, respectively (8). Extensive mutagenesis of Navs (in particular the rat neuronal isoform Nav1.2) allowed mapping of receptor site 3 to the extracellular surface of VSD IV of the channels with additional participation of the loops protruding from PD I (9–11).
Besides being the armament of venomous animals, natural toxins are perfect tools for investigation of the channel structure and function. They also represent templates for generation of pharmacologically active compounds modifying selectively Navs activity. Understanding of structure-activity and structure-selectivity relationships in toxins will serve as a firm basis for design of novel selective Nav modulators helpful in treating channelopathies in humans or, for example, generation of novel selective and safe insecticides.
Conventionally, scorpion α-toxins are divided into three groups based on taxon selectivity of their action: “mammal toxins” preferentially affecting mammals, “insect toxins” showing high insect toxicity, and “α-like toxins” affecting both mammals and insects (12). This classification is not strict, however, because insect toxins, for instance, are able to kill mammals, albeit at higher doses. Scorpion α-toxins have been the objects of structural biology research for over 25 years (13, 14), and it has been firmly established that these small proteins (∼65 residues) share a common βαββ motif, stapled by four disulfide bridges (7). Although much effort has been made to understand the structure-activity relationships in the three groups of α-toxins, no universal conclusion has been derived.
It is apparent that toxin selectivity is somehow encoded in the properties of its molecular surface and dynamics. A number of techniques have been developed for delineation of specific regions on molecular surfaces, which can be potentially involved in protein-protein recognition. Detailed mapping of surface hydrophobic/hydrophilic properties of two interacting molecules has been widely used for protein-ligand and protein-protein interactions (15–18), where molecular surface properties are projected onto a plane, cylinder, or sphere, aiding reduction of complexity and facilitating relatively straightforward comparison of projected “maps” rather than matching complex three-dimensional shapes.
To address the problem of α-toxin selectivity, we employ an original computational approach for detailed mapping and comparison of various physico-chemical properties, namely, hydrophobicity, flexibility, and electrostatic potential. This approach is based on spherical projection of the molecular properties and initially takes into account the dynamic behavior of molecules. Our results add novel atomistic description of putative mechanism of Nav-selective recognition by their peptide ligands and bring new formalism into the field of toxinology.
EXPERIMENTAL PROCEDURES
Homology Modeling
To enrich statistics, structures of several α-toxins with known pharmacological profile were modeled using related toxins as templates (see Table 1 for details). Alignments were produced with the ClustalW software (19) (Fig. 1). Homology modeling was performed with Modeler version 8.2 (20). The cis/trans configuration of the peptide bond between residues 8 and 9 was left “as is” in experimental structures; in models, the configuration was derived from templates, but it always got stabilized as trans for mammal and insect toxins and primarily cis for α-like toxins. The C termini of certain toxins were amidated where needed (see the footnotes to Table 1). 20 models were produced for each molecule; the model with the lowest value of the energy-like Modeler objective function was selected for molecular dynamics (MD) calculations.
TABLE 1.
Properties of α-toxins involved in the study
| Toxina | PDB ID or Uniprot ID/template (italics)b | Computed datac |
Toxicity (LD50)f |
References | ||||
|---|---|---|---|---|---|---|---|---|
| MHPSMd | MHPCored | RMSFRT e | RMSFCore e | Insects | Mammals | |||
| Mammal toxins | nm | nm | μg/kg | |||||
| Aah1 | P01479/1PTX | −0.027 ± 0.283 | −0.029 ± 0.017 | 0.17 ± 0.09 | 0.07 ± 0.06 | 1 900 | 0.5 (i.c.v.), 17.5 (s.c.) | 72, 73 |
| Aah2* | 1PTX | −0.109 ± 0.315 | 0.010 ± 0.013 | 0.18 ± 0.11 | 0.08 ± 0.06 | 6 500 | 0.025 (i.c.v.), 12 (s.c.) | 72–74 |
| Aah3 | P01480/1PTX | −0.017 ± 0.266 | 0.044 ± 0.015 | 0.26 ± 0.13 | 0.08 ± 0.04 | 10 200 | 0.35 (i.c.v.), 25 (s.c.) | 72, 73 |
| BmK M8 | 1SNB | −0.239 ± 0.273 | −0.015 ± 0.015 | 0.18 ± 0.09 | 0.12 ± 0.09 | 10,000 (i.v.) | 75 | |
| BmK αTx11 | 2KBH | −0.190 ± 0.311 | −0.023 ± 0.020 | 0.22 ± 0.10 | 0.12 ± 0.07 | |||
| Bot3* | P01485/1PTX | −0.138 ± 0.279 | 0.036 ± 0.017 | 0.16 ± 0.11 | 0.06 ± 0.04 | 1.25 (i.c.v.) | 76 | |
| Lqh2* | P59355/1PTX | −0.138 ± 0.294 | 0.013 ± 0.017 | 0.20 ± 0.12 | 0.09 ± 0.06 | 2 040 | 0.1 (i.c.v.) 64 (s.c.) | 74 |
| Lqq5* | P01481/1PTX | −0.148 ± 0.185 | −0.012 ± 0.015 | 0.24 ± 0.12 | 0.20 ± 0.12 | 16 900 | 0.125 (i.c.v.), 25 (s.c.) | 72–74 |
| Insect toxins | ||||||||
| Aah2 chimera | 1SEG | 0.096 ± 0.020 | 0.022 ± 0.013 | 0.13 ± 0.08 | 0.07 ± 0.05 | 60 (s.c.) | 37 | |
| BjαIT* | Q56TT9/1SN1 | 0.087 ± 0.025 | −0.021 ± 0.017 | 0.19 ± 0.10 | 0.10 ± 0.05 | 50 | >6,000 (s.c.) | 77 |
| BmK αIT1 | 1OMY | 0.068 ± 0.016 | −0.058 ± 0.017 | 0.16 ± 0.10 | 0.07 ± 0.06 | 5.2g | 4,500 (s.c.) | 78 |
| Bot IT1 | P55902/1LQQ | 0.030 ± 0.024 | 0.001 ± 0.021 | 0.17 ± 0.12 | 0.06 ± 0.03 | 600 | 50 (i.c.v.) | 79 |
| Lqh αIT | 1LQH | −0.003 ± 0.025 | 0.043 ± 0.018 | 0.23 ± 0.12 | 0.12 ± 0.08 | 18.8 | 55 (i.c.v.), 60 (s.c.) | 72–74 |
| Lqq3 | 1LQQ | 0.049 ± 0.019 | −0.008 ± 0.016 | 0.11 ± 0.09 | 0.05 ± 0.04 | 60 | 55 (i.c.v.), 55 (s.c.) | 72–74 |
| α-Like toxins | ||||||||
| BmK M1 | 1SN1 | 0.071 ± 0.021 | −0.042 ± 0.017 | 0.20 ± 0.09 | 0.14 ± 0.08 | 530 (i.v.) | 40 | |
| BmK M2 | 1CHZ | 0.020 ± 0.018 | −0.034 ± 0.014 | 0.16 ± 0.08 | 0.07 ± 0.07 | |||
| BmK M4 | 1SN4 | −0.033 ± 0.018 | −0.034 ± 0.017 | 0.13 ± 0.06 | 0.09 ± 0.06 | 4,000 (i.v.) | 80 | |
| BmK M7 | 1KV0 | 0.071 ± 0.022 | 0.032 ± 0.013 | 0.21 ± 0.11 | 0.07 ± 0.05 | |||
| BmK M10 | 2KBK | 0.022 ± 0.016 | −0.043 ± 0.014 | 0.10 ± 0.08 | 0.05 ± 0.04 | >50,000 (i.v.) | 81 | |
| Bom3 | P13488/1FH3 | 0.013 ± 0.020 | −0.052 ± 0.018 | 0.24 ± 0.12 | 0.11 ± 0.09 | 360 | 1.15 (i.c.v.), 150 (s.c.) | 72–74 |
| Bom4 | P59354/1SN1 | 0.033 ± 0.039 | −0.082 ± 0.018 | 0.17 ± 0.12 | 0.04 ± 0.03 | 140 | 1.15 (i.c.v.), 275 (s.c.) | 72–74 |
| Bot1* | P01488/1SN1 | 0.112 ± 0.016 | −0.063 ± 0.011 | 0.20 ± 0.10 | 0.10 ± 0.07 | 97 (i.v.) | 31 | |
| Bot2* | P01483/1SN1 | 0.033 ± 0.019 | −0.037 ± 0.012 | 0.10 ± 0.05 | 0.05 ± 0.04 | 160 (i.v.) | 31 | |
| Lqh3* | 1BMR | −0.071 ± 0.021 | 0.015 ± 0.015 | 0.27 ± 0.12 | 0.12 ± 0.08 | 200 | 2.5 (i.c.v.), 160 (s.c.) | 73 |
| Lqh4* | P83644/1LQQ | 0.053 ± 0.024 | −0.015 ± 0.014 | 0.22 ± 0.10 | 0.15 ± 0.08 | |||
| Lqh6* | P59356/1FH3 | 0.022 ± 0.024 | 0.010 ± 0.019 | 0.27 ± 0.11 | 0.12 ± 0.04 | 230 | 34 (i.c.v.), 96.5 (s.c.) | 74 |
a α-Toxins from venom of the following species: Aah, Androctonus australis; Bj, Buthotus judaicus; BmK, Mesobuthus martensii; Bom, Buthus occitanus mardochei; Bot, Buthus occitanus tunetanus; Lqh, Leiurus quinquestriatus hebraeus; Lqq, Leiurus quinquestriatus quinquestriatus; Od, Odontobuthus doriae. Asterisks mark toxins that have amidated C termini (according to Uniprot annotation).
b α-Toxins with unknown experimental three-dimensional structures. These molecules were modeled. The specified Uniprot accession numbers and Protein Data Bank (PDB) codes were used as a source of the modeled toxin sequence and its structural template, respectively (see “Experimental Procedures”).
c These parameters were computed from the 20–60-ns time span of MD simulations (the first 20 ns were considered as the equilibration period). The values are means ± S.D.
d Average MHP on the surface of the SMs and core modules.
e Average root mean square fluctuation calculated from the trajectory, filtered using the first eigenvector (RMSF-NM) for residues of the RT loops and core domains.
f Toxicity data were collected from the literature. Toxicity to insects was assayed on the cockroach B. germanica. Toxicity to mammals was assayed on mice by either intracerebroventricular (i.c.v.), subcutaneous (s.c.), or intravenous (i.v.) injections.
g PD50, half-paralytic dose.
FIGURE 1.

Sequence alignment of α-toxins used in the study. Residues are numbered according to the Aah2 toxin. Conserved secondary structure elements and the disulfide bridges are shown below. Residues belonging to the SM are boxed. Residues are colored as follows. Red, negatively charged (−; Asp and Glu); blue, positively charged (+; Arg and Lys); green, polar (∼; Ser, Thr, Asn, Gln, and His); orange, hydrophobic aromatic (@; Trp, Tyr, and Phe); olive, hydrophobic aliphatic (#; Ala, Val, Leu, Ile, and Met); gold background, cysteine; black, glycine and proline. A “consensus” sequence is given below each group, where symbols indicate conservation of ≥75%. Residues that are conserved within but differ between the groups are shown on a pink background and are considered as “functionally variable” (see Fig. 4A). Residues that have been hypothesized to evolve under positive selection are marked with red arrows above (66, 67).
MD Simulations
The Gromacs version 4.0.7 package (21) and a unified protocol were used. Each toxin was placed in a water box (60 × 60 × 60 Å3) together with the necessary amount of counterions and subjected to energy minimization, followed by heating to 300 K for 100 ps and 60 ns of unconstrained MD runs. The Gromos96 45a3 force field (22) and SPC water model (23) were used. MD simulations were carried out with a time step of 2 fs and imposed three-dimensional periodic boundary conditions in the isothermal-isobaric (NPT) (where N = number of particles, P = pressure, and T = temperature) ensemble with an isotropic pressure of 1 bar and a constant temperature of 300 K. The temperature and the pressure were scaled using the V-scale thermostat (24) and Berendsen barostat (25) with 0.1 and 1 ps relaxation parameters, respectively. The van der Waals and electrostatic interactions were truncated using the twin range 10/12 Å spherical cut-off.
All trajectories were fitted to a single structure to eliminate rotation and translation and permit direct comparison of hydrophobic and electrostatic properties of molecules (see “Molecular Hydrophobicity Potential (MHP) Calculations”). To perform the analysis of essential motions, mass-weighted covariance matrices and their eigenvectors were extracted from MD trajectories using standard tools from the Gromacs package. The calculations were performed for protein heavy atoms and polar hydrogen atoms (as defined by the force field) using the 20–60-ns time span of the trajectory. Root mean square fluctuation (RMSF-NM; “NM” represents “normal mode”) was calculated from the trajectory filtered using the slowest mode (first eigenvector), and expressed as a measure of molecular flexibility.
Fig. 2 was prepared with PyMOL (26); “intermediate” structures are the result of Gromacs normal mode analysis. Residue-residue maps of high amplitude correlated motions were constructed with an in-house Python script (Fig. 3). First, each residue in a toxin of length n was assigned a vector that describes its motion along the slowest mode. Then and n × n map of dot products was calculated to reveal the high amplitude correlated motions. Second, an averaged map for each toxin group was plotted, in accordance with the sequence alignment (Fig. 1) that relates structural elements. As a result of the alignment procedure, each map in Fig. 3A has dimensions of 69 × 69 residues.
FIGURE 2.
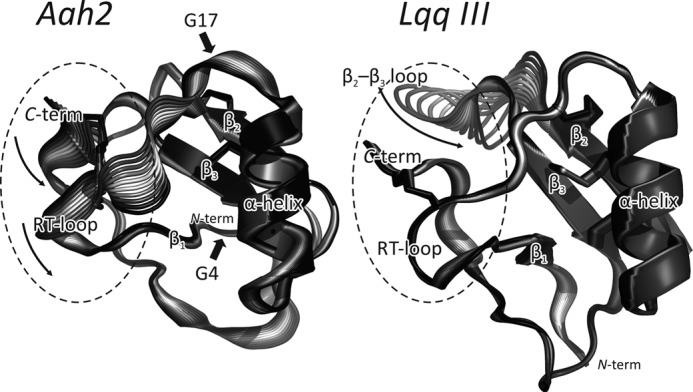
Mammal and insect α-toxins present distinct dynamic organization. Motions along the first eigenvector for typical mammal (Aah2) and insect (Lqq III) toxins are depicted (schematics show two “extreme” structures, colored gray and black, with several “intermediate” structures generated by morphing). Disulfide bridges are shown with sticks. The SMs are shown with dashed ellipses. N and C termini are marked N-term and C-term, respectively. For Aah2, the positions of Gly-4 and Gly-17 are shown with thick arrows.
FIGURE 3.

Analysis of essential dynamics in scorpion α-toxins. A, most similar modes in collective motions of scorpion α-toxins. Each of the three maps shows high amplitude correlated motions, characteristic of the corresponding toxin group. Disulfide bridge positions are shown with yellow circles; β-strands and α-helices are marked with green and pink diagonal lines, respectively. Black boxes highlight areas of the most prominent difference between the groups. The scale is the dot product of two vectors, describing the motion of each residue in the first eigenvector-related dynamics. Note that maps are symmetrical with respect to the diagonal. White strips in the maps correspond to gaps in the alignment, presented in Fig. 1. B, mammal toxins feature the most flexible RT loops. Group-averaged mean RMSF-NM values for residues in the RT loop and core modules are plotted. p values are given to emphasize the significance of difference between groups of mammal and insect toxins. Error bars, S.D.
Molecular Hydrophobicity Potential (MHP) Calculations
The MHP approach assumes that each atom in a molecule possesses its “intrinsic” value of hydrophobicity (atomic hydrophobicity constant), taking the molecular topology into account (16, 17). These constants have been determined from the database of experimental log P values for a large number of organic compounds (27); MHP at any given point is calculated as a superposition of contributions created by each atom, monotonically decaying with distance. The calculations were performed with the PLATINUM software (28).
Comparison of hydrophobic/hydrophilic properties of specificity modules (SMs) and core modules of α-toxins was accomplished by calculating the average dynamic hydrophobicity (〈MHPSM,Core〉MD) independently for molecular surface of each module. 〈MHPSM〉 and 〈MHPCore〉 are the average MHP values over all points of the Connolly surface that belong to the SMs or core modules, respectively. Collecting statistics from an MD trajectory yields the average dynamic hydrophobicity and its S.D. value: 〈MHPSM,Core〉MD ± S.D. When comparing two groups of toxins, two distributions of 〈MHP〉MD are considered; the statistical significance of the uncovered difference is determined by the two-tailed unpaired Student's test (p value less than 0.05 was considered as significant).
Mapping Hydrophobic Properties on Molecular Surface
For more detailed and descriptive assessment of the hydrophobic/hydrophilic properties of molecular surfaces, quantitative comparison, and recognition of similarity and distinction between molecules and groups of molecules, we employ a three-dimensional to two-dimensional mapping approach, which utilizes MHP spherical projection maps. The main idea is that comparison of similar, but still complex three-dimensional objects becomes more straightforward when data are presented in a regular form. Here, we applied transformation of molecular surfaces into spheres and then used one of the standard spherical projections to represent data in two dimensions as a rectangular grid. The main steps of the method are as follows. 1) The Connolly molecular surface was calculated. 2) MHP for each point of the surface was calculated (for these two steps, PLATINUM was used (28)). 3) Each point was projected onto a sphere (here, we arbitrarily chose a sphere radius of 20 Å). The center of the sphere was aligned with the geometrical center of the surface and set as the coordinate origin. Coordinates (x, y, z) could now be described by latitude = arcsin(z/|r|) and longitude = arctan(y/x), where r is the radius vector of the point, and latitude and longitude have the same meaning as in geography. 4) The MHP data were interpolated on a regular grid determined by latitude (−90°, +90°) and longitude (−180°, +180°), with a 1° step. 5) An equal-area Mollweide cartographic projection (29) was built. 6) The map was visualized using color contours.
Steps 3–6 were implemented in the Python script with step 4 performed with the module scipy.interpolate.griddata, step 5 performed with the module basemap, and step 6 performed with the module matplotlib. Interpolation of the data on a regular grid permits not only fancier visualization of the maps but also a variety of operations like addition, subtraction, averaging, calculation of S.D., etc. MD-averaged data for each toxin under study were obtained, and Fig. 4 demonstrates group averaging. To make sure that spherical projections did not bring too much distortion into the original surface-distributed data, we additionally performed comparison of surface MHP (Fig. 4B).
FIGURE 4.
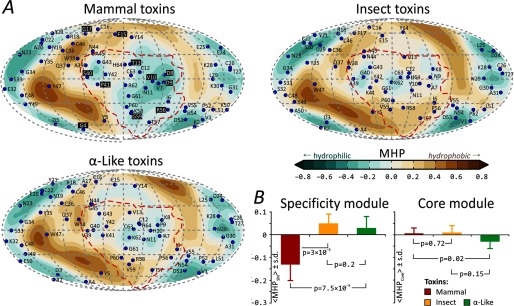
SMs are more hydrophilic in mammal than insect α-toxins. A, MHP spherical projection maps reveal alteration of hydrophobic and hydrophilic areas. The SMs are enclosed by a red border (this is a projection from the Aah2 toxin static structure). For clarity, projections of Cα atoms are shown for Aah2, Lqq III, and BmK M1 toxins on the mammal, insect, and α-like group maps, respectively. For mammal toxins, the “functionally variable” residues (see Fig. 1) are shown on a black background. B, comparison of average dynamic MHPs for the three groups of toxins. 〈MHPSM/Core〉MD values for the SMs and core modules are shown. Error bars, S.D.
Calculation of Nav Loop Hydrophobicity and Charge
An amino acid sequence alignment of seven insect (from Drosophila melanogaster, Musca domestica, Anopheles gambiae, Bombyx mori, Blattella germanica, Heliothis virescens, and Nasonia vitripennis) and 21 mammalian channels (Nav1.1–1.7 from humans, rats, and mice) was built (not shown). For estimation of hydrophobicity, the MHP-based scale was used; each residue was assigned a log P value (P representing the water/octanol distribution coefficient), equal to the sum of atomic MHP constants (27). Alternatively, the Eisenberg whole-residue hydrophobicity scale was used (30), which yielded very similar results (not shown). For each channel loop, hydrophobicity was calculated as an average value over single residue hydrophobicities (Fig. 5A). Loop charges were calculated analogously (Fig. 5B). Because α- and β-toxins have been proposed to interact with channels' VSDs at “clefts” between the S1-S2 and S3-S4 extracellular loops (10, 32), we have extended loop sequences by several residues in both directions to engage probable cleft surface.
FIGURE 5.

Net hydrophobicity and electric charge of Nav extracellular loops. Each pair of columns highlights differences between mammalian (black bars) and insect (gray bars) channels. Each bar represents the average net hydrophobicity (A) or electric charge (B) ± S.D. (error bars) of certain loops in the insect and mammalian channel groups. Asterisks mark loops that are significantly different between the groups (p < 0.05). The typical loop length for insect (I) and mammalian (M) channels are given in parentheses and in italic type.
Production of Recombinant Toxin BeM9
All steps were performed according to the published guidelines (33). DNA encoding BeM9 from the venom of the scorpion Mesobuthus eupeus (34) (Uniprot ID P09981) was assembled from synthetic oligonucleotides (Table 2) by PCR. Because no methionine residues are found in BeM9, we introduced a methionine codon upstream of the toxin-coding sequence and used cyanogen bromide (CNBr) to liberate the toxin from the carrier protein thioredoxin (Trx; see below). The toxin DNA was cloned into the expression vector pET-32b (Novagen (Madison, WI)), which was then used to transform Escherichia coli BL21(DE3). Gene expression was induced by 0.2 mm isopropyl β-d-1-thiogalactopyranoside. Trx-BeM9 fusion protein was isolated by affinity chromatography on TALON Superflow Metal Affinity Resin (Clontech, Mountain View, CA) and then hydrolyzed using CNBr as suggested (35). Recombinant BeM9 was purified by reversed-phase HPLC on a Jupiter C5 column (250 × 4.6 mm; Phenomenex, Torrance, CA).
TABLE 2.
Synthetic oligonucleotides used to construct BeM9 toxin-encoding DNA
Restriction sites (KpnI in M9f1 and BamHI in M9r) are underlined, the methionine codon is in boldface and italic type, and the stop codon is in boldface type.
| Name | Sequence |
|---|---|
| M9f1 | ATATGGTACCATGGCTCGTGACGCTTACATCGCTA |
| M9f2 | AACCGCACAACTGCGTTTACGAATGCTACAACCCGAAAGGTTCTT |
| M9f3 | ACTGCAACGACCTGTGCACCGAAAACGGTGCTGAATCTGGTTACT |
| M9f4 | GCCAGATCCTGGGTAAATACGGTAACGCTTGCTGGTGCATCCA |
| M9f5 | GCTGCCGGACAACGTTCCGATCCGTATCCCGGGTAAATGCC |
| M9r1/2 | AAACGCAGTTGTGCGGTTTAGCGATGTAAGCGTCAC |
| M9r2/3 | TGCACAGGTCGTTGCAGTAAGAACCTTTCGGGTTGT |
| M9r3/4 | ATTTACCCAGGATCTGGCAGTAACCAGATTCAGCAC |
| M9r4/5 | GAACGTTGTCCGGCAGCTGGATGCACCAGCAAGC |
| M9r | GCATGGATCCCTAGTGGCATTTACCCGGGATAC |
Electrophysiology
All stages were performed as reported previously (36). For the expression of Nav channels (rNav1.2, rNav1.4, hNav1.5, mNav1.6, the insect channel DmNav1 (Para), and the auxiliary subunits rβ1, hβ1, and TipE) in Xenopus laevis oocytes, linearized plasmids were transcribed using the T7 or SP6 mMESSAGE-mMACHINE transcription kit (Ambion, Carlsbad, CA). Stage V-VI Xenopus oocytes were injected with 50 nl of RNA solution (concentration of 1 ng/nl) using a microinjector (Drummond Scientific, Broomall, PA). The oocytes were incubated in a solution containing 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 2 mm MgCl2, and 5 mm HEPES (pH 7.4), supplemented with 50 mg/liter gentamicin sulfate.
Two-electrode voltage-clamp recordings were performed at room temperature using a Geneclamp 500 amplifier (Molecular Devices, Downingtown, PA) controlled by a pClamp data acquisition system (Axon Instruments, Union City, CA). Bath solution composition was the same as oocyte incubation solution but without gentamicin. Sodium current traces were evoked from a holding potential of −90 mV by 100-ms depolarizations to Vmax (the voltage corresponding to maximal sodium current in control conditions) with a start-to-start interval of 0.2 Hz. All data were analyzed using pClamp Clampfit version 10.0 (Molecular Devices) and Origin version 7.5 (OriginLab, Northampton, MA) software.
RESULTS
The flowchart of the study is as follows. 1) A comprehensive database of currently available three-dimensional structures of α-toxins was established and supplemented by homology models of several other well characterized members of the family. 2) A series of MD simulations was carried out, and group analysis of essential motions was performed. 3) Hydrophobic/hydrophilic properties were calculated on the molecular surface using the MHP approach and averaged over MD trajectories. 4) These properties were mapped using spherical projection. 5) Hydrophobic/hydrophilic properties of the extracellular loops of Navs were estimated based on their sequences and compared with the proposed modular organization of the toxins. 6) Activity of several α-toxins from M. eupeus venom was predicted based on the calculated properties and compared with experimental data.
Homology Modeling of Scorpion α-Toxins
To perform analysis of dynamic and physicochemical properties of α-toxins that might determine their phylum selectivity and toxicity, we created a database of available experimental structures of α-toxins and divided them into three groups, according to the published data on their toxicity: 1) “classic” mammal toxins, 2) insect toxins, and 3) α-like toxins that are active on both phyla. We should note that the boundaries between the three groups are not strict (see toxicity data in Table 1). It seems, however, that in mammals, the three groups show differential activity with respect to Nav isoforms. To make the statistics more robust, we extended the database by homology models of several toxins with unknown three-dimensional structure but clearly described pharmacological profile. The high conservation of α-toxin spatial structure makes homology modeling rather straightforward. In total, the data set included eight mammal, six insect, and 13 α-like toxins (Table 1 and Fig. 1).
MD Simulations Reveal Modular Organization of Scorpion α-Toxins
Comparison of static structures can be biased, especially in the case of homology models. To take into account the flexibility and to study the dynamic organization of scorpion α-toxins, we performed an analysis of the essential motions based on 60-ns-long MD simulations in a box with explicit water. Here, the last 40 ns of each MD trajectory were used, assuming that toxin structural parameters reach equilibrium after the first 20 ns. The total MD statistics for all toxins exceeds 1 μs.
Analysis of several “slow” (low frequency) modes (eigenvectors 1–5) reveals three regions in α-toxin structure that display relatively independent movements: 1) the N-terminal reverse turn (RT) loop (residues 8–12; throughout, the numbering is according to Aah2) coupled with the C terminus (residues 56–64) and constituting the so-called “RC domain” previously identified based on biochemical data (37–41); 2) the β2–β3 loop (residues 39–43); and 3) the rest of the molecule (its “core”). A common feature of all α-toxins is the “reciprocal” motion of the β2–β3 loop and the C terminus (see below). Based on the dynamic behavior, we rationalize that the α-toxin structure comprises two parts, or modules: the core module and the SM (Fig. 2). The latter encompasses the “RC domain” and the β2–β3 loop. Interestingly, the “classic” mammal toxins have the most conformationally flexible SMs. The RT loop of these toxins demonstrates the highest amplitude of motions along the slowest mode (an example for insect and mammal toxins is shown in Fig. 2).
We applied group analysis and built averaged residue-residue maps of high amplitude correlated motions (Fig. 3A). First, these maps allow identification of the most flexible regions that are common for all toxins in the group (diagonal elements). It is seen that mammal toxins have very mobile RT loops as compared with insect toxins (Fig. 3A, large boxes). High mobility of the RT loop in mammal toxins is also detected by analysis of RMSF along the first eigenvector (RMSF-NM) (Fig. 3B). Second, the off-diagonal elements illustrate the correlated (blue regions), anti-correlated (red regions), or non-correlated (white regions) character of motions between different parts of a protein. The small boxes in Fig. 3A highlight that in mammal toxins, the RT loop moves collectively with the C terminus of the protein, whereas in insect toxins, this is not the case. The “lake-like” pattern in mammal toxins (inside the small box) mostly corresponds to the “twisting” motion of the RT loop and C terminus. The reciprocal movements of the β2–β3 loop and C terminus are found in all toxins (red region around residues 43 and 62). In mammal toxins, the RT loop and C terminus move in a concerted manner with respect to the core module (Aah2 is shown in Fig. 2). On the contrary, in insect toxins, the “RC domain” is relatively rigid, and the most mobile part is the β2–β3 loop (Fig. 2, Lqq III).
It is worth noting that mammal toxins have conserved glycine residues in positions 4 and 17 (Fig. 1). Although distant from the RT loop, these residues might affect the loop flexibility, acting like “hinges” (Fig. 2). In contrast, insect toxins most frequently have alanine or phenylalanine residues in the same positions associated with the lower amplitude of the RT loop motions in these toxins. To test the putative role of Gly-4 and Gly-17 in toxin flexibility, we introduced respective in silico mutations in a typical mammal toxin Bot 3 (G17A and G17F) and typical insect toxins Lqq III (F17G) and BmK αIT1 (A17G) and simulated their MD using the standard protocol. As a result, it was shown that the flexibility increased in BmK αIT1 (A17G) (RMSF-NM = 0.11 ± 0.03 versus 0.04 ± 0.01 nm, mutant versus wild type) and dropped in Bot3 (G17F) (RMSF-NM = 0.09 ± 0.02 versus 0.13 ± 0.03 nm, mutant versus wild type). For the two other mutants, no apparent effect on flexibility was found. The double mutant of Lqq III toxin (A4G/F17G) was studied to see if the introduction of both “hinge” residues increases RT loop flexibility (in this case, a single substitution F17G is not enough). Accordingly, the flexibility increased (RMSF-NM = 0.10 ± 0.03 versus 0.05 ± 0.03 nm, mutant versus wild type).
Mammal α-Toxins Possess Prominently Hydrophilic Specificity Modules
To compare the average dynamic hydrophobicity in the three groups of toxins, we employed the MHP approach (16). It represents a powerful tool to calculate spatial distribution of hydrophobic/hydrophilic properties proven to be important in molecular recognition (15). Similar dynamic analysis of the surface hydrophobicity has recently been used in characterization of posttranslational modification effects in globular proteins (42, 43).
In addition, we employed an original computational method for two-dimensional mapping of properties like MHP and electrostatic potential, distributed over molecular surfaces of the toxins. This method utilizes spherical projection maps and is helpful in the delineation of a possible relationship between protein surface properties and activity. The hydrophobic/hydrophilic maps are constructed as follows (see “Experimental Materials” for further details). 1) The MHP value is calculated in each point of the Connolly surface of a molecule; 2) these values are projected onto a sphere concentric with the surface; 3) spherical MHP projection is interpolated on a rectangular coordinate grid; and 4) a Mollweide equal area projection (often used for global world or sky maps) is built. A detailed assessment of this method's potential to relate structural and dynamic features of small proteins to their activity (with a set of biologically relevant examples) will be published elsewhere.
Hydrophobicity maps for individual mammal, insect, and α-like toxins were built (not shown). Fig. 4A shows group- and MD-averaged maps that help to spot common and distinct features between these groups of toxins. A common feature for all α-toxins is the interchange of hydrophobic “isles” and hydrophilic “lakes”. At the same time, the large and “deep” hydrophilic “lake” is present only in mammal toxins. Surprisingly, this “lake” is located inside the SM (marked with a red border) and moreover perfectly superimposes with the “RC domain.” Correspondingly, quantitative analysis of averaged dynamic hydrophobicity reveals conserved properties of the core module and the variability of SM (Fig. 4B). This result is statistically significant; 〈MHPSM〉MD values (see “Experimental Procedures”) for mammal toxins and either insect or α-like toxins differ with p < 10−4. At the same time, the SMs present non-uniformly distributed MHP values for groups of mammal and especially α-like toxins (Table 1), thus suggesting that the binding sites for α-toxins in mammalian Navs may differ substantially.
Sequence-based Analysis of Extracellular Loops in Sodium Channels
To date, the three-dimensional structure of eukaryotic Navs is unknown. The recently solved structures of bacterial sodium channels (44–46) represent homotetramers, in contrast to single-chain quasitetrameric eukaryotic channels. No structures of complexes with toxins are available yet, although there is a wealth of biochemical and modeling data on toxin–channel interactions. It has been experimentally established that scorpion α-toxins interact preferentially with VSD IV of Navs (9), with additional evidence that contacts may also form with loops in PD I (47).
We find that the observed modular organization of scorpion α-toxins (particularly the “asymmetry” of hydrophobic/hydrophilic properties) is mirrored in properties of Nav extracellular loops. To assess the probable differences in Navs from diverse animals, we calculated the average net hydrophobicities of those loops that can participate in toxin binding: S1-S2 and S3-S4 loops of repeat domain IV (VSD IV) and S5-P and P-S6 loops of repeat domains I and III (PD I and PD III), using sequences of seven insect and 21 mammalian channels. The most notable feature is the conserved hydrophobic properties of VSD IV loops (Fig. 5A). This comes in contrast to all other repeat domains (not shown in Fig. 5). Taking into account the pronounced differences in the SMs between mammal and insect α-toxins (see above), it is unlikely that the toxins bind to VSD IV by these modules. From the perspective of loop hydrophobicity, it seems more probable that the conserved core module is responsible for interaction with VSD IV. Instead, the SMs may interact with the adjacent repeat domain pore loops. Both S5-P and P-S6 pore loops of repeat domain I have a hydrophobicity pattern similar to the corresponding toxins; mammalian channels possess more hydrophilic loops. The P-S6 loop in PD III is much more hydrophilic in insect channels and is unlikely to be involved in the interaction with toxins.
Analogously to hydrophobicity, the average electric charge of the same set of extracellular loops of insect and mammalian channels was calculated (Fig. 5B). The net charge of the channel loops is negative to match the positive charge of α-toxins. The largest negative charge is carried by pore loops in repeat domains I (S5-P) and IV (P-S6). Overall, loop charge in insect and mammal channels is similar, with VSDs I and IV being most conserved. Moreover, there is little difference in electrostatic properties between toxin groups (not shown). It is therefore unlikely that α-toxin selectivity is determined by electrostatic interactions with the target channels.
Validation of the Method; Activity Prediction of Toxins from M. eupeus
Although peptides BeM9, -10, and -14 from M. eupeus scorpion venom were among the first α-toxins to be described (34, 48) and the first to be studied structurally (13, 14), they were rather poorly characterized biochemically, and no exact evidence existed that they affected insect or mammalian Navs. Recent work by Zhu et al. has described the activity of four other α-toxins from this venom named MeuNaTxα-1, -2, -4, and -5 (36). Because those results were not included in our reference list (Table 1), we put our approach to the test.
We applied the developed strategy to predict the activity of M. eupeus toxins. From the analysis of dynamic and hydrophobic properties (Fig. 6, A and B), we assign BeM9 and MeuNaTxα-1 and -2 toxins to either the insect or α-like group (〈MHPSM〉MD = 0.051 ± 0.018, −0.069 ± 0.022, and −0.033 ± 0.039, respectively; compare with the values in Table 1; typical values for mammal toxins are <−0.1). Furthermore, BeM9 has dynamic features characteristic of insect toxins: a relatively rigid RT loop and rather flexible β2–β3 loop. Please note that our assignment disagrees with the phylogenic tree (Fig. 6C); MeuNaTxα-1 is suggested to belong to the mammal toxin group from homology.
FIGURE 6.
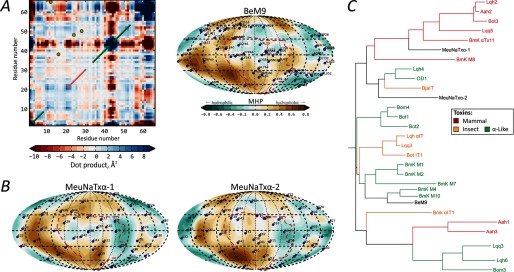
Prediction of BeM9 and MeuNaTxα-1 and -2 activity. A (left), analysis of the essential dynamics of BeM9 (analogous to Fig. 3A). A (right), MHP spherical projection map of BeM9 (analogous to Fig. 4A). B, MHP spherical projection maps of MeuNaTxα-1 and -2. C, phylogenetic tree of α-toxins built with Clustal. Red, mammal toxins; yellow, insect toxins; green, α-like toxins; black, BeM9 and MeuNaTxα-1 and -2.
BeM9 was produced recombinantly in a conventional E. coli system with Trx as the fusion partner. CNBr was used for target peptide separation from Trx, and the toxin was then recovered by HPLC (Fig. 7A); the yield was 2 mg/liter of bacterial culture. BeM9 activity was then tested against a number of mammalian Navs, and the insect Para channel expressed in X. laevis oocytes using the voltage clamp technique (Fig. 7B). Similarly to typical α-like toxins, BeM9 was found to affect both insect and mammalian Navs, with the exception of Nav1.2, the major isoform found in the central nervous system. Both MeuNaTxα-1 and -2 were characterized as α-like toxins by Zhu et al. (36), supporting our assumptions.
FIGURE 7.

Production and activity testing of toxin BeM9. A, purification of recombinant toxin after Trx-BeM9 fusion cleavage with CNBr by HPLC. The fraction containing BeM9 is marked by an asterisk. B, representative whole-cell current traces recorded from oocytes expressing cloned Nav isoforms in control and after toxin application. The dotted line indicates the zero current level. Asterisks mark traces after application of 1 μm toxin. Shown are representative traces of at least three independent experiments.
DISCUSSION
Finding essential similarities and differences in structures of biological molecules is a major challenge. In contrast to small molecules, where pharmacophore-based analyses (49) and QSAR-approaches (50) are applied routinely to mine active molecules from computer databases of chemical compounds, there are no widespread techniques that can address this task for proteins. The protein-protein docking field is coming of age, but it still has a number of limitations (51) and can hardly be applied if the spatial structure of either partner is unknown or poorly determined. One of the most essential properties of a molecule is its surface, which represents the interface of intermolecular communication. The concept of molecular surfaces persists in structural and computational biology for more than 40 years (52–54), but its potential has more to deliver to the field. Surface mapping and comparison is widely used in computational molecular biology and computer-aided drug design (18, 55–58). For example, calculation of hydrophobic “complementarity” in the binding pockets at the protein/ligand interface uncovers important principles in molecular recognition, and this idea may be applied to improve docking results (15, 28). Moreover, surface hydrophobicity is a good measure for conformational transitions (42, 43) and generally “tags” areas of intermolecular recognition (16).
Surface Mapping of Scorpion α-Toxins
In this work, we utilize a new approach to combine several computational methods for whole-molecule mapping of various physicochemical properties on the molecular surface. We intentionally simplify the complex shape of molecules by projecting their surface onto a sphere. This operation is legitimate for relatively small molecules of approximately spherical shape (like α-toxins), and it can be readily adapted for ellipsoid shape. The following advantages of the new surface mapping method should be outlined. 1) It allows for easy and clear visualization of the whole molecular surface at once. The area of interest can easily be “zoomed” with less distortion. 2) Dynamic behavior of the molecules can easily be taken into account by calculating the “average map” over individual maps for MD snapshots. Even such rigid molecules as α-toxins have intrinsic dynamics, which may be even more important for other objects. 3) Map averaging reveals the most essential common features in groups of molecules; the less important features will be “averaged out” in the resulting map. 4) Comparison of averaged maps for groups of molecules with different activity highlights functional patches (Fig. 4). Moreover, the differential maps are easily constructed to emphasize the differences.
The main shortcoming is transformation of the spherical surface to a two-dimensional coordinate grid, because it distorts data; the other way is not to build projections but to perform comparison directly in the spheroid vertices (18). Technical details of the method and assessment of its general applicability using a set of biologically active polypeptides will be published elsewhere.
Scorpion α-Toxins Possess Modular Organization
The spatial structure of scorpion α-toxins may be described in terms of a βαββ scaffold (see Fig. 2). Together with plant defensins that present the same type of fold, scorpion toxins are classified into a separate superfamily of knottins by SCOP, and into the homologous superfamily 3.30.30.10 (mixed α-β class) by CATH.
Despite their relatively small size, close inspection identifies modular organization of scorpion α-toxins. Indeed, our results support dissection of these molecules into two modules or subdomains, the core modules and SMs (see above and Fig. 1 for localization). We note that in the conventional expressions “core domain” and “RC domain” (a part of SM), the term “domain” is used quite incorrectly and prevails due to historical reasons. It is advisable instead to use the more correct terms “part,” “subdomain,” or “module.”
The dissection arises from the following observations. 1) MD reveals that the two modules of scorpion α-toxins demonstrate essential motions independent from each other and can therefore be considered as “dynamic domains” (Figs. 2 and 3A). Moreover, the high mobility of SMs reliably distinguishes mammal toxins (Fig. 3B). It is important to note that analysis of available NMR and x-ray structures of α-toxins readily shows higher mobility of the “RC domain” as compared with the core domain in each particular structure (59–61), yet only MD simulation in the same setup is suitable for comparison of dynamic features of different molecules. Two glycine residues (Gly-4 and Gly-17 in Aah2) are conserved in mammal toxins, and these are believed to act as “hinges” for the SMs. This conclusion is supported by considering the mobility of in silico mutated toxins. 2) Whereas virtually no difference in bulk physico-chemical properties is noted between the core modules in mammal, insect, and α-like toxins, the SMs of the former are significantly more hydrophilic (elegantly visualized on the spherical projection maps; Fig. 4A). Although previous studies suggested the division of α-toxin molecules into two parts (37, 39, 41, 62), never before was this clearly illustrated from the MD and surface hydrophobicity. 3) Functionally, the core module seems to determine the overall ability of α-toxins to target Navs, whereas the SM determines toxin specificity and eventually its classification as mammal, insect, or α-like. This conclusion is derived from numerous mutagenesis studies (63–65) and is illustrated by our results. Probably the most direct evidence was provided by the Gurevitz group by excision of the “RC domain” from the insect toxin Lqh αIT and its implantation into the mammal toxin Aah2; the resulting chimera featured activity of the “RC domain” donor Lqh αIT (37, 62). 4) From the evolutionary point of view, we may consider the SM as a fast changing segment, whereas the core module is more conserved (for distribution of “functionally variable” residues over the toxin surface map, see Fig. 4A). The SM seems to evolve more quickly to adjust the toxin specificity to target different Navs. Note that many of the amino acid residues that have been proposed to evolve under positive selection (66, 67) (see red arrows in Fig. 1) are located inside the SM. Two positions, 39 and 41, reside in the β2–β3 loop, thereby supporting our finding that this loop is part of the SM. It was also mentioned in the literature that the C terminus of scorpion toxins has gone through structural rearrangements during evolution, probably to adapt toward new target sites (68).
Although the three groups of toxins may be differentiated based on the functional and structural properties, we should note that they rather form a continuum, not a discrete pattern, with several toxin species presenting “intermediate” properties that are difficult to assign to a particular group. This fact is illustrated by the limited correlation between the toxicity of α-toxins to mammals or insects and the hydrophobic/hydrophilic properties of their surface (Table 1).
We should also notice that current classification of Nav toxins, which is based on animal toxicity and competitive binding data, is vague (e.g. “insect toxins” are not completely insect-specific; they are often reported active on mammalian channels (also see Table 1)). More biochemically accurate classification based on individual channel isoform recordings is needed. If a sufficiently complete body of such data were available, computational analysis would result in more straightforward structure-activity relationships.
Differences between Toxins Are Mirrored in the Structure of the Target Ion Channels
To date, the three-dimensional structure of eukaryotic Navs remains elusive. A breakthrough in the field was the recently presented crystal structures of bacterial Navs (44–46). The major difference between eukaryotic Navs and their prokaryotic counterparts is that the α-subunits of the former are monomers, whereas the latter are homotetramers. A problem of repeat domain orientation arises for eukaryotic Navs; both “clockwise” and “counterclockwise” orientations seem possible. Clockwise orientation (if viewed from the extracellular side) was predicted from analysis of interactions with a pore-blocking μ-conotoxin (69) and is currently backed by most investigators in the field. This orientation was also determined from mutant double cycle analysis of scorpion β-toxin Css4 and the rat Nav1.4 channel (70). A clockwise disposition of VSDs with respect to the S5-S6 segments of the same subunit takes place (so that VSD I, for instance, is in close proximity to PD II) in bacterial Nav (44). Scorpion α-toxins are known to interact with the so-called receptor site 3, which is believed to locate on the extracellular surface of VSD IV and PD I of Navs (9–11).
Because the SMs of insect toxins, determining their taxon specificity, are significantly more hydrophobic than those of mammal toxins, we compared extracellular loops of the respective target channels (i.e. insect versus mammalian Navs) (Fig. 5). We find that in terms of hydrophobicity, the extracellular surface of VSD IV is very similar in channels from different animals (and this is not the case for VSDs I–III). Conversely, the extracellular surface of PD I is significantly more hydrophobic in insect channels. It is therefore reasonable to conclude that the conserved core module of scorpion α-toxins binds to loops S1-S2 and S3-S4 in repeat domain IV, whereas the SM interacts with loops S5-P/P-S6 from repeat domain I of Navs (this corresponds to a “clockwise” arrangement of the repeat domains; Fig. 8). This hypothesis, based on our simple consideration of toxin and receptor properties, is supported by the results of less advanced sequence analysis (71) and computer docking simulations performed by other groups (10, 11).
FIGURE 8.
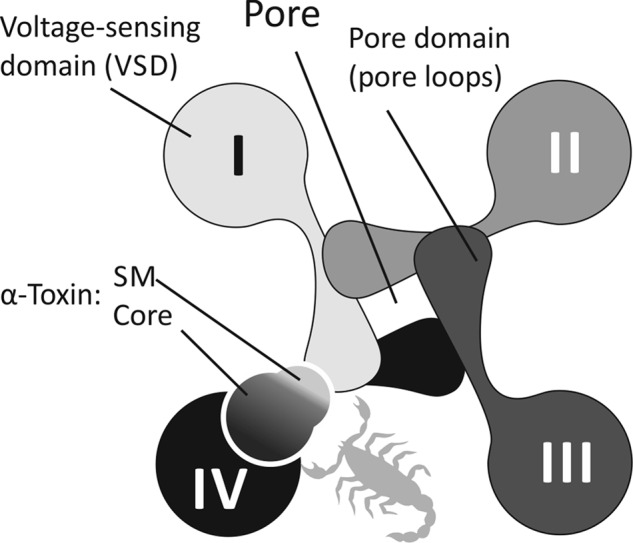
A hypothesis; modular organization of α-toxins mirrors the domain structure of Navs. Four channel repeat domains are shown with different shades of gray and marked I–IV; the pore is located in the center, and VSDs are located around it. Results of molecular modeling and biochemical experiments suggest that toxin core modules interact with conserved VSD IV, whereas the variable and flexible SMs may bind to pore loops from repeat domain I. The view is from the extracellular side of the membrane.
CONCLUSION
Our simulation results suggest that scorpion α-toxins possess modular organization, and individual modules interact with different parts of their target channels. We propose that the stable and conserved core module binds to the conserved VSD IV and provides toxin activity toward Navs per se. In contrast, the mobile and variable SM interacts with the variable loops of PD I. The latter interaction probably underlies the observed taxon specificity (mammals versus insects) of α-toxins.
Thorough comparison of structural, hydrophobic/hydrophilic, and dynamic properties of mammal, insect, and α-like toxins led us to identification of molecular determinants underlying their specificity. The SM was found considerably more hydrophilic and flexible in the mammal toxins, whereas in insect toxins, the same module was much more hydrophobic and rigid. As expected, α-like toxins feature intermediate hydrophobicity and flexibility. We hypothesize that α-toxins have acquired a modular architecture in the evolutionary arms race to effectively target the multidomain Navs.
Surface mapping serves as an alternative method to predict orphan α-toxin activity. We validated the approach by assigning the specificity of several toxins from M. eupeus venom. Importantly, the method has shown advantages compared with the conventional sequence-based predictions.
Our findings may aid the development of novel Nav ligands for treatment of channelopathies or fight against agricultural pests. Moreover, the proposed algorithm for mapping of physico-chemical properties on the molecular surface is of a general nature and may be utilized for detailed comparison of groups of proteins and protein-protein complexes.
Acknowledgments
We thank A. S. Nikolsky for the search of toxin LD50 values and N. B. Ustinov for technical assistance. Access to computational facilities of the Joint Supercomputer Center of the Russian Academy of Sciences (Moscow) and Moscow Institute of Physics and Technology is gratefully acknowledged. We thank A. L. Goldin for sharing genes encoding rNav1.2 and mNav1.6, G. Mandel for rNav1.4, R. G. Kallen for hNav1.5, S. H. Heinemann for the rat β1 subunit, S. C. Cannon for the hβ1 subunit, and M. S. Williamson for providing the para and tipE clones.
This work was supported by the Ministry of Education and Science of the Russian Federation (Contracts P818, 07.514.11.4127, and 8794), by the Russian Foundation for Basic Research (Grants 11-04-01606 and 10-04-01217), and by the “Basic Fundamental Research for Nanotechnologies and Nanomaterials” and “Molecular and Cell Biology” programs of the Russian Academy of Sciences.
- Nav
- voltage-gated sodium channel
- MD
- molecular dynamics
- MHP
- molecular hydrophobicity potential
- PD
- pore domain
- RMSF
- root mean square fluctuation
- RMSF-NM
- RMSF along the first eigenvector in the normal mode analysis
- SM
- specificity module
- Trx
- thioredoxin
- VSD
- voltage-sensing domain
- RT
- reverse turn.
REFERENCES
- 1. Hille B. (2001) Ion Channels of Excitable Membranes, 3rd Ed., pp. 1–93, Sinauer Associates Inc., Sunderland, MA [Google Scholar]
- 2. Catterall W. A. (2000) From ionic currents to molecular mechanisms. The structure and function of voltage-gated sodium channels. Neuron 26, 13–25 [DOI] [PubMed] [Google Scholar]
- 3. Andavan G. S., Lemmens-Gruber R. (2011) Voltage-gated sodium channels. Mutations, channelopathies and targets. Curr. Med. Chem. 18, 377–397 [DOI] [PubMed] [Google Scholar]
- 4. Lopreato G. F., Lu Y., Southwell A., Atkinson N. S., Hillis D. M., Wilcox T. P., Zakon H. H. (2001) Evolution and divergence of sodium channel genes in vertebrates. Proc. Natl. Acad. Sci. U.S.A. 98, 7588–7592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dong K. (2007) Insect sodium channels and insecticide resistance. Invert. Neurosci. 7, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stevens M., Peigneur S., Tytgat J. (2011) Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bosmans F., Tytgat J. (2007) Voltage-gated sodium channel modulation by scorpion α-toxins. Toxicon 49, 142–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Catterall W. A., Goldin A. L., Waxman S. G. (2005) International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409 [DOI] [PubMed] [Google Scholar]
- 9. Rogers J. C., Qu Y., Tanada T. N., Scheuer T., Catterall W. A. (1996) Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. J. Biol. Chem. 271, 15950–15962 [DOI] [PubMed] [Google Scholar]
- 10. Wang J., Yarov-Yarovoy V., Kahn R., Gordon D., Gurevitz M., Scheuer T., Catterall W. A. (2011) Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 108, 15426–15431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gur M., Kahn R., Karbat I., Regev N., Wang J., Catterall W. A., Gordon D., Gurevitz M. (2011) Elucidation of the molecular basis of selective recognition uncovers the interaction site for the core domain of scorpion α-toxins on sodium channels. J. Biol. Chem. 286, 35209–35217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gordon D., Karbat I., Ilan N., Cohen L., Kahn R., Gilles N., Dong K., Stühmer W., Tytgat J., Gurevitz M. (2007) The differential preference of scorpion α-toxins for insect or mammalian sodium channels. Implications for improved insect control. Toxicon 49, 452–472 [DOI] [PubMed] [Google Scholar]
- 13. Pashkov V. S., Maiorov V. N., Bystrov V. F., Hoang A. N., Volkova T. M., Grishin E. V. (1988) Solution spatial structure of “long” neurotoxin M9 from the scorpion Buthus eupeus by 1H-NMR spectroscopy. Biophys. Chem. 31, 121–131 [DOI] [PubMed] [Google Scholar]
- 14. Pashkov V. S., Khoang N. A., Maĭorov V. N., Bystrov V. F. (1986) [The structure of Buthus eupeus neurotoxin M9 in a solution studied by 1H-NMR spectroscopy]. Bioorg. Khim. 12, 1306–1316 [PubMed] [Google Scholar]
- 15. Pyrkov T. V., Priestle J. P., Jacoby E., Efremov R. G. (2008) Ligand-specific scoring functions. Improved ranking of docking solutions. SAR QSAR Environ. Res. 19, 91–99 [DOI] [PubMed] [Google Scholar]
- 16. Efremov R. G., Chugunov A. O., Pyrkov T. V., Priestle J. P., Arseniev A. S., Jacoby E. (2007) Molecular lipophilicity in protein modeling and drug design. Curr. Med. Chem. 14, 393–415 [DOI] [PubMed] [Google Scholar]
- 17. Efremov R. G., Gulyaev D. I., Vergoten G., Modyanov N. N. (1992) Application of three-dimensional molecular hydrophobicity potential to the analysis of spatial organization of membrane domains in proteins. I. Hydrophobic properties of transmembrane segments of Na+,K+-ATPase. J. Protein Chem. 11, 665–675 [DOI] [PubMed] [Google Scholar]
- 18. Hasegawa K., Funatsu K. (2012) New description of protein-ligand interactions using a spherical self-organizing map. Bioorg. Med. Chem. 20, 5410–5415 [DOI] [PubMed] [Google Scholar]
- 19. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 20. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2007) Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. Chapter 2, Unit 2.9 [DOI] [PubMed] [Google Scholar]
- 21. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4. Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 22. Schuler L. D., Daura X., van Gunsteren W. F. (2001) An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 22, 1205–1218 [Google Scholar]
- 23. Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., Hermans J. (1981) Interaction models for water in relation to protein hydration. in Intermolecular Forces. (Pullman B., ed) pp. 331–342, Reidel Publishing Company, Dordrecht, The Netherlands [Google Scholar]
- 24. Bussi G., Donadio D., Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 25. Berendsen HJC P. J., Vangunsteren W. F., Dinola A., Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 26. DeLano W. (2002) The PyMOL User's Manual, DeLano Scientific, San Carlos, CA [Google Scholar]
- 27. Ghose A. K., Viswanadhan V. N., Wendoloski J. J. (1998) Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragmental methods: an analysis of ALOGP and CLOGP methods. J. Phys. Chem. A 102, 3762–3772 [Google Scholar]
- 28. Pyrkov T. V., Chugunov A. O., Krylov N. A., Nolde D. E., Efremov R. G. (2009) PLATINUM. A Web tool for analysis of hydrophobic/hydrophilic organization of biomolecular complexes. Bioinformatics 25, 1201–1202 [DOI] [PubMed] [Google Scholar]
- 29. Snyder J. P. (1993) Flattening the Earth: Two Thousand Years of Map Projections, pp. 112–113, University of Chicago Press, Chicago [Google Scholar]
- 30. Eisenberg D., McLachlan A. D. (1986) Solvation energy in protein folding and binding. Nature 319, 199–203 [DOI] [PubMed] [Google Scholar]
- 31. Martin M. F., Rochat H. (1984) Purification of thirteen toxins active on mice from the venom of the North African scorpion Buthus occitanus tunetanus. Toxicon 22, 279–291 [DOI] [PubMed] [Google Scholar]
- 32. Zhang J. Z., Yarov-Yarovoy V., Scheuer T., Karbat I., Cohen L., Gordon D., Gurevitz M., Catterall W. A. (2011) Structure-function map of the receptor site for β-scorpion toxins in domain II of voltage-gated sodium channels. J. Biol. Chem. 286, 33641–33651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shlyapnikov Y. M., Andreev Y. A., Kozlov S. A., Vassilevski A. A., Grishin E. V. (2008) Bacterial production of latarcin 2a, a potent antimicrobial peptide from spider venom. Protein Expr. Purif. 60, 89–95 [DOI] [PubMed] [Google Scholar]
- 34. Volkova T. M., Garsia A. F., Telezhinskaia I. N., Potapenko N. A., Grishin E. V. (1984) [Amino acid sequence of 2 neurotoxins from the scorpion Buthus eupeus venom]. Bioorg. Khim. 10, 979–982 [PubMed] [Google Scholar]
- 35. Andreev Y. A., Kozlov S. A., Vassilevski A. A., Grishin E. V. (2010) Cyanogen bromide cleavage of proteins in salt and buffer solutions. Anal. Biochem. 407, 144–146 [DOI] [PubMed] [Google Scholar]
- 36. Zhu S., Peigneur S., Gao B., Lu X., Cao C., Tytgat J. (2012) Mol. Cell Proteomics 11, 10.1074/mcp.M111.012054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karbat I., Frolow F., Froy O., Gilles N., Cohen L., Turkov M., Gordon D., Gurevitz M. (2004) Molecular basis of the high insecticidal potency of scorpion α-toxins. J. Biol. Chem. 279, 31679–31686 [DOI] [PubMed] [Google Scholar]
- 38. He X. L., Li H. M., Zeng Z. H., Liu X. Q., Wang M., Wang D. C. (1999) Crystal structures of two α-like scorpion toxins. Non-proline cis peptide bonds and implications for new binding site selectivity on the sodium channel. J. Mol. Biol. 292, 125–135 [DOI] [PubMed] [Google Scholar]
- 39. Guan R. J., Xiang Y., He X. L., Wang C. G., Wang M., Zhang Y., Sundberg E. J., Wang D. C. (2004) Structural mechanism governing cis and trans isomeric states and an intramolecular switch for cis/trans isomerization of a non-proline peptide bond observed in crystal structures of scorpion toxins. J. Mol. Biol. 341, 1189–1204 [DOI] [PubMed] [Google Scholar]
- 40. Ye X., Bosmans F., Li C., Zhang Y., Wang D. C., Tytgat J. (2005) Structural basis for the voltage-gated Na+ channel selectivity of the scorpion α-like toxin BmK M1. J. Mol. Biol. 353, 788–803 [DOI] [PubMed] [Google Scholar]
- 41. Liu L. H., Bosmans F., Maertens C., Zhu R. H., Wang D. C., Tytgat J. (2005) Molecular basis of the mammalian potency of the scorpion α-like toxin, BmK M1. FASEB J. 19, 594–596 [DOI] [PubMed] [Google Scholar]
- 42. Petrov D., Zagrovic B. (2011) Microscopic analysis of protein oxidative damage: effect of carbonylation on structure, dynamics, and aggregability of villin headpiece. J. Am. Chem. Soc. 133, 7016–7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Polyansky A. A., Zagrovic B. (2012) Protein electrostatic properties predefining the level of surface hydrophobicity change upon phosphorylation. J. Phys. Chem. Lett. 3, 973–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Payandeh J., Scheuer T., Zheng N., Catterall W. A. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Payandeh J., Gamal El-Din T. M., Scheuer T., Zheng N., Catterall W. A. (2012) Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang X., Ren W., DeCaen P., Yan C., Tao X., Tang L., Wang J., Hasegawa K., Kumasaka T., He J., Wang J., Clapham D. E., Yan N. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gurevitz M. (2012) Mapping of scorpion toxin receptor sites at voltage-gated sodium channels. Toxicon 60, 502–511 [DOI] [PubMed] [Google Scholar]
- 48. Volkova T. M., Garsia A. F., Telezhinskaia I. N., Potapenko N. A., Grishin E. V. (1985) [Neurotoxins from the venom of the Central Asian scorpion Buthus eupeus]. Bioorg. Khim. 11, 1445–1456 [PubMed] [Google Scholar]
- 49. Tanrikulu Y., Schneider G. (2008) Pseudoreceptor models in drug design. Bridging ligand- and receptor-based virtual screening. Nat. Rev. Drug Discov. 7, 667–677 [DOI] [PubMed] [Google Scholar]
- 50. Dong X., Ebalunode J. O., Yang S. Y., Zheng W. (2011) Receptor-based pharmacophore and pharmacophore key descriptors for virtual screening and QSAR modeling. Curr. Comput. Aided Drug Des. 7, 181–189 [DOI] [PubMed] [Google Scholar]
- 51. Janin J. (2010) Protein-protein docking tested in blind predictions. The CAPRI experiment. Mol. Biosyst. 6, 2351–2362 [DOI] [PubMed] [Google Scholar]
- 52. Lee B., Richards F. M. (1971) The interpretation of protein structures. Estimation of static accessibility. J. Mol. Biol. 55, 379–400 [DOI] [PubMed] [Google Scholar]
- 53. Connolly M. L. (1983) Solvent-accessible surfaces of proteins and nucleic acids. Science 221, 709–713 [DOI] [PubMed] [Google Scholar]
- 54. Norel R., Fischer D., Wolfson H. J., Nussinov R. (1994) Molecular surface recognition by a computer vision-based technique. Protein Eng. 7, 39–46 [DOI] [PubMed] [Google Scholar]
- 55. Ben-Shimon A., Niv M. Y. (2011) Deciphering the arginine-binding preferences at the substrate-binding groove of Ser/Thr kinases by computational surface mapping. PLoS Comput. Biol. 7, e1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kozakov D., Hall D. R., Chuang G. Y., Cencic R., Brenke R., Grove L. E., Beglov D., Pelletier J., Whitty A., Vajda S. (2011) Structural conservation of druggable hot spots in protein-protein interfaces. Proc. Natl. Acad. Sci. U.S.A. 108, 13528–13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Polanski J., Walczak B. (2000) The comparative molecular surface analysis (COMSA). A novel tool for molecular design. Comput. Chem. 24, 615–625 [DOI] [PubMed] [Google Scholar]
- 58. Norel R., Lin S. L., Wolfson H. J., Nussinov R. (1995) Molecular surface complementarity at protein-protein interfaces. The critical role played by surface normals at well placed, sparse, points in docking. J. Mol. Biol. 252, 263–273 [DOI] [PubMed] [Google Scholar]
- 59. Landon C., Cornet B., Bonmatin J. M., Kopeyan C., Rochat H., Vovelle F., Ptak M. (1996) 1H-NMR-derived secondary structure and the overall fold of the potent anti-mammal and anti-insect toxin III from the scorpion Leiurus quinquestriatus quinquestriatus. Eur. J. Biochem. 236, 395–404 [DOI] [PubMed] [Google Scholar]
- 60. Tugarinov V., Kustanovich I., Zilberberg N., Gurevitz M., Anglister J. (1997) Solution structures of a highly insecticidal recombinant scorpion α-toxin and a mutant with increased activity. Biochemistry 36, 2414–2424 [DOI] [PubMed] [Google Scholar]
- 61. Krimm I., Gilles N., Sautière P., Stankiewicz M., Pelhate M., Gordon D., Lancelin J. M. (1999) NMR structures and activity of a novel α-like toxin from the scorpion Leiurus quinquestriatus hebraeus. J. Mol. Biol. 285, 1749–1763 [DOI] [PubMed] [Google Scholar]
- 62. Kahn R., Karbat I., Ilan N., Cohen L., Sokolov S., Catterall W. A., Gordon D., Gurevitz M. (2009) Molecular requirements for recognition of brain voltage-gated sodium channels by scorpion α-toxins. J. Biol. Chem. 284, 20684–20691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zilberberg N., Froy O., Loret E., Cestele S., Arad D., Gordon D., Gurevitz M. (1997) Identification of structural elements of a scorpion α-neurotoxin important for receptor site recognition. J. Biol. Chem. 272, 14810–14816 [DOI] [PubMed] [Google Scholar]
- 64. Wang C. G., Gilles N., Hamon A., Le Gall F., Stankiewicz M., Pelhate M., Xiong Y. M., Wang D. C., Chi C. W. (2003) Exploration of the functional site of a scorpion α-like toxin by site-directed mutagenesis. Biochemistry 42, 4699–4708 [DOI] [PubMed] [Google Scholar]
- 65. Sun Y. M., Bosmans F., Zhu R. H., Goudet C., Xiong Y. M., Tytgat J., Wang D. C. (2003) Importance of the conserved aromatic residues in the scorpion α-like toxin BmK M1. The hydrophobic surface region revisited. J. Biol. Chem. 278, 24125–24131 [DOI] [PubMed] [Google Scholar]
- 66. Zhu S., Bosmans F., Tytgat J. (2004) Adaptive evolution of scorpion sodium channel toxins. J. Mol. Evol. 58, 145–153 [DOI] [PubMed] [Google Scholar]
- 67. Weinberger H., Moran Y., Gordon D., Turkov M., Kahn R., Gurevitz M. (2010) Positions under positive selection. Key for selectivity and potency of scorpion α-toxins. Mol. Biol. Evol. 27, 1025–1034 [DOI] [PubMed] [Google Scholar]
- 68. Gurevitz M., Gordon D., Ben-Natan S., Turkov M., Froy O. (2001) Diversification of neurotoxins by C-tail “wiggling.” A scorpion recipe for survival. FASEB J. 15, 1201–1205 [DOI] [PubMed] [Google Scholar]
- 69. Dudley S. C., Jr., Chang N., Hall J., Lipkind G., Fozzard H. A., French R. J. (2000) μ-Conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J. Gen. Physiol. 116, 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cohen L., Ilan N., Gur M., Stühmer W., Gordon D., Gurevitz M. (2007) Design of a specific activator for skeletal muscle sodium channels uncovers channel architecture. J. Biol. Chem. 282, 29424–29430 [DOI] [PubMed] [Google Scholar]
- 71. del Río-Portilla F., Hernández-Marín E., Pimienta G., Coronas F. V., Zamudio F. Z., Rodríguez de la Vega R. C., Wanke E., Possani L. D. (2004) NMR solution structure of Cn12, a novel peptide from the Mexican scorpion Centruroides noxius with a typical β-toxin sequence but with α-like physiological activity. Eur. J. Biochem. 271, 2504–2516 [DOI] [PubMed] [Google Scholar]
- 72. Little M. J., Wilson H., Zappia C., Cestèle S., Tyler M. I., Martin-Eauclaire M. F., Gordon D., Nicholson G. M. (1998) δ-Atracotoxins from Australian funnel-web spiders compete with scorpion α-toxin binding on both rat brain and insect sodium channels. FEBS Lett. 439, 246–252 [DOI] [PubMed] [Google Scholar]
- 73. Gordon D., Martin-Eauclaire M. F., Cestèle S., Kopeyan C., Carlier E., Khalifa R. B., Pelhate M., Rochat H. (1996) Scorpion toxins affecting sodium current inactivation bind to distinct homologous receptor sites on rat brain and insect sodium channels. J. Biol. Chem. 271, 8034–8045 [DOI] [PubMed] [Google Scholar]
- 74. Hamon A., Gilles N., Sautière P., Martinage A., Kopeyan C., Ulens C., Tytgat J., Lancelin J. M., Gordon D. (2002) Characterization of scorpion α-like toxin group using two new toxins from the scorpion Leiurus quinquestriatus hebraeus. Eur. J. Biochem. 269, 3920–3933 [DOI] [PubMed] [Google Scholar]
- 75. Li H. M., Zhao T., Jin L., Wang M., Zhang Y., Wang D. C. (1999) A series of bioactivity-variant neurotoxins from scorpion Buthus martensii Karsch. Purification, crystallization and crystallographic analysis. Acta Crystallogr. D Biol. Crystallogr. 55, 341–344 [DOI] [PubMed] [Google Scholar]
- 76. Benkhadir K., Kharrat R., Cestèle S., Mosbah A., Rochat H., El Ayeb M., Karoui H. (2004) Molecular cloning and functional expression of the α-scorpion toxin BotIII. Pivotal role of the C-terminal region for its interaction with voltage-dependent sodium channels. Peptides 25, 151–161 [DOI] [PubMed] [Google Scholar]
- 77. Arnon T., Potikha T., Sher D., Elazar M., Mao W., Tal T., Bosmans F., Tytgat J., Ben-Arie N., Zlotkin E. (2005) BjalphaIT. A novel scorpion α-toxin selective for insects. Unique pharmacological tool. Insect Biochem. Mol. Biol. 35, 187–195 [DOI] [PubMed] [Google Scholar]
- 78. Wu H., Wu G., Huang X., He F., Jiang S. (1999) Purification, characterization and structural study of a neuro-peptide from scorpion Buthus martensi Karsch. Pure Appl. Chem. 71, 1157–1162 [Google Scholar]
- 79. Borchani L., Stankiewicz M., Kopeyan C., Mansuelle P., Kharrat R., Cestèle S., Karoui H., Rochat H., Pelhate M., el Ayeb M. (1997) Purification, structure and activity of three insect toxins from Buthus occitanus tunetanus venom. Toxicon 35, 365–382 [DOI] [PubMed] [Google Scholar]
- 80. Luo M. J., Xiong Y. M., Wang M., Wang D. C., Chi C. W. (1997) Purification and sequence determination of a new neutral mammalian neurotoxin from the scorpion Buthus martensii Karsch. Toxicon 35, 723–731 [DOI] [PubMed] [Google Scholar]
- 81. Cao Z. Y., Mi Z. M., Cheng G. F., Shen W. Q., Xiao X., Liu X. M., Liang X. T., Yu D. Q. (2004) Purification and characterization of a new peptide with analgesic effect from the scorpion Buthus martensi Karch. J. Pept. Res. 64, 33–41 [DOI] [PubMed] [Google Scholar]