

Abstract
Acute traumatic brain injury (TBI) is associated with long-term cognitive and behavioral dysfunction. In vivo studies have shown histone deacetylase inhibitors (HDACis) to be neuroprotective following TBI in rodent models. HDACis are intriguing candidates because they are capable of provoking widespread genetic changes and modulation of protein function. By using known HDACis and a unique small-molecule pan-HDACi (LB-205), we investigated the effects and mechanisms associated with HDACi-induced neuroprotection following CNS injury in an astrocyte scratch assay in vitro and a rat TBI model in vivo. We demonstrate the preservation of sufficient expression of nerve growth factor (NGF) and activation of the neurotrophic tyrosine kinase receptor type 1 (TrkA) pathway following HDACi treatment to be crucial in stimulating the survival of CNS cells after TBI. HDACi treatment up-regulated the expression of NGF, phospho-TrkA, phospho-protein kinase B (p-AKT), NF-κB, and B-cell lymphoma 2 (Bcl-2) cell survival factors while down-regulating the expression of p75 neurotrophin receptor (NTR), phospho-JNK, and Bcl-2–associated X protein apoptosis factors. HDACi treatment also increased the expression of the stem cell biomarker nestin, and decreased the expression of reactive astrocyte biomarker GFAP within damaged tissue following TBI. These findings provide further insight into the mechanisms by which HDACi treatment after TBI is neuroprotective and support the continued study of HDACis following acute TBI.
Acute traumatic brain injury (TBI) is a leading cause of disability and death, and results in reduced quality of life for surviving patients and prolonged economic effects on society (1). Primary injury occurs at the moment of trauma and is the direct result of shearing, tearing, and stretching of the brain parenchyma affecting neural tissue and blood vessels, and causing immediate contusion, hemorrhage, diffuse axonal injury, and ischemia (2, 3). TBI brings about cognitive and behavioral dysfunction through a complex sequence of secondary pathologic changes following injury. The mechanisms evolve over time and include excessive neurotransmitter release, mitochondrial dysfunction, increased blood–brain barrier (BBB) permeability, cerebral edema, inflammation, and seizures, causing cell death and clinical morbidity (4).
Clinical trials designed to test therapeutic agents aimed at improving residual cerebral function after TBI have not been successful because of the multifactorial nature of the disorder (2, 5–7). No pharmacological agent is currently approved for the treatment of acute TBI. The present standard of care consists of maintaining physiologic function by preserving adequate cerebral perfusion and normal intracranial pressure. Histone deacetylase inhibitors (HDACis) are potential candidates for the treatment of TBI. They are capable of inducing widespread alterations in cellular function and protein expression through posttranslational modification of histones, transcriptional factors, and heat shock chaperones (8–10).
Several studies have demonstrated HDACis to be effective neuroprotective agents following TBI in rodent models (4, 11–16). Administration of HDACis after TBI has been shown to reduce cortical contusion volume and inflammation, induce the expression of genes and proteins involved with cell survival, and improve cognitive and motor outcomes. The precise mechanisms by which HDACis produce these effects remain unclear. We developed an in vitro astrocyte scratch assay to examine the molecular effects associated with the administration of HDACis following mechanical glial cell injury (17). A TBI rat model was also established to investigate the efficacy of the pan-HDACi LB-205 for the treatment of acute TBI in vivo.
Results
LB-205 Enhances Migration of Astrocytes and Stem Cell Characteristics While Increasing Cell Proliferation Within Injury Repair Zone and Attenuating Apoptosis in Vitro.
LB-205 is a Zn2+-dependent pan-inhibitor of class I and class II HDACs with potent activity and a long half-life (12 h) in vivo (8). We sought to examine the effect of LB-205 on the injury of astrocytes in vitro. A scratch assay with human fetal astrocytes (HFAs) was used to observe morphological changes that occur during the activation of astrocytes following mechanical injury and subsequent repair with and without HDACi treatment. LB-205 increased the quantity of histones acetyl-H1, H2A.X, acetyl-H3, and acetyl-H4 postscratch (Fig. 1A), confirming the efficacy of LB-205 treatment during astrocyte scratch.
Fig. 1.

LB-205 enhances the migration of HFAs and increases stem-like cell character following scratch in vitro. (A) Western blotting for acetyl-H1, H2A.X, acetyl-H3, and acetyl-H4, demonstrating an increase in histone acetylation. (B) Time needed to repair the wound following astrocyte scratch. Percent of wound space remaining relative to the width of initial injury was measured at the most narrow point along opposing lines of astrocyte extension. LB-205–treated HFAs exhibited significantly accelerated wound closure (36.17% relative wound width remaining) at 16 h postscratch (12 h posttreatment) in comparison with DMSO (58.61%), SAHA (45.98%), or VPA (52.19%). Data are presented as the mean ± SEM (n = 3; ‡Significant difference by repeated-measures two-way ANOVA; *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001). (C) Immunofluorescence staining for nestin (red), indicating increased expression at 24 h following LB-205 treatment in comparison with scratch alone and SAHA- or VPA-treated astrocytes. (Scale bar: 50 μm.) (D) Western blot analysis demonstrates significantly increased expression of nestin at 24 h following LB-205 treatment compared with scratch alone and SAHA or VPA. Data are presented as the mean ± SEM (n = 3; ****P ≤ 0.0001).
The growth of astrocytes was observed at 4-h time intervals following scratch. HDACis were administered 4 h postscratch, and the percentage of the injury area filled in was measured. This measurement was performed to evaluate the effect of HDACis on the time needed to repair the wound. LB-205 significantly enhanced the migration of astrocytes and accelerated wound repair more effectively than suberoylanilide hydroxamic acid (SAHA), valproic acid (VPA), or DMSO at 16 h postscratch [12-h posttreatment; repeated-measures two-way ANOVA F(9,18) = 13.77, P < 0.0001; Fig. 1B]. The expression of biomarkers associated with glial activation was also evaluated. Immunofluorescence staining revealed an increased population of nestin (type-VI intermediate filament protein)-expressing cells at the edges of wounds after treatment with LB-205 compared with untreated scratched cells or scratched cells treated with SAHA or VPA (Fig. 1C). Nestin is a known marker for neural stem cell and progenitor cell populations within the CNS (17, 18). Western blot analysis of the expression of nestin 24 h postscratch further confirmed this result [one-way ANOVA F(5,12) = 1,435, P < 0.0001], indicating that administration of LB-205 elicited an increase in cells with neural stem cell characteristics within the proliferative zone of injury repair (Fig. 1D).
Analysis of cell cycle was carried out to assess cellular proliferation after astrocyte scratch. The percentage of G2/M cells at 24 h postscratch determined through fluorescence-absorption cell cycle analysis was significantly increased in scratched HFAs following treatment with LB-205 (12.5%) compared with scratched cells treated with DMSO alone (8.01%) or scratched cells treated with SAHA or VPA (Fig. 2A). The higher proportion of cells found to be in the G2/M phase indicated that treatment with LB-205 increased astrocyte proliferation during the repair of the injury. Quantification of immunofluorescence staining for Ki-67 confirmed that LB-205 significantly enhanced proliferation within the injury repair zone in scratched astrocytes (one-way ANOVA F(3,36) = 10.31, P < 0.0001; Fig. 2B). Astrocytes treated with LB-205 postscratch exhibited a decreased proportion of apoptotic cells at 24 h (15.95%) compared with those treated with DMSO alone (24.66%) or scratched cells treated with SAHA or VPA (Fig. 2C). This result demonstrated that treatment with LB-205 attenuated astrocyte apoptosis following scratch injury.
Fig. 2.

LB-205 increases G2/M transition, attenuates apoptosis, and alters gene expression of cell survival factors in HFAs following scratch in vitro. (A) Cell cycle analysis of cultured HFAs by FACS. Percentage of cells in G1, S, and G2 phases 24 h after scratch demonstrate increased percentage of G2-phase cells after LB-205 (12.5%), SAHA (8.94%), and VPA (10.1%) treatment compared with scratch alone (8.01%). (B) Quantification of Ki-67–positive nuclei calculated as percent positive per 10× microscopic field. LB-205 significantly enhanced proliferation of HFAs after scratch within the proliferation zone of injury repair (14.4%) in comparison with scratch alone (6.84%) or treatment with SAHA (8.38%) or VPA (10.5%). Data are presented as mean ± SEM (n = 10; ****P ≤ 0.0001). (C) FACS analysis of cultured HFAs demonstrated the percentage of cells undergoing apoptosis at 24 h post-scratch. Percentage of apoptotic cells was decreased following LB-205 (15.95%) treatment compared with scratch alone (24.66%) and SAHA (28.76%) or VPA (25.52%). (D) Whole-genome microarray analysis quantifying change in expression associated with astrocyte scratch with or without LB-205 treatment.
Whole-Genome Expression Microarray Analysis in Vitro.
Gene-expression profiling has the capability to identify biomarkers and elucidate complex genetic interactions that may play a mechanistic role in the molecular response and outcome following TBI with HDACi modulation (19). Whole-genome microarray analysis of scratched astrocytes was preformed with untreated samples and samples treated with LB-205. Biological functions and individual gene expressions were filtered for ≥twofold changes in induction. The expression of individual genes related to neural cell survival through the nerve growth factor (NGF) neurotrophic tyrosine kinase receptor type 1 (TrkA) pathway, NGF, NTRK1, AKT, NFKB, and BCL2 were significantly up-regulated [NGF, one-way ANOVA F(2,6) = 29.79, P < 0.0008; NTRK1, one-way ANOVA F(2,6) = 85.57, P < 0.0001; AKT, one-way ANOVA F(2,6) = 79.03, P < 0.0001; NFKB, one-way ANOVA F(2,6) = 53.65, P < 0.0001; BCL2, one-way ANOVA F(2,6) = 81.04, P < 0.0001; Fig. 2D]. The expression of genes related to the activation of apoptosis through the NGF p75 neurotrophin receptor (NTR) pathway, p75(NTR), JNK, and Bcl-2–associated X protein (BAX) were significantly down-regulated [p75(NTR), one-way ANOVA F(2,6) = 133.9, P < 0.0008; JNK, one-way ANOVA F(2,6) = 15.20, P < 0.0045; BAX, one-way ANOVA F(2,6) = 292.0, P < 0.0001; Fig. 2D].
Rats Treated with LB-205 Exhibit Decreased Functional Neurological Deficit Following TBI in Vivo, and Brain Tissue Specimens Show Decreased Contusion Volume and Enhanced Cellular Density upon Histological Examination.
Studies have shown HDACis to be neuroprotective following TBI in rodent models and to improve long-term recovery of neurological function (16). Most of this work has focused primarily on assessing the efficacy of VPA, a class I-specific HDACi with long half-life (8–15 h), although potent pan-HDACis (ITF2357) have also been shown to be efficacious. In considering our in vitro data, it being known that LB-205 possesses potent activity and long half-life (12-h) in vivo, and previous studies used in vivo TBI models for HDACi testing, we sought to develop an appropriate TBI rodent model to test the efficacy of LB-205 and to evaluate its mechanism. The right frontal cortex of rats aged 6 to 8 wk was lesioned by using a cortical impact device. i.p. injections of HDACi were administered 4 h, 24 h, and 48 h after injury. Rats treated with 10 mg/kg of LB-205 performed significantly better at standardized cross-beam balance motor testing 7 d after TBI [n = 10; repeated-measures two-way ANOVA F(6,18) = 101.9, P < 0.0001] than those that were untreated (Fig. 3A). LB-205 administration was well tolerated by the rats. Histological analysis was performed on post-TBI tissue samples from normal brain, heart, lung, liver, spleen, kidney, and pancreas in rats following LB-205 treatment and demonstrated no organ toxicity in comparison with nontreated rats. Compared with TBI alone, LB-205 treated rats killed 2 wk postinjury demonstrated less scaring and decreased contusion volume upon gross postmortem examination of brain tissues (Fig. 3B). Pathological analysis revealed enhanced gliosis and increased cellular density with LB-205 treatment following TBI (Fig. 3C).
Fig. 3.
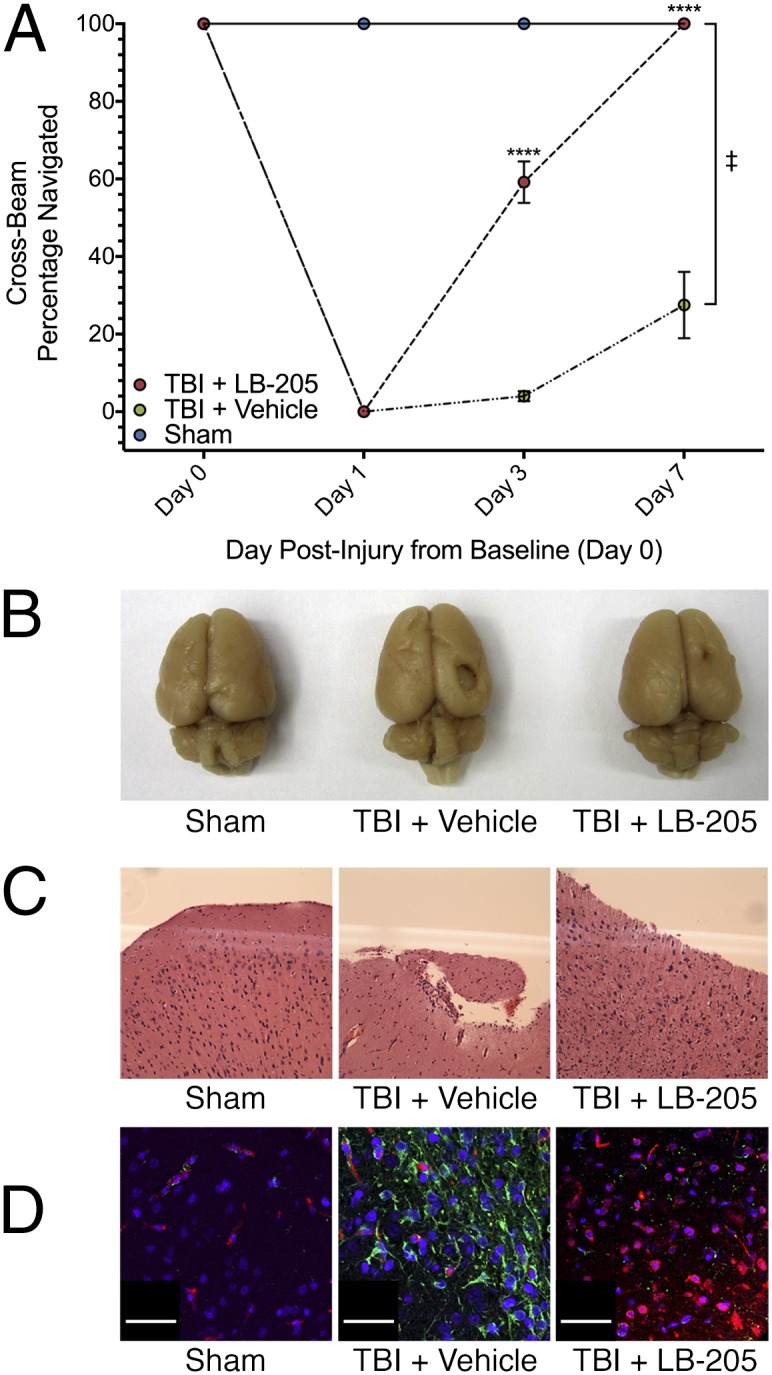
LB-205 improves clinical outcome in rats when administered following TBI while also altering injury repair response and attenuating postmortem signs of injury. (A) Measurement of cross-beam balance examination performance, recorded as percentage of beam successfully navigated at day 0 (pre-TBI) and days 1, 3, and 7 (post-TBI) in groups treated with LB-205 (10 mg/kg) or vehicle (50% DMSO) 4 h, 24 h, and 48 h following injury. LB-205 treated rats performed significantly better than vehicle treated rats at days 3 and 7 post-TBI. Data are presented as mean ± SEM (n = 3–4; ‡Significant difference by repeated-measures two-way ANOVA; ****P ≤ 0.0001). (B) Gross postmortem examination of LB-205–treated rat brains demonstrate less scaring and decreased contusion volume post-TBI than TBI alone. (C) H&E staining demonstrates enhanced gliosis and increased cellular density in brain tissues of rats treated with LB-205 compared with TBI alone. (D) Immunofluorescence staining for nestin (red) and GFAP (green), indicating increased expression of nestin and decreased expression of GFAP at 1 wk post-TBI following LB-205 treatment in comparison with TBI alone. (Scale bar: 30 μm.)
LB-205 Blunts Reactive Astrocytosis and Promotes Proliferation of Cells Expressing Neural Stem Cell Biomarkers in Vivo.
Immunofluorescence assays were performed 2 wk postinjury to assess the expression pattern of biomarkers associated with the repair of CNS injury. Expression of GFAP (type-III intermediate filament protein) in LB-205–treated rats was reduced at the sight of injury compared with nontreated rats (Fig. 3D). The expression of nestin was increased with LB-205 treatment and was most pronounced in proliferative cells adjacent to the site of injury. These results indicate that LB-205 enhanced the molecular repair process in vivo by reducing reactive astrocytosis and stimulating the formation of proliferative cells with stem cell characteristics.
LB-205 Maintains NGF Expression Post-TBI in Vivo and Postscratch in Vitro.
NGF is a member of the neurotrophin superfamily and is known to maintain the survival of neurons during development through stimulation of its high-affinity TrkA receptor (20, 21). NGF can also induce cell-mediated death through NGF withdrawal or stimulation of its low-affinity p75(NTR) receptor (21, 22). The finding of elevated activation of NGF TrkA pathway with whole-genome expression assay in vitro led us to examine lesioned rat brain tissue samples for NGF pathway modulation 1 wk following TBI with and without treatment with LB-205. Expression of NGF and its receptor TrkA were preserved after treatment with LB-205 compared with untreated TBI [NGF, one-way ANOVA F(2,6) = 1,955, P < 0.0001; phospho-TrkA (p-TrkA), one-way ANOVA F(2,6) = 203.1, P < 0.0001; Fig. 4A]. A prominent increase in p75(NTR) expression was observed following TBI, which was significantly attenuated by treatment with LB-205 [one-way ANOVA F(2,6) = 4794, P < 0.0001; Fig. 4A]. Increases in phospho-JNK (p-JNK), cleaved caspase 3, and cleaved-poly ADP ribose polymerase (PARP) proapoptotic factors were mitigated after treatment with LB-205 [p-JNK, one-way ANOVA F(2,6) = 235.8, P < 0.0001; caspase 3, one-way ANOVA F(2,6) = 252.2, P < 0.0001; PARP, one-way ANOVA F(2,6) = 29.94, P < 0.0001; Fig. 4B]. Administration of LB-205 also increased the expression of phospho-protein kinase B (p-AKT), NF-κB, and nuclear receptor co-repressor-1 (NCoR), a known regulator of stem cell development [p-AKT, one-way ANOVA F(2,6) = 4,531, P < 0.0001; NF-κB, one-way ANOVA F(2,6) = 83.29, P < 0.0001; NCoR, one-way ANOVA F(2,6) = 24.57, P < 0.0001; Fig. 4C].
Fig. 4.

LB-205 administration preserves NGF TrkA pathway activity post-TBI in vivo and after astrocyte scratch in vitro. (A) Western blot analysis demonstrated significantly increased expression of NGF and p-TrkA and attenuated p75(NTR) expression following LB-205 treatment at 7 d post-TBI compared with TBI alone. Data are presented as mean ± SEM (n = 3; ****P ≤ 0.0001). (B) Western blot analysis indicated significantly diminished expression of p-JNK, cleaved caspase-3, and PARP following LB-205 treatment post-TBI compared with TBI alone. Data are presented as mean ± SEM (n = 3; ****P ≤ 0.0001). (C) Western blot analysis demonstrated significantly increased expression of p-AKT, NF-κB, and NCoR following LB-205 treatment post-TBI compared with TBI alone. Data are presented as mean ± SEM (n = 3; **P ≤ 0.01 and ****P ≤ 0.0001). (D) Quantitative real-time PCR, with expression of factors being conveyed as fold-induction relative to control sample expression (fold-induction = 1), demonstrated 1.56-fold increased expression of NGF following LB-205 treatment post-TBI in comparison with TBI alone in vivo, and 0.87-, 1.40-, and 0.48-fold increased expression in NGF, NTRK1, and NFKB at 24 h post-scratch in vitro in comparison with scratch alone. Data are presented as mean ± SEM (n = 3; **P ≤ 0.01 and ***P ≤ 0.001).
Real-time quantitative PCR of lesioned rat brain tissues supported Western blot data and demonstrated a 1.56-fold increase in NGF expression 7 d post-TBI after treatment with LB-205 compared with TBI alone [NGF, one-way ANOVA F(2,6) = 33.19, P = 0.0006; Fig. 4D]. Real-time quantitative PCR of scratched astrocytes revealed similar 0.87-, 1.4-, and 0.48-fold increases in NGF, NTRK1 and NFKB expression, respectively, at 24 h postscratch following LB-205 administration compared with scratch alone [NGF, one-way ANOVA F(4,10) = 10.01, P = 0.0016; NTRK1 one-way ANOVA F(2,6) = 40.11, P = 0.0003; NFKB, one-way ANOVA F(2,6) = 10.01, P = 0.0123; Fig. 4D]. LB-205 was more effective than SAHA or VPA at inducing NGF expression postscratch. These results indicate that LB-205 effectively preserved NGF expression and its antiapoptotic function through the TrkA pathway in both in vitro astrocyte scratch in vivo TBI models.
Discussion
Epigenetic mechanisms are important regulators of gene–environment interactions. Histone methylation and acetylation are critically involved in cognitive function and are necessary for maintaining neural plasticity (9). Rodent models have established that neurons exhibit increased histone acetylation during the process of learning, and hindrance of this process leads to age-dependent memory impairment (23, 24). It has also been shown that histone-H3 acetylation is reduced following TBI, and that this finding is associated with measurable cognitive dysfunction in rodents (15). Translating this knowledge has led to the discovery that many human neurodegenerative disorders are also associated with relative decreases in histone acetylation. These findings have formed the basis for exploring the use of HDACis in the treatment of TBI and other forms of CNS injury.
HDACis have been shown to be efficacious in treating TBI in rodent models, with recent work predominately focused on the use of VPA. VPA is an HDACi with additional functionality in its ability to inhibit GABA transamination, reduce NMDA-mediated excitation, and block voltage-gated sodium and calcium channels (25). Rodents treated with VPA post-TBI exhibit improved performance in memory and motor dependent tasks (4). Our results demonstrate that LB-205 similarly improved functional outcome in a rat TBI model. Upon molecular examination in vitro, we found that LB-205 increased histone-H3 acetylation and the expression of Hsp70 while attenuating caspase-3 activation, analogous to the results obtained in studies testing the use of VPA in an ischemic injury rodent model (14). Furthermore, we demonstrated that LB-205 enhanced astrocyte migration and proliferation within the injury repair zone more than VPA or SAHA. VPA is a drug with known dose-dependent liver toxicity, and has been shown to require supratherapeutic dosing (400 mg/kg) to produce comparative molecular effects and clinical outcomes in rodents (4). LB-205 reduced post-TBI morbidity and enhanced the injury repair response in rats at a safe low therapeutic dose (10 mg/kg).
Here we present that HDACi treatment following TBI preserves NGF expression and TrkA survival pathway activation while attenuating expression of GFAP and inducing expression of nestin in reactive astrocytes, as insight into the mechanisms underlying the improved functional outcomes in rodent models observed with HDACi treatment post-TBI. NGF is a highly conserved neuropeptide and shares homology within different species. It plays an important role in rapid neural apoptosis and axonal guidance during embryogenesis and early development. In adult life, NGF is produced within the cortex, hippocampus, and pituitary gland while also playing an essential role in the survival of basal forebrain cholinergic neurons involved with arousal, attention, memory, and motivation (26). However, NGF may also play an critical role in the regeneration of injured neurons post-TBI (26–30). In addition, increased NGF levels in cerebral spinal fluid following TBI are known to be correlated with improved clinical outcome (31). NGF expression may be necessary to promote neuron survival and function post-TBI during the injury repair process.
NGF stimulates cell survival or cell death through independent signaling mechanisms (Fig. 5) (21). When NGF binds to its high-affinity cellular receptor TrkA, it stimulates the p-AKT pathway through PI3K, which, in turn, through NF-κB, induces the transcription of genes responsible for cell survival. Conversely, when NGF binds its low-affinity p75(NTR) caspase activity is induced, and apoptosis is triggered. NGF is elevated within the penumbra in rats following ischemic injury, and might be necessary to prevent low-affinity p75(NTR) receptor activation and neuronal apoptosis (29). Additionally, neurotrophic factor treatment in rats following TBI has been shown to attenuate reductions in cortical volume and enhance functional recovery following ischemic injury and TBI (28, 30). We found that LB-205 up-regulated the expression of NGF, p-TrkA, p-AKT, NF-κB, and Bcl-2 cell survival factors, while diminishing the expression of p75(NTR), p-JNK, and BAX apoptosis factors. This result effectively modulated the injury repair process by maintaining a balance of NGF activity in favor of cell survival in vivo and in vitro. Determining the modification of which specific HDAC(s) and histones are responsible for these effects requires further study. It should also be considered that modulation of temporal and spatial patterns of NGF expression after HDACi treatment within in areas of the brain not directly lesioned could affect functional outcomes post-TBI.
Fig. 5.
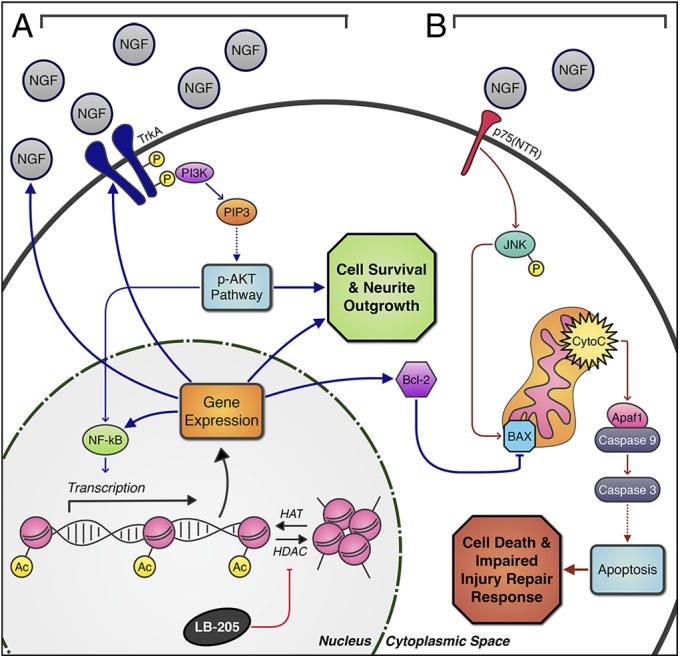
Schematic depiction of LB-205 effects on NGF stimulated pathways. (A) LB-205 inhibits HDACs and increases acetylation of histones by histone acetyltransferases (HATs), which play a key role in regulating gene expression in the nucleus. Activated PI3K initiates the p-AKT pathway, which acts through NF-κB transcriptional modification and other targets to alter expression of NGF, TrkA, and NF-κB to promote cell survival, growth, and proliferation. In addition, BAX is blocked through increased Bcl-2 expression, inhibiting the apoptosis pathway. (B) Without LB-205 treatment, decreased NGF and increased p75(NTR) expression post-TBI induce p-JNK through the p75(NTR) receptor-mediated pathway and subsequent release of apoptosis associated factors from the mitochondria, triggering the caspase cascade through BAX activation, inducing cell apoptosis, and impairing the CNS injury repair response.
Expression of nestin was significantly enhanced by LB-205 in vivo and in vitro within the zone of injury repair, suggesting that increased proliferation of cells with stem cell characteristics might play an important role in enhancing CNS injury repair and preserving neural cell function post-TBI. Expression of GFAP was diminished by LB-205 treatment at 2 wk postinjury and may represent an additional mechanism by which HDACi treatment post-TBI blunts CNS scar formation and significant disruption of the remaining neural network. GFAP, a biomarker of reactive astrocytosis and axonal damage, is elevated in the CSF of patients post-TBI (32). The hallmark of reactive gliosis following injury is increased GFAP expression and subsequent hypertrophy of astrocyte processes and repression nestin expression (33). Following scratch injury, GFAP expression is induced in reactive astrocyte populations migrating to fill in and repair the injury zone (17). Reactive astrocytes therefore play an important role in the acute stage of CNS injury, although they may inhibit neural regeneration and CNS repair with prolonged activation. The specific mechanisms by which expression of nestin is up-regulated and GFAP expression is diminished following HDACi treatment post-TBI require further study. However, NGF is known to stimulate the expression of biomarkers associated with neural stem cell characteristics, and may be responsible for the increased expression of nestin and attenuated GFAP expression observed following treatment with LB-205 (34).
The complete mechanisms underlying how HDACis modulate the injury repair response in the CNS remain unknown. Gene chip analysis revealed significant up-regulation of noteworthy genes, including HSPA1A, EDN3 and IL6, that were not investigated in depth. Heat shock protein 70, coded for by HSPA1A, a molecular chaperone (protein) also induced after treatment with VPA and ITF2357, is neuroprotective and improves protein salvage by presumably serving to expand the availability of proteins necessary for cell survival following injury (16, 35). The role of endothelin-3 (EDN3), coded for by EDN3, vasoactive peptide induction following HDACi is less clear. Blocking the EDN3 receptor triggers apoptosis in glioma cancer stem cells and a functional reduction in cell migration (36). EDN3 therefore may have a role in maintaining cell survival and improving glial cell motility post-TBI. The deficiency of proinflammatory cytokine IL-6, coded for by IL6, in KO mice has been demonstrated to diminish the capacity to cope with brain injury (37). Furthermore, increased BBB permeability has been shown to be an important mechanism involved with neural cell loss following TBI, although this process likely improves CNS drug delivery (38). HDACi effects on BBB permeability and other factors mentioned require further study.
HDACis likely produce much of their beneficial effects post-TBI by buffering the cellular response to injury. We have established the potential significance of HDACi-mediated preservation of NGF TrkA pathway activation and augmentation of proliferation of nestin-expressing cells with stem cell characteristics post-TBI. We have also shown that LB-205 reduces post-TBI morbidity in rats at lower therapeutic concentrations than VPA, and with no observed toxicity. Our experimental findings support growing evidence for the neuroprotective effects of HDACis and continued research into their use following acute TBI.
Materials and Methods
Animals and in Vivo HDACi Treatment.
Animal experiments were approved for use and care of animals under the protocol guidelines of the National Institutes of Health (NIH) Animal Care and Use Committee. Sprague–Dawley rats (aged 6–8-wk, male) were purchased from Harlan Laboratories. Animals were randomized into three groups: sham (n = 6), TBI (n = 6), and TBI treated with LB-205 (n = 8). Animals were subjected to controlled cortical impact TBI as previously described (39). Briefly, animals were anesthetized with isoflurane (1.5%) in 1:1 oxygen:nitrous oxide gas mixture and placed in a stereotactic device. A 5 × 5-mm craniotomy was placed in the right parietal bone with a handheld trephine. The impactor tip was regulated down to the dural surface, and a penetrating contusion was made over the right frontal cortex at 2.1 to 2.2 mm depth at 5 m/s velocity. The burr hole was covered with a bone flap and sealed with Jet Denture Repair. Following standard postsurgical analgesia, each animal was given buprenorphine at 0.01 to 0.05 mg/kg (i.p. injection). All rats were allowed to recuperate after surgery before return to cages. Animals were observed postsurgery for appropriate incision healing and monitored daily for clinical and behavioral signs. Further details are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. Paul E. Gallant [National Institute of Neurological Disorders and Stroke (NINDS) Light Microscopy Facility] for assistance with confocal microscopy, Dr. Dragan Maric (NINDS Flow Cytometry Core Facility) for assistance with flow cytometry, and Lixte Biotechnology Holdings for providing LB-205. This work was supported by the Department of Defense in the Center for Neuroscience and Regenerative Medicine, Intramural Research Programs of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), NINDS, Imaging Sciences Training Program (sponsored by Radiology and Imaging Sciences in the Clinical Center and Intramural Research Programs of the NIBIB) (J.L.), and Howard Hughes Medical Institute–NIH Medical Research Scholars Program (J.M.F.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1308950110/-/DCSupplemental.
References
- 1.Coronado VG, et al. Centers for Disease Control and Prevention (CDC) Surveillance for traumatic brain injury-related deaths—United States, 1997-2007. MMWR Surveill Summ. 2011;60(5):1–32. [PubMed] [Google Scholar]
- 2.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26(8):1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Vink R, Nimmo AJ. Multifunctional drugs for head injury. Neurotherapeutics. 2009;6(1):28–42. doi: 10.1016/j.nurt.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dash PK, et al. Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS ONE. 2010;5(6):e11383. doi: 10.1371/journal.pone.0011383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Margulies S, Hicks R. Combination Therapies for Traumatic Brain Injury Workshop Leaders Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26(6):925–939. doi: 10.1089/neu.2008.0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narayan RK, et al. Clinical trials in head injury. J Neurotrauma. 2002;19(5):503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shein NA, Shohami E. Histone deacetylase inhibitors as therapeutic agents for acute central nervous system injuries. Mol Med. 2011;17(5-6):448–456. doi: 10.2119/molmed.2011.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu J, et al. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc Natl Acad Sci USA. 2011;108(52):21200–21205. doi: 10.1073/pnas.1119181109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer A, Sananbenesi F, Mungenast A, Tsai L-H. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31(12):605–617. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 11.Dash PK, Orsi SA, Moore AN. Histone deactylase inhibition combined with behavioral therapy enhances learning and memory following traumatic brain injury. Neuroscience. 2009;163(1):1–8. doi: 10.1016/j.neuroscience.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, et al. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 2008;1226:181–191. doi: 10.1016/j.brainres.2008.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim HJ, et al. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321(3):892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 14.Ren M, Leng Y, Jeong M, Leeds PR, Chuang D-M. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: Potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89(6):1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- 15.Gao W-M, et al. Immunohistochemical analysis of histone H3 acetylation and methylation—evidence for altered epigenetic signaling following traumatic brain injury in immature rats. Brain Res. 2006;1070(1):31–34. doi: 10.1016/j.brainres.2005.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shein NA, et al. Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 2009;23(12):4266–4275. doi: 10.1096/fj.09-134700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang C, et al. β-Catenin signaling initiates the activation of astrocytes and its dysregulation contributes to the pathogenesis of astrocytomas. Proc Natl Acad Sci USA. 2012;109(18):6963–6968. doi: 10.1073/pnas.1118754109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rietze RL, et al. Purification of a pluripotent neural stem cell from the adult mouse brain. Nature. 2001;412(6848):736–739. doi: 10.1038/35089085. [DOI] [PubMed] [Google Scholar]
- 19.Barr TL, Alexander S, Conley Y. Gene expression profiling for discovery of novel targets in human traumatic brain injury. Biol Res Nurs. 2011;13(2):140–153. doi: 10.1177/1099800410385671. [DOI] [PubMed] [Google Scholar]
- 20.Johnson D, et al. Expression and structure of the human NGF receptor. Cell. 1986;47(4):545–554. doi: 10.1016/0092-8674(86)90619-7. [DOI] [PubMed] [Google Scholar]
- 21.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407(6805):802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 22.Jing S, Tapley P, Barbacid M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron. 1992;9(6):1067–1079. doi: 10.1016/0896-6273(92)90066-m. [DOI] [PubMed] [Google Scholar]
- 23.Levenson JM, et al. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279(39):40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- 24.Peleg S, et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328(5979):753–756. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 25.Rosenberg G. The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees? Cell Mol Life Sci. 2007;64(16):2090–2103. doi: 10.1007/s00018-007-7079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aloe L, Rocco ML, Bianchi P, Manni L. Nerve growth factor: From the early discoveries to the potential clinical use. J Transl Med. 2012;10(239):239. doi: 10.1186/1479-5876-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mahmood A, Lu D, Chopp M. Intravenous administration of marrow stromal cells (MSCs) increases the expression of growth factors in rat brain after traumatic brain injury. J Neurotrauma. 2004;21(1):33–39. doi: 10.1089/089771504772695922. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, et al. Improvement in functional recovery with administration of Cerebrolysin after experimental closed head injury. J Neurosurg. 2013;2013(Apr):12. doi: 10.3171/2013.3.JNS122061. [DOI] [PubMed] [Google Scholar]
- 29.DeKosky ST, et al. Upregulation of nerve growth factor following cortical trauma. Exp Neurol. 1994;130(2):173–177. doi: 10.1006/exnr.1994.1196. [DOI] [PubMed] [Google Scholar]
- 30.Holtzman DM, Sheldon RA, Jaffe W, Cheng Y, Ferriero DM. Nerve growth factor protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 1996;39(1):114–122. doi: 10.1002/ana.410390117. [DOI] [PubMed] [Google Scholar]
- 31.Chiaretti A, et al. NGF, DCX, and NSE upregulation correlates with severity and outcome of head trauma in children. Neurology. 2009;72(7):609–616. doi: 10.1212/01.wnl.0000342462.51073.06. [DOI] [PubMed] [Google Scholar]
- 32.Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76(5):886–899. doi: 10.1016/j.neuron.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 33.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50(4):427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 34.Chen L-W, Zhang J-P, Kwok-Yan Shum D, Chan Y-S. Localization of nerve growth factor, neurotrophin-3, and glial cell line-derived neurotrophic factor in nestin-expressing reactive astrocytes in the caudate-putamen of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57/Bl mice. J Comp Neurol. 2006;497(6):898–909. doi: 10.1002/cne.21014. [DOI] [PubMed] [Google Scholar]
- 35.Yang C, Huntoon K, Ksendzovsky A, Zhuang Z, Lonser RR. Proteostasis modulators prolong missense VHL protein activity and halt tumor progression. Cell Rep. 2013;3(1):52–59. doi: 10.1016/j.celrep.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, et al. Autocrine endothelin-3/endothelin receptor B signaling maintains cellular and molecular properties of glioblastoma stem cells. Mol Cancer Res. 2011;9(12):1668–1685. doi: 10.1158/1541-7786.MCR-10-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulsen CB, et al. Brain response to traumatic brain injury in wild-type and interleukin-6 knockout mice: A microarray analysis. J Neurochem. 2005;92(2):417–432. doi: 10.1111/j.1471-4159.2004.02877.x. [DOI] [PubMed] [Google Scholar]
- 38.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6(7):393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoskison MM, et al. Persistent working memory dysfunction following traumatic brain injury: Evidence for a time-dependent mechanism. Neuroscience. 2009;159(2):483–491. doi: 10.1016/j.neuroscience.2008.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.