

Abstract
In this study, a new class of duplex microfluidic device is described that utilizes a dual perifusion network to simultaneously perform live-cell optical imaging of physiological activities and study insulin release kinetics on two islet populations. In addition, this device incorporates on-chip staggered herringbone mixers (SHMs) to increase mixing efficiency and facilitate the generation of user-defined chemical gradients. Using mouse islets, we simultaneously measure dynamic insulin release, changes in mitochondrial potentials, and calcium influx in responses to insulin secretagogues (glucose and tolbutamide), showing a high signal-to-noise ratio and spatiotemporal resolution of all measured parameters for both perifusion chambers. This system has many potential applications for studying β-cell physiology and pathophysiology, as well as for therapeutic drug screening. This dual perifusion device is not limited to islet studies and could easily be applied to other tissues and cells without major modifications.
Keywords: Microfluidic, perifusion, staggered herringbone mixer (SHM), islets of Langerhans, insulin secretion, and fluorescence imaging
1. INTRODUCTION
Type 1 diabetes is an autoimmune disorder which involves the destruction of β-cells (insulin-producing cells in the islets of Langerhans) leading to hyperglycemia and long-term health complications. In order to study β-cell physiology and pathophysiology in vitro, a laboratory setup that has the capability to more closely mimic the dynamic physiological in vivo microenvironment is required. Islet studies have traditionally been conducted in a static culture well or plate, which has limited physiological relevance since islets, in vivo, experience a dynamically changing environment. In particular, it has been demonstrated that insulin release kinetics, in response to glucose challenge, are biphasic and oscillatory in nature (Bergsten et al., 1994; Gilon et al., 2002; Ravier et al., 1999). Since the 1970s the macroscale perifusion approach has revolutionized how islets can be studied. Several types of macroscale perifusion apparatuses have been established with the capability to regulate temperature, pH, perfusate flow rate, and switch between perfusate streams of various stimuli (Hoshi et al., 1973; Lacy et al., 1972; Sturis et al., 1994). However, these systems face several challenges, including high reagent consumption, difficult operation, limited flow control, and a lack of integration with conventional analytical techniques and live-cell imaging. In recent years, microfluidic technology has emerged as a valuable tool for islet study, mainly due to its versatility, enhanced efficiency, and minimal consumption of reagents and bioanalytes. In addition, microfluidic perifusion devices offer a superior platform over macroscale platforms in that they: (i.) more closely mimic the islet in vivo perfusing microenvironment; (ii.) have the ability to precisely control physiologically relevant parameters; (iii.) can effectively integrate analysis tools; and (iv.) allow for multiplex tasking (Chen et al., 2008; Dishinger et al., 2007; Dishinger et al., 2009; Rocheleau et al., 2004; Roper et al., 2003; Shackman et al., 2005).
It is well-known that islets compose 1% of the pancreas mass but receive 2-10% of the blood flow perfusion (Carlsson et al., 1996; Lifson et al., 1985). Many β-cell cellular processes, including proliferation, exocytosis, metabolism, protein biosynthesis, and gene expression, are influenced by dynamic environmental changes in perfusing blood (Henquin, 2000; Lee et al., 2009). Under normal in vivo conditions, pancreatic islets experience dynamic changes in the perfusing environment including, but not limited to: glucose, hormones, neural transmitters, and corresponding changes in shear forces from vasodilation and vasoconstriction. Therefore, efficient fluid control in microfluidics that can mimic the dynamically changing in vivo chemical and physical environment is necessary to achieve a better understanding of hormone secretion kinetics, metabolism, and ion signaling of β-cells.
There are several methods to create a desired concentration gradient in microfluidic networks (Atencia et al., 2009; Du et al., 2009). Yet, the majority of methods generate chemical gradients with respect to position and not with respect to time. Additionally, these existing gradient production methods are plagued by inconsistency, fabrication complexity, and operational difficulties. Despite the importance of chemical gradient generation in understanding β-cell kinetics, there are few existing microfluidic systems capable of generating and maintaining time-varying gradients using either two or more on-chip diaphragm pumps (Zhang et al., 2010; Zhang et al., 2009) or off-chip mixers in concert with pumps (Adewola et al., 2010).
We have previously described two three-layer polydimethylsiloxane (PDMS) microfluidic islet perifusion devices that are capable of immobilizing multiple islets of variable size (50-400 μm) and imaging islet physiological status through single or multiple channel fluorescence (Adewola et al., 2010; Mohammed et al., 2009). While these devices demonstrate a moderate spatiotemporal resolution of the measured parameters there are several limitations that hinder their application for more complex research purposes. Although multiple chambers can be integrated in parallel into a single microchip for islet perifusion, only one of these chambers can be optically monitored at a given time. In addition to this relatively low throughput, only one type of microenvironment can be applied in this device. Moreover, macroscale off-chip mixers constrain the device applicability, especially for those experiments requiring rapid mixing at low Reynolds numbers. Therefore, we sought to address these limitations by designing a discrete dual perifusion network integrated with an on-chip chaotic mixer. This device will enable simultaneous perifusion and optical detection of two distinct islet populations or alternatively, the same islet population within two different microenvironments such as treated vs. non-treated under a single field of view at the same time.
2. METHODS AND MATERIALS
2.1 Microfluidic device design and fabrication
The PDMS-based microfluidic perifusion device was designed and fabricated according to previously described methods (Mohammed et al., 2009). Briefly, a 3-inch silicon wafer was sequentially cleaned with acetone, methanol, and isopropanol. Then the wafer was dried with nitrogen gas and further treated with oxygen plasma at 100 W for 30 s (Plasma Preen), to oxidize any residual organic molecules. SU-8 photoresist was then spin coated on the wafer, patterned, and further developed using previously established protocols. These steps were repeated for each layer. PDMS was then poured over each photoresist mold and cured at 80 °C for 1 h and 45 min. The inlets and outlets were made into the cured PDMS using a No. 11 gauge (~2 mm) hole-punch. All of the layers were then bound together using an oxygen plasma corona surface treater (BD-20A) and further annealed on a hot plate for 2 h at 75 °C to form the final device.
2.2 Evaluation of chaotic mixer efficiency
The mixing efficiency of the integrated chaotic mixer in the inlet channel was first experimentally characterized by mixing two color food dyes. It was further confirmed by fluorescence imaging of mixtures of either fluorescein isothiocyanate (FITC) and deionized (DI) water or albumin conjugated with FITC and DI water mixing. For the food dye experiment, syringe pumps (Harvard apparatus, model 22) containing 1% (vol/vol) blue and yellow dyes dissolved in DI water were delivered into two separate inlets into the serpentine chaotic mixer channel at a speed of 250 μL/min. A representative mixing image of the whole mixing channel was recorded using a stereomicroscope (Olympus MVX10, Olympus) with a high-speed and a high-resolution charged couple device (CCD) (Retiga, SRV, Fast 1394, QImaging).
All quantitative mixing assessments were done with fluorescent dyes (FITC or FITC-albumin) dissolved in DI water. For the fluorescence mixing, 30 μM FITC and DI water or 25% albumin-FITC and DI water were simultaneously perifused via two separate inlets at either different flow rates or different ratios. FITC fluorescence images were collected at various inlet and outlet positions, with the same CCD used in the food dye mixing experiment, and analyzed off-line by ImageJ software.
The degree of advective mixing (convection plus diffusion) was quantified as a function of axial channel mixer distance. For fluorescence image intensity calculations, images were taken from the bottom of the device and an area (flow area) was selected including the whole channel width. For the inlet and outlet, length and width dimensions were 1 mm by 0.1 mm. For spaces in between SHMs, the length and width dimensions were 2 mm by 0.1 mm. Fluorescent microscopy images were taken from flow areas parallel to flow from the bottom of the device, and intensity was measured using ImageJ software.
Adapting the same equation from Stroock et al. (Stroock et al., 2002): The standard deviation of the intensity distribution in the flow areas was calculated using the equation: , where I is the grayscale value (between 0 and 1) of a pixel, and ⟨⟩ is the average over all the pixels in the image. The grayscale value at a pixel was normalized from 0 to 1. To quantitatively express mixing efficiency, σ of the intensity distribution of the flow area fluorescence imaging was calculated, with mixing efficiency expressed from a range of 0.0 (complete mixing) to 0.5 (no mixing). The mixing efficiencies (σ) were expressed as either a function of the channel mixer axial distance or as a function of increasing flow ratios.
2.3 Evaluation of perifusion chamber fluid exchange
Fluid exchange in single perifusion chamber was characterized experimentally with, 30 μM FITC dye in both water and 25% FITC-albumin solution. In comparing simultaneous flow dynamics between perifusion chambers, only 25% FITC-albumin solution was used. At a speed of 250 μL/min, DI water was first perifused for 60 s, followed with either FITC or FITC-albumin for 120 s, and finally washed out with DI water for additional 120 s. The protocol was repeated three times. Time-lapse images were taken at several different positions in the chamber with the CCD and analyzed using ImageJ software to quantify fluid exchange in the chamber.
2.4 Evaluation of chemical temporal gradient creation
The ability of the microfluidic network to create and maintain chemical gradients was characterized by mixing two different concentrations of Krebs’s Ringer Buffer (KRB: 119 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2•2H2O, 1.2 mM MgSO4•7H2O, 1.2 mM KH2PO4, 25 mM NaHCO3) containing either high (25 mM) or low (2 mM) glucose concentrations. The glucose solutions were delivered via two separate inlets using syringe pumps (Harvard Apparatus, model 22). The flow rate and volume of each solution were modulated by in-house programmed LabView software, as described previously (Adewola et al., 2010). The glucose solutions were mixed by the chaotic mixer before entering each respective perifusion chamber and perifusates were collected via the outlet at an interval of 1 mL/min. Glucose concentrations of perifusates were determined using a glucometer (Ascensia, Elite XL). Simultaneous creation of different glucose profiles in the dual perifusion chambers were then analyzed and compared to expected values: linear vs. stair; low to high vs. high to low; ʌ-shape vs. V-shape, respectively. Coefficient of determination (R2) was used to compare with theoretical values.
2.5 Islet isolation and tissue culture
8-12 week old male C57/B6 mice (Jackson Laboratory, MA) were sacrificed and pancreatic islets were isolated, as previously described (Lacy et al., 1972). Briefly, in a retrograde manner, the pancreas was injected and distended through the pancreatic duct with 2 mL of Collagenase P (Roche, Applied Science, IN) at a concentration of 0.375 mg/dL. The distended pancreas was incubated at 37 °C for 15 min, shaken vigorously for 5 s to dissociate the pancreatic tissue, and washed twice with cold HBSS. Islets were purified using a discontinuous Ficoll gradient (Mediatech, VA) and were then cultured at 37 °C with 5% CO2 in RPMI-1640 medium, supplemented with 10% fetal bovine serum (Hyclone Inc, MA), 100 unit/mL of penicillin and 100 μg/mL of streptomycin. All experiments were performed after overnight culture of the isolated islets.
2.6 Microfluidic chip operation
2.6.1 Device preconditioning
Prior to use, the microfluidic device was first flushed with 70% ethanol for 10 min to sterilize the device and minimize bubble formation and accumulation. The ethanol solution was then flushed out with DI water for 10 min. Finally, the device was preconditioned with KRB containing 2 mM glucose and 0.3% bovine serum albumin (BSA). BSA was used to prevent absorption of insulin secreted by β-cells into the interior surfaces of the device and tubings.
2.6.2 Islet loading
Prior to islet loading, all tubings were disconnected and PDMS-filled stoppers were removed from the islet loading port to prevent rapid and unstable pressure changes inside the perifusion chambers. Under a dissecting microscope, islets were manually picked using a 20 μL micropipette and gently injected into the perifusion chambers. In order to facilitate cell loading into the imaging zone, the device was tilted so that islets were directed inward towards the center of the device near the barrier separating the adjacent perifusion chambers. Then the device was tilted to ensure the rest of the islets gently settled down into an array of circular wells defined as the perifusion zone. After islet loading, all ports were completely closed using the PDMS-filled stoppers and all tubings were reconnected to the device.
2.6.3 Simultaneous islet perifusion and fluorescence imaging
In vivo, glucose-induced insulin release is a complex and dynamic process (Lefebvre et al., 1987; Polonsky et al., 1988). The process begins by the facilitated diffusion of glucose into β-cells by the glucose transporter GLUT2 resulting in an increase in glycolytic activity and, subsequently, the synthesis of ATP through the mitochondrial Tricarboxylic Acid Cycle (TCA cycle). The rise in ATP closes ATP-sensitive K+ (KATP) channels, thus initiating plasma membrane depolarization and the rapid influx of calcium ions via voltage-dependent calcium channels (VDCCs). Finally, the increase in intracellular calcium ([Ca2+]c) triggers the fusion of the insulin granules with the plasma membrane, which results in the exocytosis of insulin, C-peptide, and proinsulin (Ashcroft et al., 1984; Keizer et al., 1989).
For all perifusion and imaging experiments conducted in this study, mouse islets were labeled with the ratiometric fluorescent dye Fura-2/AM (Fura-2, Molecular Probes, CA) to determine intracellular calcium levels and fluorescent Rhodamine 123 dye (Rh123, Sigma, MO) to determine changes in mitochondrial potentials. In brief, the islets were incubated with 5 μM Fura-2 and 2.5 μM Rh123 for 30 min at 37 °C in KRB containing 2 mM glucose. The islets were then introduced into the temperature equilibrated microfluidic device through the loading port and the device was mounted on an inverted epifluorescence microscope (Leica DMI 4000B). The loaded islets were perifused by continuous flow of KRB2 at 37 °C (pH 7.4) for 10 min. KRBs containing high glucose (14 mM) or tolbutamide (250 μM) were then administered to the islets using a peristaltic pump. Dual-wavelength Fura-2/AM was excited at 340 and 380 nm and fluorescent emission was detected at 510 nm. Intracellular [Ca2+]c was expressed as a ratio of fluorescent emission intensity F340/F380 (%). Rh123 is a lipophilic cation that partitions selectively into the negatively-charged mitochondrial membrane. Glucose-induced hyperpolarization of the mitochondrial membrane causes uptake of Rh123 into the mitochondria and a decrease in Rh123 fluorescence via fluorescence quenching. Rh123 was excited at 490 nm ± 10, and emission was measured at 530 nm ± 10. Fura-2 and Rh123 fluorescence emission spectra were filtered using a Fura-2/FITC polychroic beamsplitter and double band emission filter (Chroma Technology. Part number: 73100bs). These images were collected with a CCD (Retiga-SRV, Fast 1394, QImaging). SimplePCI software (Hamamatsu Corp.) was used for image acquisition and analysis. Both fluourescence signals were expressed as ‘change-in-percentage’ after being normalized against basal intensity levels established before stimulation. The perifusates were collected from device outlets using an automated fraction collector (Gilson, model 203B, WI) at an interval of 1 mL/min to determine β-cell insulin secretory kinetics.
2.6.4 Insulin ELISA quantification
Collected perifusate samples were frozen at −80 °C prior to insulin quantification. All samples were then completely thawed and mixed well at room temperature prior to the determination of insulin levels via ELISA kit (Mercodia AB, Uppsala, Sweden), according to the manufacturer protocol.
3 RESULTS AND DISCUSSION
3.1 Construction of the dual perifusion system
Fig.1 illustrates the design and dimensions of the device used in this study. The dual perifusion networks, which are mirror images of each other, each contain three layers. The top-layer consists of two inlets, a chaotic mixer composed of a serpentine channel embossed with eight staggered herringbone mixers (SHMs), an outlet, and an islet-loading port corresponding to each perifusion chamber. Solutions from the two inlets (1 mm in width and 1 mm in height) meet at a T-channel intersection and then move downstream into the SHMs. First described by Stroock et al. (Stroock et al., 2002) and further geometrically optimized by Lynn et al. (Lynn et al., 2007), the SHM is a chaotic advection mixer that uses alternating cycles of asymmetric herringbone grooves to generate solution mixing by increasing diffusion time with the advantages of high efficiency, simple fabrication, and easy operation. We applied the same design and integration principles in our device. Each SHM is composed of eight linear grooves (6 mm in length) with a channel aspect ratio of 0.2 (h/w = 0.2), a groove depth ratio of 1.6 (d/h = 1.6), and a ridge length of 145 μm embossed within the bottom of the serpentine channel. In addition, the winding-shape serpentine channel design was used in this device to allow for further fluid flow stretching, folding, splitting, and recombination, which are all key processes in chaotic advection induction.
Fig. 1. A 2-D design rendering and dimensions of the dual perifusion platform.

In each perifusion network, the top-layer consists of two inlets, SHMs, a loading port, and one outlet. The middle-layer consists of a hexagonal-shape perifusion chamber. The bottom-layer has two zones of circular wells, perifusion zone and imaging zone, for islet immobilization and imaging. The image insert shows the black and white converted image of Rh123 labeled islets sitting in the imaging zones of chambers A and B, separated by a thin barrier. Scale bar = 100 μm.
The middle-layer is composed of a hexagonal-shape perifusion chamber with a total chamber volume of 50 μL. In our previous apparatus, a circular perifusion chamber was designed with total volume of 100 μL. To allow dual perifusion with concurrent optical imaging, a hexagonal-shape perifusion chamber was designed in this study. The dual chambers are arranged in parallel and are fluidically separated by a barrier that is 500 μm in height and 200 μm in width. With the assistance of the chaotic mixer in the inlet channel with a smaller chamber volume, flow dynamics in perifusion chamber were shown similar to previous design as confirmed by a software simulation (data not shown).
The bottom-layer consists of circular wells, or pockets, for islet immobilization. These wells have a diameter of 350 μm and a depth of 250 μm. There were designed to accommodate for the variability in islet size, which ranges between 50-400 μm, both across and within different species. The space between each circular well is 100 μm horizontally and vertically. When designing a microfluidic-based perifusion system for islet study, a suitable design for islet immobilization is considered to be one of most important requirements, as well as one of the greatest challenges, for the success of the device due to the unique spatial distribution and cellular composition of islets. As previously described (Adewola et al., 2010; Mohammed et al., 2009), a major advantage of this pocket design is that it allows islets to passively sit and interface with the perifusing medium in a manner that is more analogous to the in vivo physiological environment with minimized mechanical stress and shear forces. This is particularly important for islet cells because they are vulnerable to mechanical stress, in that even minimal stress can cause cell damage and insulin secretion. Furthermore, this pocket design allows multiple islets to be easily immobilized, under various flow rates, and analyzed simultaneously.
There are two groups of circular wells in our dual perifusion device: the imaging-zone and a perifusion-zone. The imaging-zone consists of 2 wells per chamber located adjacent to the separation barrier (shown in Fig.1 imaging insert) this facilitates optical monitoring under a single objective lens at 10X or 20X. The perifusion-zone contains an array of circular wells with identical dimensions to that of the imagine zone. The intention of designing these two areas was to increase device applicability and throughput for either single or multiple islet studies while most of the current microfluidic devices used for islet study can only immobilize a single islet (Chen et al., 2008; Easley et al., 2009; Rocheleau et al., 2004).
3.2 Mixing efficacy using on-chip integrated chaotic mixer
Conventional food dyes were added to the device to confirm the mixing efficacy (Fig. 2a). At a flow rate of 500 μL/min, the flow dynamics in the inlet and mixing channel can be grossly divided into three stages: stage 1: independent laminar flow streams from position of inlet (0 mm) to 16 mm, where both laminar flow streams (blue and yellow dye) remained quite stable and formed independent streams at the proximal region with partial mixing near the distal region; stage 2: partial to complete mixing from 32 mm to 48 mm, where partial mixing occurred at 16 mm and became more complete downstream; and stage 3: steady state mixing from 56 mm to the position of outlet (64 mm). At 48 mm, mixing was complete after passing through the fifth or sixth SHM grove mixers and fluid mixing continued through 64 mm to ensure full homogeneous mixing.
Fig.2. Mixing efficacy of SHMs in the winding-shape serpentine channel using food dyes and fluorescence.

(a) A representive image of the mixing efficacy using 1% blue and 1% yellow food dyes in the inlet serpentine channel at a flow rate of 500 μL/min (n=3). (b) A plot of standard deviation of the FITC intensity as a function of microchannel distance at three flow rates, under constant flow ratio (1:1); n=3. (c) A plot of standard deviation of the FITC intensity as a function of microchannel distance at three flow ratios, under constant total flow rate of 250 μL/min; n=3. (d) A plot of standard deviation of the albumin-FITC intensity as a function of microchannel distance at three flow rates, under constant flow ratio (1:1); n=3. (e) A plot of standard deviation of the albumin-FITC intensity as a function of microchannel distance at three flow ratios, under constant total flow rate of 250 μL/min; n=3. The horizontal dotted line indicates the value of σ that corresponds to 90% mixing (0.05). Data was reported as Mean ± SD.
In order to quantify mixing efficiency and understand the influence of flow rate and flow mixing ratio, a serial of mixing experiments using 30 μM FITC and DI water or 25% FITC-albumin and DI water at different flow rates and different mixing ratios were conducted (Fig.2b-2e). Without the assistance of an on-chip mixer, the length of the microchannel required for complete mixing is directly dependent on both the diffusion time as measured by flow stream width, and the mixing ratio of the two mixing streams as a function of flow width under constant flow. In our design, the widths of both inlet channels were the same. Therefore, the mixing distance in the microchannel would be largely controlled by changes to the mixing ratio. In theory, the expected microchannel length that is needed for complete mixing can be reduced by either increasing the flow rate ratio, while keeping the total flow constant or decreasing the flow rate, while keeping the mixing ratio constant.
Our results for the integrated SHMs and serpentine channel demonstrate that under various flow rates (100 to 500 μL/min); all mixing profiles were similar (Fig.2b and 2d). Complete mixing occurred between 40 mm and 48 mm regardless of the flow rate. At a constant flow rate of 250 μL/min, the increasing mixing ratio of FITC or FITC-albumin to water enhanced stage 2 mixing in the microchannel. But under the three different ratios, complete mixings were almost identical and were achieved by approximately 48 mm (Fig.2c and 2e). The above results strongly support the design feature of eight SHMs embossed within the serpentine channel predominated in facilitating the mixing process over passive diffusion mixing across the microchannel length. Of note, the inlet fluorescence intensities were slighter lower than 0.5 (no mixing). The deviations were caused from selection of the whole channel width of flow area, instead of subtracting ¼ of channel width laterally in contrast to Stroock et al. However, the mixing pattern and efficacy are similar and are effectively equivalent when compared to each other.
3.3 Flow dynamics in the perifusion chamber
Both FITC and FITC-albumin mixing experiments were conducted to confirm fluid dynamics in the perifusion chamber. As shown in Fig.3a at a flow rate 50 μL/min, the three FITC intensity profiles taken from the imaging zone, perifusion zone, and near-outlet were found to be almost identical, reaching maximum intensities between 50-60 s and having matching washing-out profiles. Some time differences were observed in that FITC reached maximum intensity at the imaging zone with a small delay of 10-20 s and a bigger delay in washing-out period (20-30 s). However, the FITC-albumin experiments showed identical fluourescence profiles among the three areas (Fig.3b). The deviation between FTIC alone and FITC-albumin is presumably caused by different diffusivity constants of the two compounds. Under the current experimental protocol, the potential impact of such a deviation on insulin kinetics would be minimal since the majority of the loaded islets are located in the perifusion zone. In similar, the impact on fluorescence imaging would be also small since fluorescence intensities are typically recorded at a frequency of 5-10 s. We further compared the flow dynamics of the imaging zones between two perifusion chambers, showing similar fluorescence profiles (Fig.3c).
Fig.3. The evaluation of perifusion flow dynamics using FITC and albumin-FITC.

(a) A plot of FITC fluorescence was recorded at three locations in the perifusion chamber: the imaging zone, perifusion zone, and outlet. (b) A plot of albumin-FITC fluorescence was recorded at three locations in the perifusion chamber: the imaging zone, perifusion zone, and outlet. (c) A plot of albumin-FITC fluorescence was recorded in imaging zones of both perifusion chambers. Each imaging cycle was repeated three times over 15-17 min period.
3.4 The creation of temporal glucose gradients in the microfluidic network
To examine the glucose-stimulation profiles obtainable in this device, we used two solutions of glucose and a pumping regiment followed by the measurement of the final glucose concentration using a glucometer. As shown in Fig.4a, a linear glucose profile was established from low (2 mM) to high (25 mM) concentration versus a linear glucose profile from high (25 mM) to low (2 mM) concentration in chambers A and B, each with R2 of 0.9963 and 0.9947, respectively. Fig.4b shows a ʌ-shaped glucose profile (2-25-2 mM) vs. V-shaped glucose profile (25-2-25 mM) with R2 values from 0.98 and 0.99, respectively. A linear glucose temporal gradient from 2 to 25 mM glucose was created at a rate Δ [Glu] of 0.92 mM/min in chamber A with an R2 value of 0.9886. Meanwhile in chamber B, a three-stair glucose ramp (2, 12.5, and 25 mM) was created with a maintenance period of 8 min per stair (Fig.4c).
Fig. 4. The creation of glucose temporal gradients in the dual perifusion networks.
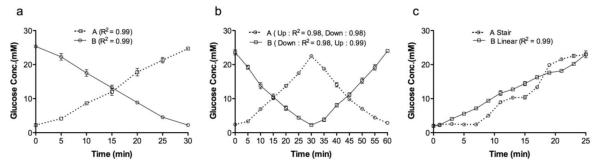
(a) A linear gradient from low (2 mM) to high (25 mM) vs. a linear gradient from high (25 mM) to low (2 mM) concentration. (b) A ʌ-shaped gradient (2-25-2 mM) vs. a v-shaped glucose profile (25-2-25 mM). (c) A linear gradient from 2 to 25 mM glucose vs. a three-stair glucose ramp (2, 12.5, and 25 mM). For each glucose profile, n=3; data was reported as Mean ± SD. The various glucose gradients were created by mixing two solutions of glucose controlled by an automated pumping regiment and glucose concentrations were then determined using a glucometer.
In this study, we successfully demonstrated that through the simple integration of microscale SHMs with off-chip syringe pumps, the creation and maintenance of user-defined temporal concentration gradients is possible. In addition, the hexagonal-shaped perifusion chamber allows for temporal gradient synchronization across the imaging zone, perifusion zone, and outlet zone. Moreover, this method demonstrates high consistency and flexibility without major fabrication hurdles or operational difficulties.
3.5 Concurrent islet perifusion and optical imaging
In this study, we applied Fura-2AM and Rh123 fluorescence probes to simultaneously measure change intracellular calcium levels and mitochondrial potentials in response to various insulin secretagogues during islet perifusion. In Fig.5a and 5c, the representative traces of calcium influx and mitochondrial electrical potentials in response to either 14 mM glucose or 250 μM tolbutamide were recorded simultaneously from chambers A and B. In response to 14 mM glucose stimulation, the islets initially exhibit the canonical hyperpolarization of mitochondrial membrane potentials and subsequent biphasic calcium signaling pattern with a primary initial maximal influxive pulse and a secondary sustained lower pulse period followed by slow calcium oscillatory patterns. In contrast, the islets stimulated with tolbutamide showed a monophasic calcium signaling pattern with no detectable calcium oscillations and no significant decrease in mitochondrial membrane potentials preceding recorded changes in [Ca2+]c. Unlike glucose-induced calcium influx, tolbutamide directly closes KATP channels without the involvement of ATP production, causing membrane depolarization and VDCC activation. This is evident with the more rapidly observed tolbutamide-stimulated calcium influx juxtaposed against the relatively constant mitochondrial potentials as compared to normal glucose challenge observed here.
Fig.5. Simultaneous islet perifusion and multi-channel fluorescence imaging in response to glucose and tolbutamide.

(a) A representive fluorescence record of [Ca2+]c and ΔΨM of a mouse islet in response to 14 mM glucose (n=15-20 islets) (b) A representive trace of the insulin kinetics from 20 mouse islets in response to 14 mM glucose (n=3). (c) A representive fluorescence record of [Ca2+] c and ΔΨM of a mouse islet in response to 250 μM tolbutamide (n=15-20 islets). (d) A representive trace of the insulin kinetics from 20 mouse islets in response to 250 μM tolbutamide (n=3). The signals were normalized against basal intensity established before the stimulation and expressed in percentage changes. Flow rate of delivered insulin secretagogues was set at 250 μL/min. The hashmarks indicated time of insulin secretagogues stimulation.
A high signal-to-noise ratio is required for valid real-time fluorescent detection of active β-cell metabolic and ion signaling events. Our results demonstrate that both fluorescence signals show a good signal-to-noise ratio with change greater than 20%. The insulin secretion profiles from 20 islets in response to glucose and tolbutamide demonstrated that the system had sufficient sensitivity and temporal resolution (Fig. 5b and 5d).
Islet immobilization and shear force on insulin secretion kinetics have to be considered when designing a microfluidic device. In this device, we designed pockets or wells for immobilizing islets in manner consistent to our previous device, but with different dimensions. The immobilization capability of the pocket has been tested at different speeds from 50 to 1,000 μl/mL and no obvious islet ejection has been observed (data not shown). A stabilized basal insulin level within a 5-minute wash period and typical biphasic insulin kinetics induced by high glucose suggest that the effect of shear force on β-cells was minimal in this apparatus. Intracellular calcium signaling and cell energetic status as observed in mitochondrial potentials data further support this observation.
CONCLUSION
This report outlines the development of a dual microfluidic perifusion network for concurrent islet perifusion and optical imaging. This novel microfluidic platform represents the first of its kind, consisting of two separated perifusion chambers on a single chip. In addition to enabling simultaneous islet perifusion and optical imaging, the integration of an on-chip SHM mixer on a serpentine inlet channel allows for more efficient mixing and easier flow control. Practically, this microfluidic device can be used in the future where real-time concurrent monitoring of either two different types of islets (e.g., rodent islets vs. human islets) under the same microenvironment or the same types of islets under two different microenvironments (e.g., control, vs. insulin secretagogues). The dual perfusion device design constitutes an improvement over our previously reported microfluidic chip. This device design allows users to have superior control over the desired islet microenvironment and culture conditions without compromising the spatiotemporal resolution of the measured parameters. While this device was originally designed for whole islet study, it is feasible to extend it to dissociated islet cells or other type cells through minor modification of the microwell “pocket” dimensions.
ACKNOWLEDGEMENTS
This work was supported by a start up grant by the University of Illinois at Chicago College of Medicine (J.O), the American Association of University Women International Fellowship (A. A), and the Chicago Diabetes Project (CDP).
REFERENCE CITED
- Adewola AF, Lee D, et al. Biomed Microdevices. 2010;12 doi: 10.1007/s10544-010-9398-1. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Harrison DE, et al. Nature. 1984;312 doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- Atencia J, Morrow J, et al. Lab Chip. 2009;9 doi: 10.1039/b902113b. [DOI] [PubMed] [Google Scholar]
- Bergsten P, Grapengiesser E, et al. J Biol Chem. 1994;269 [PubMed] [Google Scholar]
- Carlsson PO, Andersson A, et al. Am J Physiol. 1996;271 doi: 10.1152/ajpendo.1996.271.6.E990. [DOI] [PubMed] [Google Scholar]
- Chen D, Du W, et al. Proc Natl Acad Sci U S A. 2008;105 doi: 10.1073/pnas.0807916105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dishinger JF, Kennedy RT. Anal Chem. 2007;79 doi: 10.1021/ac061425s. [DOI] [PubMed] [Google Scholar]
- Dishinger JF, Reid KR, et al. Anal Chem. 2009;81 doi: 10.1021/ac900109t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Shim J, et al. Lab Chip. 2009;9 doi: 10.1039/b815990d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley CJ, Benninger RK, et al. Lab Chip. 2009;9 doi: 10.1039/b816575k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilon P, Ravier MA, et al. Diabetes. 2002;51(Suppl 1) doi: 10.2337/diabetes.51.2007.s144. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Diabetes. 2000;49 doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- Hoshi M, Shreeve WW. Diabetes. 1973;22 doi: 10.2337/diab.22.1.16. [DOI] [PubMed] [Google Scholar]
- Keizer J, Magnus G. Biophys J. 1989;56 doi: 10.1016/S0006-3495(89)82669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy PE, Walker MM, et al. Diabetes. 1972;21 [Google Scholar]
- Lee YC, Nielsen JH. Mol Cell Endocrinol. 2009;297 doi: 10.1016/j.mce.2008.08.033. [DOI] [PubMed] [Google Scholar]
- Lefebvre PJ, Paolisso G, et al. Diabetologia. 1987;30 doi: 10.1007/BF00279610. [DOI] [PubMed] [Google Scholar]
- Lifson N, Lassa CV, et al. Am J Physiol. 1985;249 doi: 10.1152/ajpendo.1985.249.1.E43. [DOI] [PubMed] [Google Scholar]
- Lynn NS, Dandy DS. Lab Chip. 2007;7 doi: 10.1039/b700811b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed JS, Wang Y, et al. Lab Chip. 2009;9 doi: 10.1039/b809590f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polonsky KS, Given BD, et al. J Clin Invest. 1988;81 doi: 10.1172/JCI113339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravier MA, Gilon P, et al. Diabetes. 1999;48 doi: 10.2337/diabetes.48.12.2374. [DOI] [PubMed] [Google Scholar]
- Rocheleau JV, Walker GM, et al. Proc Natl Acad Sci U S A. 2004;101 doi: 10.1073/pnas.0405149101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper MG, Shackman JG, et al. Anal Chem. 2003;75 doi: 10.1021/ac0346813. [DOI] [PubMed] [Google Scholar]
- Shackman JG, Dahlgren GM, et al. Lab Chip. 2005;5 doi: 10.1039/b404974h. [DOI] [PubMed] [Google Scholar]
- Stroock AD, Dertinger SK, et al. Science. 2002;295 doi: 10.1126/science.1066238. [DOI] [PubMed] [Google Scholar]
- Sturis J, Pugh WL, et al. Am J Physiol. 1994;267 doi: 10.1152/ajpendo.1994.267.2.E250. [DOI] [PubMed] [Google Scholar]
- Zhang X, Grimley A, et al. Anal Chem. 2010;82 doi: 10.1021/ac101461x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Roper MG. Anal Chem. 2009;81 doi: 10.1021/ac802579z. [DOI] [PMC free article] [PubMed] [Google Scholar]