

Abstract
Cardiac excitation–contraction coupling occurs primarily at the sites of transverse (T)-tubule/sarcoplasmic reticulum junctions. The orderly T-tubule network guarantees the instantaneous excitation and synchronous activation of nearly all Ca2+ release sites throughout the large ventricular myocyte. Because of the critical roles played by T-tubules and the array of channels and transporters localized to the T-tubule membrane network, T-tubule architecture has recently become an area of considerable research interest in the cardiovascular field. This review will focus on the current knowledge regarding normal T-tubule structure and function in the heart, T-tubule remodelling in the transition from compensated hypertrophy to heart failure, and the impact of T-tubule remodelling on myocyte Ca2+ handling function. In the last section, we discuss the molecular mechanisms underlying T-tubule remodelling in heart disease.
Keywords: T-tubules, Excitation-contraction coupling, Calcium, Heart failure, Junctophilin-2
1. Introduction
Cardiac excitation–contraction (E–C) coupling is the central mechanism governing the heart muscle to generate sufficient contractile force and pump adequate blood to the whole body.1 At the cellular level in ventricular myocytes, normal E–C coupling involves precise communication, i.e. local control of Ca2+-induced Ca2+ release (CICR), between voltage-gated L-type Ca2+ channels (LTCCs) located mainly on the T-tubule membrane and Ca2+ release channels/ryanodine receptor channels (RyRs) on the sarcoplasmic reticulum (SR).2–5 The highly organized T-tubule network forms tight physical couplings with the terminal cisternae of SR, termed dyads, at Z-line regions throughout the entire myocyte.6 These organized fine dyadic Ca2+ release apparatus are essential for local control of CICR, synchrony of SR Ca2+ release, and Ca2+ release stability during each heartbeat. The synchronized Ca2+ release during each membrane excitation allows co-ordinated contraction among the many contractile units within each large working ventricular myocyte. Ultimately, synchronized myofilament contraction within and among millions of working ventricular myocytes will permit the heart muscle to contract and generate the maximal contractile force with the least energy cost. Thus, the organized T-tubule structure is critical for normal E–C coupling and cardiac function.
2. T-tubule structure
The first evidence that clearly showed the existence of transversely oriented continuous tubules in mammalian heart muscle was from light microscopy studies by Nyström in 1897 (see Huxley, The Croonian Lecture, 19677). In that pioneering work, India ink was injected into the heart muscle to track the extracellular space (analogous to Sulphorhodamine B that is used with fluorescence microscopy). Nyström detected dark lines transversely crossing the cardiac fibres at an interval equal to that of the striations. More than 60 years passed until, using electron microscopy (EM), Lindner (1957) identified T-tubules in canine ventricular myocardial cells.8 Further studies clearly demonstrated the presence of the T-tubule system in myocardial cells of the rat, guinea pig, rabbit, cat, sheep, and human.6,9–12 Concomitantly, the existence of T-tubules was documented in the skeletal muscle. Among the ultrastructural studies that were conducted in skeletal muscle in the 1960s, Franzini-Armstrong and Porter13 provided the most convincing evidence, using improved fixation methods and fish muscle fibres, clearly defining that the T-tubule system is an entity of the fine membrane continuous with the sarcolemma, distinct from the SR but uniquely associated with the SR. Her landmark findings indicated that the T-tubule system might play a prominent role in the fast intracellular conduction of the excitatory impulse, providing clear ultrastructural evidence that supported observations made by the Nobel Laureate Huxley and his colleague Taylor in 1955.14 In Huxley and Taylor's seminal work on an isolated intact fibre of frog striated muscle, they used a micropipette electrode to achieve local sub-threshold depolarization of the cell membrane and observed fast, transverse, two-dimensional conduction of excitation towards the central axis, which they concluded to be too fast to be mediated by simple chemical diffusion.14 This work led to their hypothesis that a special membrane structure continuous with surface sarcolemma must be involved in relaying the signal to the SR, which we now define as E–C coupling. Since these early endeavours, we have garnered an understanding of the structure and function of T-tubules in the skeletal and cardiac muscle.
It is now well known that T-tubules are physical extensions and orderly invaginations of the surface membrane that are continuous with the extracellular space and extend deep into the interior of mammalian ventricular myocytes.15–19 They are perpendicular to the surface or external sarcolemma, are transverse to the longitudinal axis of myocytes, and appear to have a radial ‘spoke-like’ organization in transverse sections.20,21 They are regularly arrayed along the Z-line regions, forming a highly organized, elaborate tubular network. The majority of T-tubules are within 0.5 μm from the Z-lines.19,20,22 Besides the primary transversal components of T-tubule membrane, longitudinal elements running across from one Z-line to another are also present, with some reports that longitudinal elements occur at a much lower proportion in normal healthy ventricular cells,15,16,23,24 and others that longitudinal elements are frequent in healthy myocytes.22,25 Quantitative measurements based on confocal microscopy approximate T-tubule diameters to be ∼250 nm in rat myocytes22 and ∼400 nm in rabbit and human.26,27 A recent study using stimulated emission depletion (STED) imaging technique indicated that the T-tubule diameter is ∼200 nm in murine myocytes.25 (Note: the smaller T-tubule diameter detected by STED imaging could be due to a better spatial resolution offered by this technique than confocal microscopy). The lumens of T-tubules have notable variations in size (even within the same species).22,26 Although the volume density of the T-tubule system is only 1–3%,22,28 it represents about one-third of the entire cell-membrane area1,29,30 (Figure 1).
Figure 1.

Ultrastructure and organization of T-tubules in cardiac muscle (myocytes). (A) Electron micrograph of a transverse section of cat cardiac muscle showing four T-tubules extending inward from the periphery of the fibre (×32 000). (B) Electron micrograph of a longitudinal section of cat papillary muscle showing two T-tubules and multiple junctional couplings between T-tubules and terminal cisternae of SR, as indicated by the arrows (×40 000; reproduced with permission from Fawcett and McNutt6). (C) A confocal fluorescence image of the T-tubules in an isolated living rat ventricular myocyte stained with lipophilic membrane marker Di-8 ANEPPS. (D) 3D projection of the T-tubule network from 30 sequential sections (at 0.2 μm per section) of confocal images from the same myocyte. (E) Schematic drawing of the T-tubule system in a ventricular myocyte, viewed from the transverse section.
It is believed that T-tubule density varies among ventricular myocytes from different species. Early EM work and recent confocal imaging studies suggested that rodents (mouse, rat), which have faster heart rates, have a higher T-tubule density in ventricular myocytes relative to larger mammalian myocytes (i.e. rabbit, pig, and human), but the difference is not related to cell or heart size.31,32 In contrast, mammalian atrial myocytes have a heterogeneous distribution of the tubular system, with the longitudinal components more prominent,33–36 (see review by Katherine Dibb and colleagues in this issue). In Purkinje fibres of the conduction system, early EM studies found a lack of T-tubules.37–40 However, work by Franzini-Armstrong and colleagues as well as others demonstrated that T-tubules are occasionally present in all Purkinje fibres and form dyadic associations with the SR, with the frequency of T-tubules varying depending on the size of the animal.41,42 As an additional note, the cardiac myocytes of reptiles, birds, and fish are devoid of a T-tubule system in both atria and ventricles.16 The following discussion will focus on mammalian ventricular myocytes.
3. T-tubule function: synchronous Ca2+ release
Critical to E–C coupling function is the spatial relationship between T-tubules and the SR where Ca2+ release channels or RyRs are located (Figure 2). In normal ventricular myocytes, about 80% of LTCCs localize to the T-tubules.43 T-tubules and the junctional components of SR (jSRs) juxtapose and form the dyadic junctions. Both ultrastructural and functional studies have suggested that dyadic junctions are much more abundant in the T-tubules than at the surface membrane, e.g. in rat ∼75% of dyads are in T-tubule region compared with ∼25% at the surface sarcolemma,31,44 indicating the importance of the T-tubule membrane in controlling CICR and E–C coupling function. The much lower density of dyadic junctions in the external or surface sarcolemma may be associated with other Ca2+-dependent processes.45
Figure 2.

Cartoon of local Ca2+ micro-domain and major proteins concentrated in the dyadic junction. The local Ca2+ micro-domain includes primarily (L-type) Ca2+ channels and opposing RyRs within a 12–15 nm distance between T-tubule and SR membrane, forming functional Ca2+ release units (CRU). Other important components such as Na+/Ca2+ exchanger, Na+/K+-ATPase, and β-adrenergic receptor (β-AR) are also condensed on the T-tubules.
In ventricular myocytes, CICR is an elaborate ‘local control’ process between LTCCs on the T-tubules and RyRs on the SR. Under normal conditions, CICR occurs synchronously throughout the myocytes, producing whole-cell Ca2+ transients. Due to the spatial and temporal summation of thousands of synchronously firing local release events (i.e. Ca2+ sparks)5,46 and rapid diffusion of Ca2+ ions, the whole-cell Ca2+ transients usually manifest as uniform, evenly distributed Ca2+ signals.46 This synchronization of Ca2+ release from individual Ca2+ release sites during E–C coupling was not observed until a new fluorescent technique was developed in combination with high spatiotemporal resolution confocal microscopy.47,48 With an admixture of a fast, low-affinity Ca2+ indicator (Oregon Green 488 BAPTA 5N) and a high affinity, but slow Ca2+ chelator (EGTA) in the pipette recording solution, localized discrete SR Ca2+ release events, dubbed ‘Ca2+ spikes’, were visualized at individual T-tubule/SR junctions. At a full-blown depolarization (e.g. depolarization to 0 mV from resting membrane potential), Ca2+ spikes occur almost at the same time from all different T-tubule/SR junctions, revealing the highly synchronous nature of Ca2+ release events during E–C coupling (Figure 3A and B).47 In combination with loose seal patch clamp and confocal Ca2+ imaging techniques, it has been further demonstrated that Ca2+ sparks can be activated by Ca2+ influx from a single L-type Ca2+ channel, namely ‘Ca2+ sparklets’ (Figure 3C and D).3 This work provided the first direct visual evidence validating the ‘local control theory’ of cardiac E–C coupling, that is, local Ca2+ entry via LTCCs across the T-tubule membrane triggers local Ca2+ release from adjacent RyRs. The highly organized T-tubule network and intricate coupling between T-tubules and jSRs provide the ultrastructural basis for local control of CICR. (For a complete view of various terms on local Ca2+ events, please refer to Table 1 of Cheng and Lederer5.)
Figure 3.

Synchrony of local Ca2+ release during E–C coupling. (A) Ca2+ spikes recorded in ventricular myocyte under whole-cell voltage-clamp condition, and 4 mM EGTA and 1 mM Oregon Green 488 BAPTA 5N in patch pipette solution. The confocal scan line was placed along the longitudinal axis of myocyte. Discrete Ca2+ spikes were evoked synchronously from all T-tubule/SR junctions upon membrane depolarization to 0 mV from a holding potential of −70 mV. (B) Surface plot of Ca2+ spikes shown in (A). (C and D) Sparklet–spark coupling—a direct visualization of local control of CICR. (C) LTCC Ca2+ influx mediated sparklets (the low-amplitude events, pink arrows) and triggered sparks (the high-amplitude events) recorded under loose-seal patch-clamp conditions. Note that not every sparklet can trigger a spark. (D) Surface plot of sparklet–spark coupling. The arrow indicates a sparklet foot that triggered a spark (from Wang et al.3).
Table 1.
T-tubule remodelling examined in human heart failure or experimental animal models (in chronological order)
| Species/model of disease | Samples studied | Methods | Findings on T-tubule remodelling | Reference |
|---|---|---|---|---|
| Rat, pressure overload hypertrophy (aortic constriction) | Frozen LV tissues | EM | Increased T-tubule membrane area | Page and McCallister,49 1973 |
| Human, HCM of varied causes (aortic stenosis, etc.) | Fixed LV or ventricular septum biopsy samples | EM | Irregularly shaped or dilated T-tubules in hypertrophied cells; loss of T-tubules in degenerating cells | Maron et al.,50, 1975 |
| Human, end-stage DCM | Fixed LV tissues (frozen sections) | EM | Numerous, dilated T-tubules in hypertrophied, or T-tubule loss in degenerative cells | Schaper et al.,51 1991 |
| Human, end-stage DCM | Fixed LV tissues (frozen sections) | EM/Confocal immunofluoresence | T-tubule dilation | Kostin et al.,20 1998 |
| Human, end-stage DCM/ICM | Frozen LV tissues | EM/confocal immunofluorescence | Increase in size and number of T-tubules More longitudinal orientated elements |
Kaprielian et al.52, 2000 |
| Dog, rapid pacing-induced HF (ventricular pacing, 4–5 weeks) | Isolated LV myocytes | Confocal | Regional loss, with normally organized pattern | He et al.,53, 2001 |
| Dog, rapid pacing-induced HF (ventricular pacing, 4–6 weeks) | Isolated LV myocytes | Confocal | Regional loss, with normally organized pattern | Balijepalli et al.,54 2003 |
| Rat, spontaneously hypertension (∼20 months old) | Isolated LV myocytes | Confocal | T-tubule disorganization, loss in transverse elements and increase in longitudinal elements Overall chaotic appearance |
Song et al.,23 2006 |
| Mouse, MI (ligation of LCA, 1 week or 3 weeks) | Isolated myocytes (septum, remote from MI zone). | Confocal | Slightly disorganized 1 week post-MI Profound disorganization 3 weeks post-MI |
Louch et al.,55 2006 |
| Rat, MI (ligation of LCA, 6 weeks) | Isolated myocytes Fixed tissues (from LV non-infarcted area) | Confocal/EM | Disorganized pattern, decreased T-tubule density, fewer T-tubule regions associated with SR | Swift et al.,56 2008 |
| Pig, Regional MI (severe stenosis Of circumflex LCA, 6 weeks) | Isolated myocytes from MI adjacent (border) region | Confocal | Reduction in T-tubule density | Heinzel et al.,32 2008 |
| Human, end-stage DCM, ICM, and HCM; Rat MI (16 weeks) | Isolated myocytes from human or rat HF hearts | Confocal and ion conductance microscope | Loss of T-tubule openings; decrease in T-tubule density | Lyon et al.,57 2009 |
| Sheep, rapid pacing-induced HF (RV-apex pacing, 4 weeks) | Isolated left-atrial myocytes | Confocal | Extensive disruption and loss of atrial T-tubules | Dibb et al.,58 2009 |
| Sheep, persistent AF (129±39 days, induced by intra-atrial pacing) | Isolated right atrial myocytes | Confocal | Reduction in atrial T-tubule density and loss of T-tubule organization | Lenaerts et al.,35 2009 |
| Human, end-stage DCM, ICM, and HCM | Isolated LV myocytes | Two-photon | Only small, but not significant changes in T-tubule network | Ohler et al.,68 2009 |
| Mouse, diabetic cardiomyopathy (db/db, 20 weeks old) | Isolated LV myocytes | Confocal | Decrease in T-tubule density | StØlen et al.,67 2009 |
| Rat, mechanical unloading (4 weeks) | Isolated LV myocytes | Confocal, ion conductance microscope | T-tubule disorganization, disruption of T-tubule openings | Ibrahim et al.,59 2010 |
| Rat, pressure overload (aortic constriction, ∼8–12 weeks) | Langendorff-perfused intact hearts | Confocal | Discrete T-tubule loss in hypertrophied hearts, progressive loss and disorganization from hypertrophy to HF, penetration from LV to RV | Wei et al.,60 2010 |
| Rat with metabolic syndrome: exercise induces concentric hypertrophy (8 weeks); or normal rats post-MI/exercise | Isolated LV myocytes | Confocal | Loss of T-tubules in pathological remodelling; intact T-tubule structure in physiological hypertrophy | Kemi et al.,61 2011 |
| Mouse, HF (inducible Junctophilin-2 knockdown) | Isolated LV myocytes or fixed LV tissues (for EM) | Confocal/EM | Severe T-tubule disorganization; disrupted T- tubule/SR junctions | van Oort et al.,62 2011 |
| Human, DCM | Fixed, frozen LV tissues | Confocal | Reduction in orderly pattern, less uniform with more transverse components; dilation | Crossman et al.,21 2011 |
| Mouse, HF (PI3K p110 α/β single or double knockout) | Langendorff-perfused intact hearts or isolated myocytes | Confocal | Severe T-tubule loss and disorganization in both LV and RV myocytes | Wu et al.,63 2011 |
| Rat, RV failure (induced by monocrotaline, 25 days) | Langendorff-perfused intact hearts | Confocal | Severe T-tubule loss and disorganization of RV myocytes | Xie et al.,114 2012 |
| Mouse, MI (5 weeks) | Langendorff-perfused intact hearts | Confocal | Remarkable remodelling in MI border zone; moderate remodelling in MI remote zone (LV) | Chen et al.,115 2012 |
| Dog, dyssynchronous HF (6 weeks after right atrial pacing with ablation of left bundle branch) | Fixed, isolated LV myocytes (anterior and lateral) | Confocal immunofluorescence | Dramatic remodelling in lateral myocytes (depletion in the centre, more longitudinal components in the cell periphery) | Sachse et al.,64 2012 |
| Rat, MI (12–16 weeks) | Isolated LV myocytes, fixed LV tissues | EM, confocal, and ion conductance microscope | T-tubule density reduction; T-tubule regularity Disruption; dilation |
Ibrahim et al.,65 2012; Lyon et al.,662012 |
| Mouse, MI (4 or 8 weeks) | Isolated myocytes | STED and confocal | Increased area of T-tubule cross-sections; increase in longitudinal and decrease in transversal elements | Wagner et al.,25 2012 |
| Mouse, HF (Serca2 knockout, 7 weeks after tamoxifen induction) | Isolated LV/septum myocytes, fixed hearts | EM, confocal | Increased T-tubule density, increased abundance of longitudinal T-tubules (newly grown T-tubules) | Swift et al.,24 2012 |
| Mouse, HF (chronic Gaq over-expression) | Langendorff-perfused intact hearts | Confocal | Heterogeneous spatial disorganization: loss of transverse elements and gain in longitudinal elements, overall mesh-like appearance | Tao et al.,103 2012 |
| Rat, pressure overload HF (Aortic constriction, 9–11 weeks) | Fixed, isolated myocytes | EM | Reduction in both volume density and surface area of total T-tubules, and T-tubules coupled with junctional SR | Wu et al.,118 2012 |
| Mouse, teletholin (Tcap) knockout (3–8 months) | Isolated myocytes | Confocal and ion conductance microscope | Progressive T-tubule loss and irregularity with ageing; profound T-tubule loss and disorganization following pressure overload | Ibrahim et al.,125 2013 |
AF, atrial fibrillation; EM, electron microscopy; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; HF, heart failure; ICD, ischaemic cardiomyopathy; LCA, left coronary artery; LV, left ventricle; MI, myocardial infarction; RV, right ventricle.
4. T-tubule remodelling in heart failure
T-tubule alterations were first observed in diseased hearts by EM. Earlier work by Page and McCallister49 showed that, in a rat pressure overload hypertrophy model (10 days after aortic constriction), the area of T-tubule membrane is increased in hypertrophied myocytes and accordingly the ratio of total sarcolemmal area to cell volume remains constant. These findings were extended by subsequent EM studies from Maron et al.,50 which included analysis of biopsies from human hypertrophied cardiomyopathies of various causes, including aortic stenosis, aortic regurgitation, obstructive LV outflow, etc. They identified irregularly shaped or dilated T-tubules in hypertrophied cells and loss of T-tubules in degenerating cells.50 Similar T-tubule alternations (dilation and loss of T-tubules) were also revealed in patient hearts with end-stage dilated or ischaemic cardiomyopathy.20,51,52 Although these early EM studies provided high-resolution visualization of myocyte ultrastructure, application of EM is limited both by the sophisticated technique, including sample processing, and by the inability to view in a single image the complete T-tubule system of a myocyte.
The appreciation of T-tubule remodelling in heart disease was boosted during the last decade by the application of laser scanning confocal microscopy and the use of fluorescent lipophilic membrane markers. Among these, He et al.53 were the first to identify and quantify the significant loss of T-tubule density in failing ventricular myocytes in an experimental pacing-induced canine heart failure model, though the overall organized pattern of the T-tubule network is not altered in this model. Similar findings using the same model were reported from this group.54 Interestingly, in a spontaneously hypertensive rat model, distinct changes in the T-tubule system in failing myocytes were found, specifically, a dramatic re-organization of the T-tubule system characterized by a loss of transverse elements but a gain in longitudinal elements, giving rise to an overall chaotic appearance of the T-tubule network (Figure 4).23 These visual observations from confocal images were confirmed by computational analysis including 2D Fourier analysis of the power of the T-tubule structure, which represents the global regularity of the T-tubule network, and calculation of the densities of transversal and longitudinal elements of the T-tubules.23 Subsequently, many other groups using other animal heart failure models have also reported profound T-tubule remodelling in single isolated failing myocytes (Table 1).21,24,25,32,55–67 Similar alterations were also observed in atrial myocytes following rapid pacing.35,58 Taken together, these studies provide clear evidence that T-tubule remodelling is a common pathological alteration in failing myocytes of almost all origins examined to date, including different animal heart failure models of different species/aetiologies, and human heart failure patients with different background diseases. The major common characteristics of T-tubule alterations from these studies include the following: (i) loss of T-tubules (or reduction in T-tubule density); (ii) disorganization or disruption of the orderly arrayed T-tubule network; (iii) a decrease in transversal elements and an increase in longitudinal elements; and (iv) an increase in T-tubule diameter (or T-tubule dilation). These studies led to the current paradigm that T-tubule remodelling is a principal problem in many forms of cardiac disease that share the common end-stage of heart failure. It should be noted that one study found minor and insignificant changes in T-tubule structure in single myocytes isolated from end-stage human failing hearts compared with control samples from rejected healthy donor hearts.68
Figure 4.

Ca2+ handling defects and T-tubule remodelling in failing myocytes. (A and B) Field-stimulated Ca2+ transients (1 Hz) in isolated myocytes from a control Wistar-Kyoto (WKY) rat and failing spontaneously hypertension rat (SHR/HF), respectively. Control healthy myocyte exhibits uniform, synchronous and stable Ca2+ transients from beat to beat. The failing myocyte displays dyssynchronous Ca2+ release at different regions of the cell. As shown by these arrows, Ca2+ releases are delayed at fixed locations on every beat. (C–F) T-tubule disorganization in a ventricular myocyte from a failing SHR heart (SHR/HF, D and F), compared with the organized T-tubule network from a WKY control myocyte (C and E). (From Song et al.23)
5. T-tubule remodelling in the transition from hypertrophy to heart failure
Although a majority of studies agree that there is remarkable remodelling within the T-tubule system in myocytes isolated from failing hearts, it is arguable that the disruptive T-tubule remodelling might be a consequence of complex molecular and biochemical changes during heart failure. It is therefore imperative to investigate the evolution of T-tubule remodelling during the development of heart failure, in other words, how early T-tubule remodelling initiates in the process of progression from hypertrophy to heart failure. Previous studies from other groups have been performed in isolated single myocytes, though enzymatic dissociation of myocytes may impair the T-tubule membrane of healthy cells.69 Myocytes at the hypertrophied stage may undergo subtle changes in the T-tubule network, which could be indistinguishable from controls in isolated myocytes. Therefore, it was necessary to develop a new imaging technique to detect T-tubule structure with higher sensitivity and less damage. With this in mind, we developed an in situ T-tubule confocal imaging technique by combining the Langendorff perfusion system with laser scanning confocal microscopy (Figures 5 and 6). In doing so, a fluorescent dye-loaded intact heart can be imaged in situ for T-tubule ultrastructure or other structures that can be visualized using fluorescent dyes in living cells, with minimal damage to the myocytes. In a pressure overload hypertrophy – heart failure rat model, this novel advanced in situ confocal imaging provided convincing evidence that T-tubule remodelling is a real phenomenon in failing hearts,60 rather than the previously postulated experimental artefact related to the isolation of failing cardiomyocytes.69 Moreover, this in situ imaging approach revealed that T-tubule remodelling is not just a manifestation of end-stage heart failure.60 Instead, the remodelling of T-tubules starts much earlier in the disease spectrum, even prior to echocardiographically detectable LV systolic dysfunction. The T-tubule system in the LV undergoes progressive deterioration from compensated hypertrophy through early heart failure to advanced heart failure.60 With progression from compensated hypertrophy to early and late heart failure, T-tubule remodelling spreads from the LV to the RV. These findings that T-tubule remodelling occurs prior to the onset of heart failure suggest that T-tubule remodelling is not a secondary modification after heart failure, but instead is an important early event during heart-failure progression. Data from this study also showed that T-tubule integrity highly correlates with cardiac ejection fraction of diseased hearts, indicating T-tubule integrity is a crucial determinant of cardiac function.60 Taken together, these studies strongly suggest that maladaptive T-tubule remodelling is a causal event that drives the transition from compensated hypertrophy to heart failure.
Figure 5.

3D reconstruction of epicardial myocyte T-tubule network in situ. Confocal images (25 confocal stacks at 0.2 μm interval) were acquired in situ from Langendorff-perfused intact healthy heart, demonstrating the periodically organized T-tubule structure in normal myocytes. (From Wei et al.60)
Figure 6.
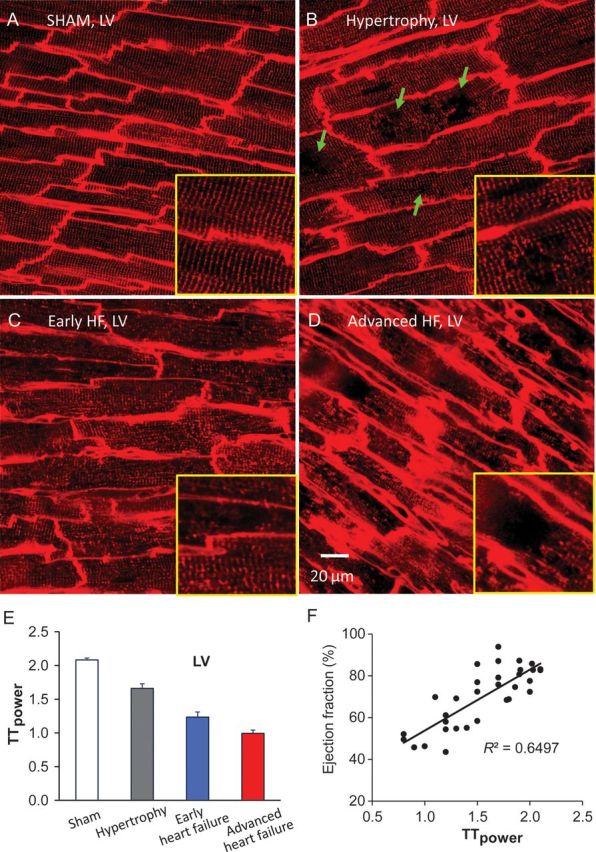
Progressive T-tubule remodelling of left-ventricular myocytes in pressure overload rat cardiomyopathy. (A–D) Representative T-tubule images from left ventricle (LV) of age-matched sham-operated heart (A), hypertrophy (B), early HF (C), and advanced HF (D). At hypertrophy stage, discrete T-tubule loss (green arrows) was often observed with slight T-tubule disorganization (B). In moderately decompensated heart, LV myocytes exhibited widely impaired T-tubule system (C). At advanced HF stage, myocytes lost majority of T-tubules with striated pattern almost vanished (D). Each yellow framed inset is a zoom-in view of an area 40×40 μm2 from associated images. (E) A gradual reduction in TTpower (an index of T-tubule regularity) with heart disease progression. (F) Cardiac global function (ejection fraction) correlates well with T-tubule integrity. LV, left ventricle; EF, ejection fraction. (From Wei et al.60)
6. T-tubule remodelling leads to defective E–C coupling in heart failure
To date, accumulating evidence supports that alterations in T-tubule structure are an important factor in Ca2+ handling dysfunction in cardiomyocytes. Under non-diseased conditions, such as in ventricular myocytes with chemically induced detubulation, electrical stimulation-elicited Ca2+ release is severely hampered. When T-tubules are almost completely depleted, the rise of Ca2+ upon stimulation begins at the cell periphery and is gradually propagated to the centre of myocytes, resulting in a loss of synchronous Ca2+ release.70,71 In another case, ventricular myocytes in culture have been shown to progressively lose T-tubules with time, which subsequently causes spatially non-uniform or dyssynchronous SR Ca2+ release.72,73 These studies provided crucial insights into the influence of T-tubule alterations on myocyte Ca2+ handling function in disease.
Impaired E–C coupling, characterized by a reduction in the amplitude of Ca2+ release and slowed kinetics, including both time to peak and decay rate of Ca2+ transients, is a hallmark of heart failure.74–77 Decreased SR Ca2+ content due to SERCA down-regulation,78–85 reduction in Ca2+ influx through LTCCs,86,87 NCX upregulation,88–94 and metabolic inhibition95–98 are among the mechanisms responsible for altered E–C coupling and Ca2+ homeostasis during heart failure. The contribution of structural alterations to cardiac E–C coupling dysfunction was not appreciated until the seminal work led by Lederer et al.99 In that study, it was first proposed that defective E–C coupling is likely due to a change in the relationship between RyRs on the SR and LTCCs on T-tubules, although no direct evidence was provided.99 Later, dyssynchronous Ca2+ sparks were documented in failing myocytes following myocardial infarction; however, the mechanism causing dyssynchrony of SR Ca2+ release was not completely understood.87 As summarized in Table 1, in the last 10 years, evidence from isolated ventricular myocytes and intact hearts suggests that T-tubule loss and/or disorganization is a significant and common event in advanced heart failure of different aetiologies and results in dyssynchronous Ca2+ release and impaired contraction.21,23,32,35,53–55,57,58,63,64
The re-organization of T-tubule structure alters the spatial organization between LTCCs and RyRs, leading to a reduction in co-localization between RyRs and LTCCs, an increase in orphaned RyRs, the loss of local control of RyRs by LTCCs, and therefore decreased E–C coupling efficacy and increased dyssynchrony of SR Ca2+ release in failing myocytes.23 Specifically, evidence from the study using spontaneously hypertensive rats revealed that, when uncoupled RyRs due to T-tubule disruption are not activated by LTCCs upon membrane depolarization, those uncoupled RyRs can be activated later by Ca2+ diffused from neighbouring functionally normal release sites, causing propagative or secondary CICR. The secondary CICR is much slower compared with the primary Ca2+ release directly triggered by Ca2+ influx across the 12 nm gap, thus producing dyssynchronous Ca2+ release (Figure 4B).23 Similar findings supporting the above interpretation/mechanism have been observed in larger animal models. For example, work from Sipido's group in an ischaemic cardiomyopathy pig model reported that T-tubule loss is associated with reduced synchrony of Ca2+ release and reduced efficiency of the coupling of Ca2+ influx to Ca2+ release.32 T-tubule remodelling and Ca2+ release dysfunction have also been observed in atrial myocytes from a rapid-pacing-induced sheep heart-failure model.58 These experimental findings are supported by computational modelling in which T-tubule re-organization reduces the synchrony of Ca2+ spark production, leading to the appearance of late Ca2+ sparks and a greater non-uniformity of intracellular Ca2+.27 In addition to its influence on the activation process of Ca2+ release, T-tubule remodelling also affects Na+/Ca2+ exchanger (NCX)-mediated Ca2+ removal process, slowing the kinetics of Ca2+ decay during diastole and thus impairing myocardial relaxation. This occurs because NCX is preferentially located to T-tubules (Figure 2) and is the major sarcolemmal Ca2+ efflux mechanism in cardiomyocytes (Figure 2).32,73,100,101
T-tubule integrity is crucial for instantaneous action potential propagation across the whole membrane system of the large myocyte. Recent investigations in rat failing myocytes post-myocardial infarction indicate that structural disorganization of the T-tubule system worsens the electrical coupling between the T-tubule system and the surface sarcolemma, leading to the failure of action potential propagation from the surface membrane to T-tubule system and ensuing triggering of SR Ca2+ release.102 It is then suggested that, in addition to orphaned RyRs, Ca2+ release units that are coupled to the dysfunctional T-tubule domain may also fail to be recruited to release Ca2+. Interestingly, in a mouse hypertrophy model of Gαq overexpression, Rubart and colleagues103 found that T-tubule remodelling is associated with spatially non-uniform action potential prolongation and alterations in spatial dispersion of epicardial repolarization, but found no changes in electrical coupling. Nevertheless, these studies provide new insights into our understanding of the contribution of T-tubule remodelling to defective E–C coupling in heart failure.
The T-tubule system harbours many important ion channels or transporters. In addition to those depicted in Figure 2, brain-type sodium channels and potassium channels (especially the steady-state component, IKss) are also concentrated at T-tubules.104 T-tubule remodelling could change the distribution or organization of ion channels and transporters, alter ion exchange between the restricted T-tubule lumen and the bulk extracellular space, and therefore alter the shape and duration of action potentials, which in turn would disturb the synchrony and efficacy of SR Ca2+ release and Ca2+ removal.104–107
In addition, T-tubule remodelling causes redistribution and loss of β-adrenergic receptors in T-tubules in cardiomyocytes. The impact of this profound change on myocyte E–C coupling is extensively discussed in the review by Gorelik and colleagues in this issue.108 Briefly, myocyte T-tubule remodelling leads to alterations at multiple levels, including (ultra)structural, electrical, and signal transduction, collectively contributing to the progression of cardiac failure and the genesis of fatal arrhythmias (Figure 7).
Figure 7.
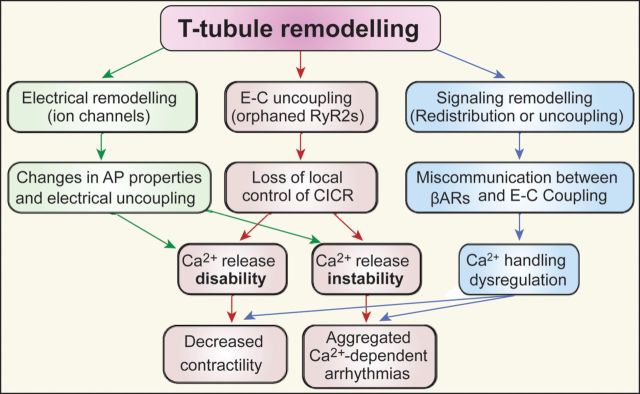
Schematic chart depicting the consequences of T-tubule remodelling and the relationship with heart failure and Ca2+-dependent arrhythmogenesis. Myocyte T-tubule remodelling leads to alterations at multiple levels, including ultrastructural, electrical, and signal transduction changes, collectively contributing to the progression of cardiac failure and the genesis of fatal arrhythmias.
7. Molecular mechanisms of T-tubule remodelling
Junctophilin-2 (JP2) bridges the physical gap between the plasma membrane and the SR in excitable cells and plays an important role in the formation of the junctional membrane complex (i.e. the cardiac dyad–T-tubule/SR couplings).109 JP2 is one of the four members of the junctophilin protein family (JP1–4), and the only junctophilin protein expressed in cardiac muscle.109–111 Conventional knockout of JP2 is embryonically lethal, and studies in embryonic myocytes with JP2 deficiency define a critical requirement for JP2 in normal cardiac function. Specifically, JP2-deficient embryonic myocytes have defective cardiac dyads, including more SR segments with no T-tubule couplings as well as reduced and unstable intracellular Ca2+ transients.109 These pioneering studies identified JP2 as a critical structural factor for normal E–C coupling and cardiac function. Very recent work by Sachse and colleagues112,113 suggests that cellular strain (including passive stretch and active myocyte contraction) could affect the geometry of T-tubules. Thus, maintaining a stable physical relationship between T-tubules and the jSR is critical for normal E–C coupling function during active myocyte contraction–relaxation cycle. From this point of view, we consider JP2 as an anchoring protein that helps maintain both the static and dynamic stability of the nano-positioning between T-tubule and jSR.
Given the clear role of JP2 in normal cardiac function, recent work has evaluated how expression of JP2 changes in response to cardiac stress, and if changes in JP2 mechanistically contribute to pathological T-tubule remodelling. Loss of JP2 expression has been documented in failing human hearts and a variety of heart failure models. For example, in rat pressure overload models,60,77,114 murine myocardial infarction model,115 murine hypertrophic or dilated cardiomyopathy models,116 and human failing hearts,117 expression of JP2 is severely down-regulated in response to cardiac stress. This loss of JP2 expression correlates with loss of T-tubule structural integrity.60 Recent studies in which JP2 was knocked down in either cultured ventricular myocytes or by transgenic expression of a JP2 shRNA in vivo suggest that loss of JP2 expression is a key mechanism underlying T-tubule remodelling in failing myocytes. Since JP2 structurally connects the T-tubules and SR and maintains the physical stability of T-tubule/SR junctions, JP2 down-regulation likely leads to T-tubule dissociation from the SR and ensuing disruption of cardiac dyads. Indeed, the effects of knockdown of JP2 mimic the loss of T-tubule/SR organization observed in response to cardiac stress.60,118 Towards understanding the mechanism by which JP2 is down-regulated in failing hearts, microRNA-24 (miR-24) has been identified as direct regulator of JP2 homeostasis in the heart.119 miRNAs are now recognized as important regulators of both normal and pathophysiological processes. Expression of miR-24 is increased in failing hearts, and overexpression of miR-24 in cultured myocytes results in JP2 down-regulation, alterations in cardiac dyads, and changes in E–C coupling function.119 Taken together, compelling evidence specifically identifies JP2 as a key mediator of stress-induced cardiac T-tubule remodelling.
In addition to JP2, other proteins have been implicated in T-tubule formation or remodelling, such as caveolin-3, amphyphisin-2 (Bin1), telethonin (Tcap), particularly in skeletal muscle.18,120–124 However, the roles of these proteins in T-tubule biogenesis and pathogenesis in cardiomyocytes remain to be determined. Newly published work (2013) from the Terracciano group using Tcap knockout mice began to shed new light into these curiosities, indicating that Tcap is a critical, load-sensitive regulator of T-tubule structure and function.125
8. Conclusions
In summary, data from electron microscopy and confocal imaging of isolated myocytes and, more recently, in situ confocal imaging of myocytes in intact hearts have provided a clear understanding of the structure of T-tubules. We now recognize T-tubules as an important structural component of E–C coupling function that, when perturbed, results in loss of co-ordinated contraction of ventricular myocytes, and T-tubule remodelling is a key player in the pathogenesis of heart failure. While the composition of the T-tubule network is fairly well-understood, we are beginning to discover the mechanistic underpinnings of T-tubule remodelling, with JP2 emerging as a clear factor regulating T-tubule integrity. Future studies are warranted to answer the following questions: (i) What is the role and pathological significance of longitudinal elements in the process of heart disease? (ii) What is the role of JP2 in T-tubule development? (iii) How does JP2 become dysregulated at both the transcriptional and post-translational levels in response to cardiac stress? (iv) JP2 is very likely not the sole molecule responsible for T-tubule integrity in health and disease. What other players are critically involved? And how? Is there any interplay between JP2 and others factors in development, healthy adults, and disease? By achieving a complete mechanistic understanding of how T-tubule integrity is developed and maintained in health and how it is lost with disease progression, we are one step closer to identifying novel strategies that improve cardiac function and decrease mortality and morbidity associated with heart failure.
Funding
This work was supported by National Heart Lung Blood Institute R01 HL090905 (L.-S.S.) and American Heart Association Scientific Development Grant 0635056N (L.-S.S.).
Acknowledgement
The authors are grateful to Shawn Roach for the graphic design.
Conflict of interest: none declared.
References
- 1.Bers DM. Excitation–Contraction Coupling and Cardiac Contractile Force. Boston: Kluwer Academic Publishers; 2001. [Google Scholar]
- 2.Stern MD. Theory of excitation–contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. doi:10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. doi:10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 4.Sun XH, Protasi F, Takahashi M, Takeshima H, Ferguson DG, Franzini-Armstrong C. Molecular architecture of membranes involved in excitation-contraction coupling of cardiac muscle. J Cell Biol. 1995;129:659–671. doi: 10.1083/jcb.129.3.659. doi:10.1083/jcb.129.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. doi:10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 6.Fawcett DW, McNutt NS. The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J Cell Biol. 1969;42:1–45. doi: 10.1083/jcb.42.1.1. doi:10.1083/jcb.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huxley AF. The activation of striated muscle and its mechanical response. Proc R Soc Lond B Biol Sci. 1971;178:1–27. doi: 10.1098/rspb.1971.0049. doi:10.1098/rspb.1971.0049. [DOI] [PubMed] [Google Scholar]
- 8.Lindner E. Submicroscopic morphology of the cardiac muscle. Z Zellforsch Mikrosk Anat. 1957;45:702–746. [PubMed] [Google Scholar]
- 9.Simpson FO, Oertelis SJ. The fine structure of sheep myocardial cells; sarcolemmal invaginations and the transverse tubular system. J Cell Biol. 1962;12:91–100. doi: 10.1083/jcb.12.1.91. doi:10.1083/jcb.12.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson DA, Benson ES. On the structural continuities of the transverse tubular system of rabbit and human myocardial cells. J Cell Biol. 1963;16:297–313. doi: 10.1083/jcb.16.2.297. doi:10.1083/jcb.16.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simpson FO. The transverse tubular system in mammalian myocardial cells. Am J Anat. 1965;117:1–17. doi: 10.1002/aja.1001170102. doi:10.1002/aja.1001170102. [DOI] [PubMed] [Google Scholar]
- 12.Sperelakis N, Rubio R. An orderly lattice of axial tubules which interconnect adjacent transverse tubules in guinea-pig ventricular myocardium. J Mol Cell Cardiol. 1971;2:211–220. doi: 10.1016/0022-2828(71)90054-x. doi:10.1016/0022-2828(71)90054-X. [DOI] [PubMed] [Google Scholar]
- 13.Franzini-Armstrong C, Porter KR. Sarcolemmal invaginations constituting the T system in fish muscle fibers. J Cell Biol. 1964;22:675–696. doi: 10.1083/jcb.22.3.675. doi:10.1083/jcb.22.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huxley AF, Taylor RE. Function of Krause's membrane. Nature. 1955;176:1068. doi: 10.1038/1761068a0. doi:10.1038/1761068a0. [DOI] [PubMed] [Google Scholar]
- 15.Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD. doi:10.1161/01.RES.0000074908.17214.FD. [DOI] [PubMed] [Google Scholar]
- 16.Song LS, Guatimosim S, Gomez-Viquez L, Sobie EA, Ziman A, Hartmann H, et al. Calcium biology of the transverse tubules in heart. Ann NY Acad Sci. 2005;1047:99–111. doi: 10.1196/annals.1341.009. doi:10.1196/annals.1341.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louch WE, Sejersted OM, Swift F. There goes the neighborhood: pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J Biomed Biotechnol. 2010;2010:503906. doi: 10.1155/2010/503906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibrahim M, Gorelik J, Yacoub MH, Terracciano CM. The structure and function of cardiac t-tubules in health and disease. Proc Biol Sci. 2011 doi: 10.1098/rspb.2011.0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McNutt NS. Ultrastructure of the myocardial sarcolemma. Circ Res. 1975;37:1–13. doi: 10.1161/01.res.37.1.1. doi:10.1161/01.RES.37.1.1. [DOI] [PubMed] [Google Scholar]
- 20.Kostin S, Scholz D, Shimada T, Maeno Y, Mollnau H, Hein S, et al. The internal and external protein scaffold of the T-tubular system in cardiomyocytes. Cell Tissue Res. 1998;294:449–460. doi: 10.1007/s004410051196. doi:10.1007/s004410051196. [DOI] [PubMed] [Google Scholar]
- 21.Crossman DJ, Ruygrok PN, Soeller C, Cannell MB. Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One. 2011;6:e17901. doi: 10.1371/journal.pone.0017901. doi:10.1371/journal.pone.0017901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. doi:10.1161/01.RES.84.3.266. [DOI] [PubMed] [Google Scholar]
- 23.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. doi:10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swift F, Franzini-Armstrong C, Oyehaug L, Enger UH, Andersson KB, Christensen G, et al. Extreme sarcoplasmic reticulum volume loss and compensatory T-tubule remodeling after Serca2 knockout. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1120172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res. 2012;111:402–414. doi: 10.1161/CIRCRESAHA.112.274530. doi:10.1161/CIRCRESAHA.112.274530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savio-Galimberti E, Frank J, Inoue M, Goldhaber JI, Cannell MB, Bridge JH, et al. Novel features of the rabbit transverse tubular system revealed by quantitative analysis of three-dimensional reconstructions from confocal images. Biophys J. 2008;95:2053–2062. doi: 10.1529/biophysj.108.130617. doi:10.1529/biophysj.108.130617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cannell MB, Crossman DJ, Soeller C. Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered t-tubule structure in human heart failure. J Muscle Res Cell Motil. 2006;27:297–306. doi: 10.1007/s10974-006-9089-y. doi:10.1007/s10974-006-9089-y. [DOI] [PubMed] [Google Scholar]
- 28.Page E, McCallister LP, Power B. Sterological measurements of cardiac ultrastructures implicated in excitation-contraction coupling. Proc Natl Acad Sci USA. 1971;68:1465–1466. doi: 10.1073/pnas.68.7.1465. doi:10.1073/pnas.68.7.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart JM, Page E. Improved stereological techniques for studying myocardial cell growth: application to external sarcolemma, T system, and intercalated disks of rabbit and rat hearts. J Ultrastruct Res. 1978;65:119–134. doi: 10.1016/s0022-5320(78)90050-3. doi:10.1016/S0022-5320(78)90050-3. [DOI] [PubMed] [Google Scholar]
- 30.Severs NJ, Slade AM, Powell T, Twist VW, Jones GE. Morphometric analysis of the isolated calcium-tolerant cardiac myocyte: organelle volumes, sarcomere length, plasma membrane surface folds, and intramembrane particle density and distribution. Cell Tissue Res. 1985;240:159–168. doi: 10.1007/BF00217570. [DOI] [PubMed] [Google Scholar]
- 31.Page E, Surdyk-Droske M. Distribution, surface density, and membrane area of diadic junctional contacts between plasma membrane and terminal cisterns in mammalian ventricle. Circ Res. 1979;45:260–267. doi: 10.1161/01.res.45.2.260. doi:10.1161/01.RES.45.2.260. [DOI] [PubMed] [Google Scholar]
- 32.Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, et al. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–346. doi: 10.1161/CIRCRESAHA.107.160085. doi:10.1161/CIRCRESAHA.107.160085. [DOI] [PubMed] [Google Scholar]
- 33.Kirk MM, Izu LT, Chen-Izu Y, McCulle SL, Wier WG, Balke CW, et al. Role of the transverse-axial tubule system in generating calcium sparks and calcium transients in rat atrial myocytes. J Physiol. 2003;547:441–451. doi: 10.1113/jphysiol.2002.034355. doi:10.1113/jphysiol.2002.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tidball JG, Cederdahl JE, Bers DM. Quantitative analysis of regional variability in the distribution of transverse tubules in rabbit myocardium. Cell Tissue Res. 1991;264:293–298. doi: 10.1007/BF00313966. doi:10.1007/BF00313966. [DOI] [PubMed] [Google Scholar]
- 35.Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D'Hooge J, et al. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–885. doi: 10.1161/CIRCRESAHA.109.206276. doi:10.1161/CIRCRESAHA.109.206276. [DOI] [PubMed] [Google Scholar]
- 36.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, et al. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am J Physiol Heart Circ Physiol. 2011;301:H1996–2005. doi: 10.1152/ajpheart.00284.2011. doi:10.1152/ajpheart.00284.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sommer JR, Johnson EA. Purkinje fibers of the heart examined with the peroxidase reaction. J Cell Biol. 1968;37:570–574. doi: 10.1083/jcb.37.2.570. doi:10.1083/jcb.37.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez-Palomo A, Alanis J, Benitez D. Transitional cardiac cells of the conductive system of the dog heart. Distinguishing morphological and electrophysiological features. J Cell Biol. 1970;47:1–17. doi: 10.1083/jcb.47.1.1. doi:10.1083/jcb.47.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Legato MJ. Ultrastructure of the atrial, ventricular, and Purkinje cell, with special reference to the genesis of arrhythmias. Circulation. 1973;47:178–189. doi: 10.1161/01.cir.47.1.178. doi:10.1161/01.CIR.47.1.178. [DOI] [PubMed] [Google Scholar]
- 40.Boyden PA, Albala A, Dresdner KP., Jr Electrophysiology and ultrastructure of canine subendocardial Purkinje cells isolated from control and 24-hour infarcted hearts. Circ Res. 1989;65:955–970. doi: 10.1161/01.res.65.4.955. doi:10.1161/01.RES.65.4.955. [DOI] [PubMed] [Google Scholar]
- 41.Di Maio A, Ter Keurs HE, Franzini-Armstrong C. T-tubule profiles in Purkinje fibres of mammalian myocardium. J Muscle Res Cell Motil. 2007;28:115–121. doi: 10.1007/s10974-007-9109-6. doi:10.1007/s10974-007-9109-6. [DOI] [PubMed] [Google Scholar]
- 42.Page E. Tubular systems in Purkinje cells of the cat heart. J Ultrastruct Res. 1966;17:72–83. doi: 10.1016/s0022-5320(67)80021-2. doi:10.1016/S0022-5320(67)80021-2. [DOI] [PubMed] [Google Scholar]
- 43.Brette F, Salle L, Orchard CH. Differential modulation of L-type Ca2+ current by SR Ca2+ release at the T-tubules and surface membrane of rat ventricular myocytes. Circ Res. 2004;95:e1–e7. doi: 10.1161/01.RES.0000135547.53927.F6. [DOI] [PubMed] [Google Scholar]
- 44.Brette F, Salle L, Orchard CH. Quantification of calcium entry at the T-tubules and surface membrane in rat ventricular myocytes. Biophys J. 2006;90:381–389. doi: 10.1529/biophysj.105.069013. doi:10.1529/biophysj.105.069013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Houser SR. Ca(2+) signaling domains responsible for cardiac hypertrophy and arrhythmias. Circ Res. 2009;104:413–415. doi: 10.1161/CIRCRESAHA.109.193821. doi:10.1161/CIRCRESAHA.109.193821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. doi:10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 47.Song LS, Sham JS, Stern MD, Lakatta EG, Cheng H. Direct measurement of SR release flux by tracking 'Ca2+ spikes' in rat cardiac myocytes. J Physiol (Lond) 1998;512:677–691. doi: 10.1111/j.1469-7793.1998.677bd.x. doi:10.1111/j.1469-7793.1998.677bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sham JS, Song LS, Chen Y, Deng LH, Stern MD, Lakatta EG, et al. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci USA. 1998;95:15096–15101. doi: 10.1073/pnas.95.25.15096. doi:10.1073/pnas.95.25.15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Page E, McCallister LP. Quantitative electron microscopic description of heart muscle cells: application to normal, hypertrophied and thyroxin-stimulated hearts. Am J Cardiol. 1973;31:172–181. doi: 10.1016/0002-9149(73)91030-8. doi:10.1016/0002-9149(73)91030-8. [DOI] [PubMed] [Google Scholar]
- 50.Maron BJ, Ferrans VJ, Roberts WC. Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am J Pathol. 1975;79:387–434. [PMC free article] [PubMed] [Google Scholar]
- 51.Schaper J, Froede R, Hein S, Buck A, Hashizume H, Speiser B, et al. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83:504–514. doi: 10.1161/01.cir.83.2.504. doi:10.1161/01.CIR.83.2.504. [DOI] [PubMed] [Google Scholar]
- 52.Kaprielian RR, Stevenson S, Rothery SM, Cullen MJ, Severs NJ. Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation. 2000;101:2586–2594. doi: 10.1161/01.cir.101.22.2586. doi:10.1161/01.CIR.101.22.2586. [DOI] [PubMed] [Google Scholar]
- 53.He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, et al. Reduction in density of transverse tubules and L-type Ca(2+) channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49:298–307. doi: 10.1016/s0008-6363(00)00256-x. doi:10.1016/S0008-6363(00)00256-X. [DOI] [PubMed] [Google Scholar]
- 54.Balijepalli RC, Lokuta AJ, Maertz NA, Buck JM, Haworth RA, Valdivia HH, et al. Depletion of T-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc Res. 2003;59:67–77. doi: 10.1016/s0008-6363(03)00325-0. doi:10.1016/S0008-6363(03)00325-0. [DOI] [PubMed] [Google Scholar]
- 55.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, et al. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–533. doi: 10.1113/jphysiol.2006.107227. doi:10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swift F, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, Louch WE, et al. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc Res. 2008;78:71–78. doi: 10.1093/cvr/cvn013. doi:10.1093/cvr/cvn013. [DOI] [PubMed] [Google Scholar]
- 57.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, et al. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci USA. 2009;106:6854–6859. doi: 10.1073/pnas.0809777106. doi:10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dibb KM, Clarke JD, Horn MA, Richards MA, Graham HK, Eisner DA, et al. Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ Heart Fail. 2009;2:482–489. doi: 10.1161/CIRCHEARTFAILURE.109.852228. doi:10.1161/CIRCHEARTFAILURE.109.852228. [DOI] [PubMed] [Google Scholar]
- 59.Ibrahim M, Al Masri A, Navaratnarajah M, Siedlecka U, Soppa GK, Moshkov A, et al. Prolonged mechanical unloading affects cardiomyocyte excitation-contraction coupling, transverse-tubule structure, and the cell surface. FASEB J. 2010;24:3321–3329. doi: 10.1096/fj.10-156638. doi:10.1096/fj.10-156638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. doi:10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kemi OJ, Hoydal MA, Macquaide N, Haram PM, Koch LG, Britton SL, et al. The effect of exercise training on transverse tubules in normal, remodeled, and reverse remodeled hearts. J Cell Physiol. 2011;226:2235–2243. doi: 10.1002/jcp.22559. doi:10.1002/jcp.22559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Oort RJ, Garbino A, Wang W, Dixit SS, Landstrom AP, Gaur N, et al. Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation. 2011;123:979–988. doi: 10.1161/CIRCULATIONAHA.110.006437. doi:10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu CY, Jia Z, Wang W, Ballou LM, Jiang YP, Chen B, et al. PI3Ks maintain the structural integrity of T-tubules in cardiac myocytes. PLoS One. 2011;6:e24404. doi: 10.1371/journal.pone.0024404. doi:10.1371/journal.pone.0024404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sachse FB, Torres NS, Savio-Galimberti E, Aiba T, Kass DA, Tomaselli GF, et al. Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ Res. 2012;110:588–597. doi: 10.1161/CIRCRESAHA.111.257428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ibrahim M, Navaratnarajah M, Siedlecka U, Rao C, Dias P, Moshkov AV, et al. Mechanical unloading reverses transverse tubule remodelling and normalizes local Ca(2+)-induced Ca(2+)release in a rodent model of heart failure. Eur J Heart Fail. 2012;14:571–580. doi: 10.1093/eurjhf/hfs038. doi:10.1093/eurjhf/hfs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lyon AR, Nikolaev VO, Miragoli M, Sikkel MB, Paur H, Benard L, et al. Plasticity of surface structures and beta(2)-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail. 2012;5:357–365. doi: 10.1161/CIRCHEARTFAILURE.111.964692. doi:10.1161/CIRCHEARTFAILURE.111.964692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stolen TO, Hoydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, et al. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res. 2009;105:527–536. doi: 10.1161/CIRCRESAHA.109.199810. doi:10.1161/CIRCRESAHA.109.199810. [DOI] [PubMed] [Google Scholar]
- 68.Ohler A, Weisser-Thomas J, Piacentino V, Houser SR, Tomaselli GF, O'Rourke B. Two-photon laser scanning microscopy of the transverse-axial tubule system in ventricular cardiomyocytes from failing and non-failing human hearts. Cardiol Res Pract. 2009;2009:802373. doi: 10.4061/2009/802373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Houser SR. Reduced abundance of transverse tubules and L-type calcium channels: another cause of defective contractility in failing ventricular myocytes. Cardiovasc Res. 2001;49:253–256. doi: 10.1016/s0008-6363(00)00305-9. doi:10.1016/S0008-6363(00)00305-9. [DOI] [PubMed] [Google Scholar]
- 70.Brette F, Komukai K, Orchard CH. Validation of formamide as a detubulation agent in isolated rat cardiac cells. Am J Physiol Heart Circ Physiol. 2002;283:H1720–H1728. doi: 10.1152/ajpheart.00347.2002. [DOI] [PubMed] [Google Scholar]
- 71.Brette F, Despa S, Bers DM, Orchard CH. Spatiotemporal characteristics of SR Ca(2+) uptake and release in detubulated rat ventricular myocytes. J Mol Cell Cardiol. 2005;39:804–812. doi: 10.1016/j.yjmcc.2005.08.005. doi:10.1016/j.yjmcc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 72.Lipp P, Huser J, Pott L, Niggli E. Spatially non-uniform Ca2+ signals induced by the reduction of transverse tubules in citrate-loaded guinea-pig ventricular myocytes in culture. J Physiol. 1996;497:589–597. doi: 10.1113/jphysiol.1996.sp021792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, et al. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. doi:10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 74.Bito V, Heinzel FR, Biesmans L, Antoons G, Sipido KR. Crosstalk between L-type Ca2+ channels and the sarcoplasmic reticulum: alterations during cardiac remodelling. Cardiovasc Res. 2008;77:315–324. doi: 10.1093/cvr/cvm063. doi:10.1093/cvr/cvm063. [DOI] [PubMed] [Google Scholar]
- 75.Gomez AM, Guatimosim S, Dilly KW, Vassort G, Lederer WJ. Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation. 2001;104:688–693. doi: 10.1161/hc3201.092285. doi:10.1161/hc3201.092285. [DOI] [PubMed] [Google Scholar]
- 76.Houser SR, Piacentino V, III, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. doi:10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- 77.Xu M, Zhou P, Xu SM, Liu Y, Feng X, Bai SH, et al. Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biol. 2007;5:e21. doi: 10.1371/journal.pbio.0050021. doi:10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, et al. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. doi:10.1161/01.RES.75.3.434. [DOI] [PubMed] [Google Scholar]
- 79.Kiss E, Ball NA, Kranias EG, Walsh RA. Differential changes in cardiac phospholamban and sarcoplasmic reticular Ca2+-ATPase protein levels—effects on Ca2+ transport and mechanics in compensated pressure-overload hypertrophy and congestive heart failure. Circ Res. 1995;77:759–764. doi: 10.1161/01.res.77.4.759. doi:10.1161/01.RES.77.4.759. [DOI] [PubMed] [Google Scholar]
- 80.O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. doi:10.1161/01.RES.84.5.562. [DOI] [PubMed] [Google Scholar]
- 81.del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100:2308–2311. doi: 10.1161/01.cir.100.23.2308. doi:10.1161/01.CIR.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, et al. Chronic phospholamban-sarcoplasmic reticulum calcium ATPase interaction is the critical calcium cycling defect in dilated cardiomyopathy. Cell. 1999;99:313–322. doi: 10.1016/s0092-8674(00)81662-1. doi:10.1016/S0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 83.Hobai IA, O'Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. doi:10.1161/01.CIR.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 84.Feldman AM, Weinberg EO, Ray PE, Lorell BH. Selective changes in cardiac gene expression during compensated hypertrophy and the transition to cardiac decompensation in rats with chronic aortic banding. Circ Res. 1993;73:184–192. doi: 10.1161/01.res.73.1.184. doi:10.1161/01.RES.73.1.184. [DOI] [PubMed] [Google Scholar]
- 85.Hasenfuss G. Alterations of calcium-regulatory proteins in heart failure. Cardiovasc Res. 1998;37:279–289. doi: 10.1016/s0008-6363(97)00277-0. doi:10.1016/S0008-6363(97)00277-0. [DOI] [PubMed] [Google Scholar]
- 86.Yao A, Su Z, Nonaka A, Zubair I, Spitzer KW, Bridge JH, et al. Abnormal myocyte Ca2+ homeostasis in rabbits with pacing-induced heart failure. Am J Physiol. 1998;275:H1441–H1448. doi: 10.1152/ajpheart.1998.275.4.H1441. [DOI] [PubMed] [Google Scholar]
- 87.Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca(2+) sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. doi:10.1161/01.RES.87.11.1040. [DOI] [PubMed] [Google Scholar]
- 88.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. doi:10.1161/01.RES.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 89.Hobai IA, O'Rourke B. Enhanced Ca(2+)-activated Na(+)-Ca(2+) exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. doi:10.1161/01.RES.87.8.690. [DOI] [PubMed] [Google Scholar]
- 90.Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97(Suppl. 1):I36–I42. doi: 10.1007/s003950200027. [DOI] [PubMed] [Google Scholar]
- 91.Weisser-Thomas J, Piacentino V, III, Gaughan JP, Margulies K, Houser SR. Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc Res. 2003;57:974–985. doi: 10.1016/s0008-6363(02)00732-0. doi:10.1016/S0008-6363(02)00732-0. [DOI] [PubMed] [Google Scholar]
- 92.Piacentino V, III, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, et al. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. doi:10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 93.Dipla K, Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84:435–444. doi: 10.1161/01.res.84.4.435. doi:10.1161/01.RES.84.4.435. [DOI] [PubMed] [Google Scholar]
- 94.Sipido KR, Volders PG, Vos MA, Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc Res. 2002;53:782–805. doi: 10.1016/s0008-6363(01)00470-9. doi:10.1016/S0008-6363(01)00470-9. [DOI] [PubMed] [Google Scholar]
- 95.O'Neill SC, Valdeolmillos M, Smith GL, Eisner DA. The effects of metabolic inhibition on intracellular pH and Ca. Mol Cell Biochem. 1989;89:199–203. doi: 10.1007/BF00220776. [DOI] [PubMed] [Google Scholar]
- 96.Goldhaber JI, Parker JM, Weiss JN. Mechanisms of excitation-contraction coupling failure during metabolic inhibition in guinea-pig ventricular myocytes. J Physiol (Lond) 1991;443:371–386. doi: 10.1113/jphysiol.1991.sp018838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Overend CL, Eisner DA, O'Neill SC. Altered cardiac sarcoplasmic reticulum function of intact myocytes of rat ventricle during metabolic inhibition. Circ Res. 2001;88:181–187. doi: 10.1161/01.res.88.2.181. doi:10.1161/01.RES.88.2.181. [DOI] [PubMed] [Google Scholar]
- 98.Fukumoto GH, Lamp ST, Motter C, Bridge JH, Garfinkel A, Goldhaber JI. Metabolic inhibition alters subcellular calcium release patterns in rat ventricular myocytes: implications for defective excitation-contraction coupling during cardiac ischemia and failure. Circ Res. 2005;96:551–557. doi: 10.1161/01.RES.0000159388.61313.47. doi:10.1161/01.RES.0000159388.61313.47. [DOI] [PubMed] [Google Scholar]
- 99.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. doi:10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 100.Yang Z, Pascarel C, Steele DS, Komukai K, Brette F, Orchard CH. Na+-Ca2+ exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ Res. 2002;91:315–322. doi: 10.1161/01.res.0000030180.06028.23. doi:10.1161/01.RES.0000030180.06028.23. [DOI] [PubMed] [Google Scholar]
- 101.Sipido KR, Acsai K, Antoons G, Bito V, Macquaide N. T-tubule remodelling and ryanodine receptor organization modulate sodium-calcium exchange. Adv Exp Med Biol. 2013;961:375–383. doi: 10.1007/978-1-4614-4756-6_32. doi:10.1007/978-1-4614-4756-6_32. [DOI] [PubMed] [Google Scholar]
- 102.Sacconi L, Ferrantini C, Lotti J, Coppini R, Yan P, Loew LM, et al. Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc Natl Acad Sci USA. 2012;109:5815–5819. doi: 10.1073/pnas.1120188109. doi:10.1073/pnas.1120188109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tao W, Shi J, Dorn GW, II, Wei L, Rubart M. Spatial variability in T-tubule and electrical remodeling of left ventricular epicardium in mouse hearts with transgenic Galphaq overexpression-induced pathological hypertrophy. J Mol Cell Cardiol. 2012;53:409–419. doi: 10.1016/j.yjmcc.2012.06.006. doi:10.1016/j.yjmcc.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Orchard CH, Pasek M, Brette F. The role of mammalian cardiac t-tubules in excitation-contraction coupling: experimental and computational approaches. Exp Physiol. 2009;94:509–519. doi: 10.1113/expphysiol.2008.043984. doi:10.1113/expphysiol.2008.043984. [DOI] [PubMed] [Google Scholar]
- 105.Pasek M, Simurda J, Christe G, Orchard CH. Modelling the cardiac transverse-axial tubular system. Prog Biophys Mol Biol. 2008;96:226–243. doi: 10.1016/j.pbiomolbio.2007.07.021. doi:10.1016/j.pbiomolbio.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 106.Sah R, Ramirez RJ, Backx PH. Modulation of Ca(2+) release in cardiac myocytes by changes in repolarization rate: role of phase-1 action potential repolarization in excitation-contraction coupling. Circ Res. 2002;90:165–173. doi: 10.1161/hh0202.103315. doi:10.1161/hh0202.103315. [DOI] [PubMed] [Google Scholar]
- 107.Harris DM, Mills GD, Chen X, Kubo H, Berretta RM, Votaw VS, et al. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res. 2005;96:543–550. doi: 10.1161/01.RES.0000158966.58380.37. doi:10.1161/01.RES.0000158966.58380.37. [DOI] [PubMed] [Google Scholar]
- 108.Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, et al. Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–1657. doi: 10.1126/science.1185988. doi:10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 109.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 110.Nishi M, Mizushima A, Nakagawara K, Takeshima H. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun. 2000;273:920–927. doi: 10.1006/bbrc.2000.3011. doi:10.1006/bbrc.2000.3011. [DOI] [PubMed] [Google Scholar]
- 111.Komazaki S, Nishi M, Takeshima H. Abnormal junctional membrane structures in cardiac myocytes expressing ectopic junctophilin type 1. FEBS Lett. 2003;542:69–73. doi: 10.1016/s0014-5793(03)00340-5. doi:10.1016/S0014-5793(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 112.McNary TG, Bridge JH, Sachse FB. Strain transfer in ventricular cardiomyocytes to their transverse tubular system revealed by scanning confocal microscopy. Biophys J. 2011;100:L53–55. doi: 10.1016/j.bpj.2011.03.046. doi:10.1016/j.bpj.2011.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McNary TG, Spitzer KW, Holloway H, Bridge JH, Kohl P, Sachse FB. Mechanical modulation of the transverse tubular system of ventricular cardiomyocytes. Prog Biophys Mol Biol. 2012;110:218–225. doi: 10.1016/j.pbiomolbio.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xie YP, Chen B, Sanders P, Guo A, Li Y, Zimmerman K, et al. Sildenafil prevents and reverses transverse-tubule remodeling and Ca2+ handling dysfunction in right ventricle failure induced by pulmonary artery hypertension. Hypertension. 2012;59:355–362. doi: 10.1161/HYPERTENSIONAHA.111.180968. doi:10.1161/HYPERTENSIONAHA.111.180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen B, Li Y, Jiang S, Xie YP, Guo A, Kutschke W, et al. beta-Adrenergic receptor antagonists ameliorate myocyte T-tubule remodeling following myocardial infarction. FASEB J. 2012;26:2531–2537. doi: 10.1096/fj.11-199505. doi:10.1096/fj.11-199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, Chien KR, et al. Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun. 2004;325:852–856. doi: 10.1016/j.bbrc.2004.10.107. doi:10.1016/j.bbrc.2004.10.107. [DOI] [PubMed] [Google Scholar]
- 117.Landstrom AP, Kellen CA, Dixit SS, van Oort RJ, Garbino A, Weisleder N, et al. Junctophilin-2 expression silencing causes cardiocyte hypertrophy and abnormal intracellular calcium-handling. Circ Heart Fail. 2011;4:214–223. doi: 10.1161/CIRCHEARTFAILURE.110.958694. doi:10.1161/CIRCHEARTFAILURE.110.958694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wu HD, Xu M, Li RC, Guo L, Lai YS, Xu SM, et al. Ultrastructural remodelling of Ca2+ signalling apparatus in failing heart cells. Cardiovasc Res. 2012;95:430–438. doi: 10.1093/cvr/cvs195. doi:10.1093/cvr/cvs195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xu M, Wu HD, Li RC, Zhang HB, Wang M, Tao J, et al. Mir-24 regulates junctophilin-2 expression in cardiomyocytes. Circ Res. 2012;111:837–841. doi: 10.1161/CIRCRESAHA.112.277418. doi:10.1161/CIRCRESAHA.112.277418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, et al. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276:21425–21433. doi: 10.1074/jbc.M100828200. doi:10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- 121.Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 2010;8:e1000312. doi: 10.1371/journal.pbio.1000312. doi:10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang R, Yang J, Zhu J, Xu X. Depletion of zebrafish Tcap leads to muscular dystrophy via disrupting sarcomere-membrane interaction, not sarcomere assembly. Hum Mol Genet. 2009;18:4130–4140. doi: 10.1093/hmg/ddp362. doi:10.1093/hmg/ddp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee E, Marcucci M, Daniell L, Pypaert M, Weisz OA, Ochoa GC, et al. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297:1193–1196. doi: 10.1126/science.1071362. doi:10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- 124.Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17:720–725. doi: 10.1038/nm.2374. doi:10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- 125.Ibrahim M, Siedlecka U, Buyandelger B, Harada M, Rao C, Moshkov A, et al. A critical role for Telethonin in regulating t-tubule structure and function in the mammalian heart. Hum Mol Genet. 2013;22:372–383. doi: 10.1093/hmg/dds434. doi:10.1093/hmg/dds434. [DOI] [PMC free article] [PubMed] [Google Scholar]