

Abstract
Aims
To evaluate the presence of peripapillary retinal nerve fibre layer (RNFL) defects in patients with Stargardt disease by using spectral-domain optical coherence tomography (SD-OCT).
Methods
Fifty-two eyes of 27 patients with Stargardt disease underwent peripapillary RNFL thickness measurements using SD-OCT.
Results
Twenty-seven patients with Stargardt disease were enrolled. Their mean (±SD) age was 38.3 (14.7) years. Fourteen patients (51.9%) showed a thinning of the peripapillary RNFL in one or more quadrants in at least one eye, and four patients (14.8%) in both eyes. Five patients (18.5%) showed a thickening of the peripapillary RNFL in at least one eye, and four patients (14.8%) in both eyes.
Conclusion
This study demonstrated the presence of defects in the peripapillary RNFL thickness in patients with Stargardt disease by using SD-OCT. It would be clinically prudent that Stargardt patients considered for various treatment options be considered for RNFL thickness measurements.
INTRODUCTION
Stargardt disease is an inherited retinal dystrophy characterised by the presence of yellow-white, deep retinal fundus flecks either exclusively in the posterior pole or extending to the mid-peripheral retina. Most patients will develop overt atrophic-appearing macular lesions.1–3
Functional abnormalities in Stargardt disease include loss of macular function with or without loss of generalised cone, or cone and rod function.4 The disease is inherited as an autosomal recessive trait, and it is caused by mutations in the ABCA4 gene on chromosome 1.5,6 Histopathological studies found high levels of lipofuscin accumulation in the retinal pigment epithelium (RPE) in patients with Stargardt disease.7,8
Spectral-domain OCT (SD-OCT) is an often-used device in the clinical setting that can provide detailed cross-sectional images and quantitative information on thickness changes in the retina. Improvements in OCT technology have recently been introduced, including three-dimensional (3D), high-resolution and high-speed OCT that uses Fourier-domain (FD) detection to provide increased resolution. Previous reports showed retinal nerve fibre layer (RNFL) thickness abnormalities in hereditary retinal diseases such as juvenile-onset X linked retinoschisis, retinitis pigmentosa and cone–rod dystrophy.9–11 It has also been used to evaluate RNFL thickness in patients with glaucoma.12,13
The aim of the current study was to use high-speed, high-resolution SD-OCT to evaluate the presence of peripapillary retinal nerve fibre layer (RNFL) defects in patients with Stargardt disease.
MATERIALS AND METHODS
Participants
Fifty-two eyes of 27 patients with a diagnosis of Stargardt disease were included in a prospective investigational study. The participants were recruited for examination during a period from 9 October 2009, through 10 March 2010 at the Department of Ophthalmology at the University of Illinois at Chicago. The study was conducted in accordance with the ethical standards stated in the 1964 Declaration of Helsinki and was approved by an institutional review board at the University of Illinois. Informed consent was obtained on all participants.
Inclusion criteria included patients with Stargardt disease with different stages of severity as previously described.14 The phenotype was classified as stage I, which is typically characterised by a localised atrophic-appearing foveal lesion surrounded by parafoveal or perifoveal yellowish-white lesions (flecks). In stage II, the retinal flecks appear throughout the posterior pole, anterior to the vascular arcades and/or nasal to optic disc, while in stage III, the flecks are almost entirely resorbed. In stage-IV, there is extensive atrophy of the RPE and choroid. Patients aged ≥18 years old, with no clinically significant ocular media opacities were included in the study. Exclusion criteria were patients with a refractive error greater than ±6 dioptre (D) sphere or ±2 D cylinder, uveitis, optic neuropathy, any retinal diseases other than Stargardt disease, past history of glaucoma, increased intraocular pressure (IOP) of ≥21 mm Hg, prior history of intraocular surgery, except for uncomplicated cataract extraction, or poor OCT image quality.
All subjects underwent a complete ocular examination, including best-corrected visual acuity using an early treatment diabetic retinopathy screening chart (ETDRS) in most of the patients, except two where a Snellen chart was used and converted to logMAR visual acuity during the data analysis. A slit-lamp biomicroscopic examination and IOP measurement with Goldmann applanation tonometry were also obtained. Dilated fundus examination was performed using both direct and indirect ophthalmoscopy as well as with stereoscopic biomicroscopy of the optic nerve head with the use of a non-contact 78 dioptre lens. Any optic disc pallor was recorded and graded as mild, moderate or severe by two of the authors (MAG and GAF).
SD-OCT examinations
All subjects included in the study underwent peripapillary RNFL measurements by using an OPKO spectral-domain OCT/SLO instrument (OPKO Instrumentations, Miami, Florida), which utilises a combination OCT and a confocal scanning laser ophthalmoscope (SLO). The system employs a confocal fundus image for alignment, orientation and registration of the OCT topographic map images for diagnostic purposes. The system has a 5 μm axial resolution and scans at 27 000 A-scans per second.
For the peripapillary RNFL measurements, the RNFL exam protocol was used for scan acquisition, which was measured automatically by the existing software at a diameter of 3.45 mm around the centre of the optic disc after dilation of the pupil with 2.5% phenylephrine and 1% tropicamide. The peripapillary RNFL thickness parameters calculated by OPKO software and evaluated in this study were average thickness (360° measure), temporal quadrant thickness (316–45°), superior quadrant thickness (46–135°), nasal quadrant thickness (136–225°) and inferior quadrant thickness (226–315°). OCT software automatically measured three smaller segments within each quadrant (total of 12 segments and four quadrants).
The peripapillary RNFL thickness was measured at least three times in each eye, and data were used only if the scans were reproducible, within either a normal or abnormal range, in at least two-thirds of the acquisitions. Also, eyes with measurements inconsistent on the first set of three scans were rescanned with another set of three measurements. We then used the criteria that two-thirds of these repeat scans should be consistent in order to be included in the analysis.
Data analysis
The peripapillary total circumferential RNFL thickness was considered abnormally thin if its value was less than the fifth percentile of the age-related normal value provided by the manufacturer and thicker if it was more than the 95th percentile. Abnormal thinning or thickening of the RNFL in at least one quadrant, consistently in at least two-thirds of the RNFL OCT scans, was considered significant and therefore abnormal.
We measured the average of RNFL thickness±SD in all four individual quadrants and compared these values with normative data provided by the manufacturer that was corrected for age and which was retrieved from 245 eyes of 123 visually normal subjects aged from 19 to 85 years old (mean (SD) age was 43.1 (14.9) years).
The data were presented as mean±SD. The SPSS program package version 15.0 (SPSS) was used for statistical analysis. We analysed any differences in the mean peripapillary total circumferential RNFL thickness in our patient cohort and that from normative control data by using the two-tailed Student t test. Statistical significance was accepted at p<0.05. The Fisher exact test was used to evaluate the influence of age on the overall peripapillary RNFL thickness measurements.
RESULTS
Of the 27 patients included in the study, 17 were females (63.0%), and 10 were males (37.0%). Of the 52 eyes in these 27 patients, there were 25 right eyes (48.1%) and 27 left eyes (51.9%) that were included. In two patients (7.4%), only one eye was included due to failure in obtaining consistent and reliable RNFL OCT scans by using the criteria mentioned previously in the ‘Methods’ section.
The mean (±SD) age of the patients was 38.3 (±14.7) years with a range from 19 to 72 years. There were 15 Caucasian (55.6%), nine African–American (33.3%), two Asian (7.4%) and one Hispanic (3.7%) patients enrolled in the study. In 19 patients (70.4%), disease-causing mutations in the ABCA4 gene were previously identified (table 1).
Table 1.
Demographics and clinical characteristics of the study cohort
| Patient no/age (years) | Gender/race* | ABCA4 gene mutations | Stargardt disease staging | Visual acuity logMAR right eye/left eye |
|---|---|---|---|---|
| 1/19 | F/C | Pending results | II | 1.0/1.0 |
| 2/39 | M/AA | Pending results | II | 1.0/1.0 |
| 3/27 | F/Asian | arg1108cys exon 22 | I | 1.0/1.0 |
| 4/59 | F/C | Pending results | III | 1.3/1.3 |
| 5/20 | F/C | val849 ala exon 16 GTC>GCC | I | 1.28/1.3 |
| 6/28 | F/C | gly1961glu exon 42 | I | 0.68/0.38 |
| 7/46 | F/AA | val849ala exon 16 GTC>GCC | III | 1.05/1.0 |
| 8/38 | F/AA | Not checked | II | 0.28/0.32 |
| 9/21 | M/AA | gly607arg exon 13 and leu1201arg exon 24 | II | 1.3/1.3 |
| 10/45 | F/C | gly1961glu exon 42 | II | 0.52/0.62 |
| 11/22 | F/Asian | thr901arg exon 18 | III | 1.08/1.3 |
| 12/33 | F/C | Not checked | II | 0.76/0.6 |
| 13/35 | F/C | trp1408arg | I | 1.0/1.0 |
| 14/42 | M/AA | pro1380 leu and leu2027phe | III | 1.3/1.0 |
| 15/42 | M/C | Pending results | III | 1.3/1.3 |
| 16/20 | F/C | lys524thr and donar splice site G>A exon 13 | III | 1.0/1.3 |
| 17/43 | M/C | ala1038val exon 21 and leu541pro exon 12 and G>T donor splice exon 31 | III | 1.0/1.3 |
| 18/52 | F/C | gly991arg exon 20, arg1300gln exon 27 and IVS 28+4 C>T | I | 1.0/1.0 |
| 19/61 | F/AA | pending results | I | 1.0/1.0 |
| 20/72 | F/AA | leu729pro exon 36 and val989ala | III | 1.3/1.3 |
| 21/30 | F/AA | gly991arg exon 20 | II | 1.3/1.3 |
| 22/24 | M/C | ala1038val exon 21 and leu541pro, both heterozygous on the same allele | II | 1.3/1.05 |
| 23/61 | F/AA | arg220cys exon 6, val643met exon 13 and arg2107his exon 46, compound heterozygous | II | 1.3/1.3 |
| 24/46 | M/C | gly863ala exon 17, homozygous | I | 0.7/064 |
| 25/60 | M/C | thr IVS-10 cys exon 39 | II | 1.0/1.0 |
| 26/19 | M/C | Negative results | III | –/0.94† |
| 27/42 | M/Hispanic | his2128arg, exon 46 | III | –/1.48† |
AA, African–American; C, Caucasian; F, female; M, male.
Only left eyes were included, due to a failure to obtain consistent optical coherence tomography scans in their right eyes.
The average best-corrected visual acuity in the right eyes was 1.03 logMAR units with a SD of 0.27 (range 0.28–1.30), equivalent to 20/30–20/400. The average best-corrected visual acuity in the left eyes was 1.04 logMAR units with a SD of 0.30 (range 0.32–1.48), equivalent to 20/40–20/600.
The range of IOP for all patients was 10 to 17 mm Hg (mean 13.8 (±1.9) mm Hg). Three patients (11.1%) had mild temporal disc pallor clinically in both eyes and two patients (7.4%) in one eye.
The number of patients with various stages of Stargardt disease14 at their most recent follow-up visit was as follows: seven patients (26.0%) showed a stage I phenotype, 10 a stage II (37.0%) and 10 a stage III (37.0%). The patients’ demographics and clinical characteristics are shown in (table 1).
Among our 27 patients with Stargardt disease, 24 eyes (46.2%) of 14 patients (51.9%) showed a thinning of the peripapillary RNFL in one or more quadrants in at least one eye, and four patients (14.8%) in both eyes. Thinning of the peripapillary RNFL in one quadrant was seen in 15 eyes (28.8%) of 12 patients (44.4%), and thinning in two or more quadrants was seen in six eyes (11.5%) of five patients (18.5%) (figure 1). Among the five patients who showed optic disc temporal pallor on clinical fundus exam, we found thinning of the peripapillary RNFL in four patients in at least one quadrant (superior and/or nasal) on SD-OCT exam. Regarding the distribution for abnormal thinning of the peripapillary RNFL, thinning was seen more commonly in the superior and inferior quadrants with an equal distribution in eight eyes (33.3%) of six patients (42.9%), followed by the nasal quadrant in four eyes (16.7%) of four patients (28.6%), and the temporal quadrant in four eyes (16.7%) of three patients (21.4%).
Figure 1.
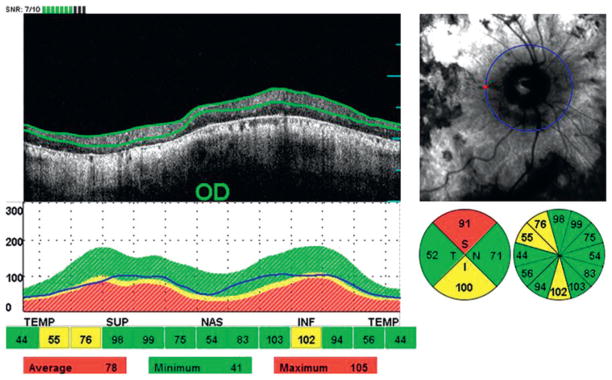
Abnormal thinning of the peripapillary retinal nerve fibre layer in two out of four quadrants in the right eye (OD) of patient 20.
Of note, the presence of RNFL thinning was present in nine (60.0%) of 15 patients who were ≥35 years of age. All of those nine patients had thinning of RNFL in at least one quadrant in all studied eyes. For those who were <35 years old, thinning of RNFL in at least one quadrant was observed in five (41.7%) of 12 patients. There was no statistically significant difference for the presence of RNFL thinning in at least one quadrant in the older (≥35 years old) compared with the younger age group (<35 years old) (p=0.74).
Our cohort of patients also showed a thickening of the peripapillary RNFL in eight eyes (15.4%) of five patients (18.5%) in at least one eye and four patients (14.8%) in both eyes (figure 2). Thickening of the peripapillary RNFL was equally distributed in the superior, nasal and temporal quadrants of five eyes (31.3%) of three patients (60.0%), followed by the inferior quadrant in one eye (6.3%) of one patient (20.0%).
Figure 2.
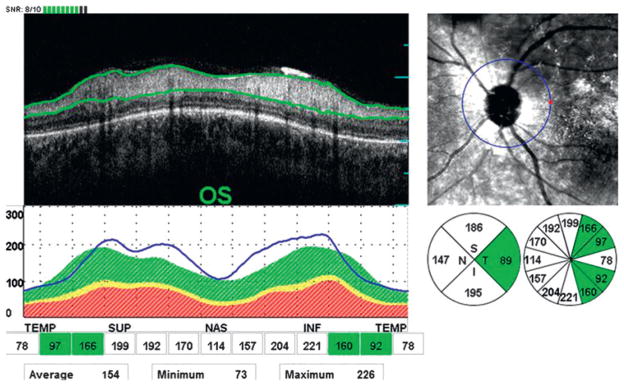
Abnormal thickening of the peripapillary retinal nerve fibre layer in three out of four quadrants in the left eye (OS) of patient 16.
We determined that the overall average total circumferential RNFL thickness, including quadrants with either increased or decreased RNFL thickness, in the 52 eyes of our 27 patients, was 101.9 μm with a SD±16.2, compared with normative data, where the average RNFL thickness was 105.2 μm with a SD±10.8. There was no statistically significant difference in the overall average circumferential RNFL thickness when compared with the normative data (p=0.20), considering both groups of patients on whom the RNFL thickness measurements were either thinner or thicker than normal.
There were eight eyes (15.4%) of six patients (22.2%), in which the overall average circumferential RNFL thickness was thinner than normal. The mean±SD was found to be 80.9±7.4 μm, which was a highly statistically significant difference when compared with the normative data (mean±SD= 105.2±10.8 μm) (p=0.001). There were three eyes (5.8%) of two patients (7.4%), in which the overall average RNFL thickness was thicker than normal. The actual values were 148, 149 and 144 μm (mean±SD=147±2.6 μm), compared with the normative data (mean±SD=105.2±10.8 μm) (p=0.001). However, the number of eyes with increased RNFL thickness was too small for the statistical analysis to be considered meaningful.
DISCUSSION
To our knowledge, our current study is the first to demonstrate the presence of peripapillary defects in RNFL thickness by using SD-OCT in patients with Stargardt disease. Among our 27 patients, these data showed that 14 patients (51.9%) had thinning of the RNFL in one or more quadrants in at least one eye, and five patients (18.5%) showed thickening of the overall circumferential peripapillary RNFL. Abnormal thinning was found more commonly in the superior and inferior quadrants, while the abnormal thickening was equally distributed in the nasal, temporal and superior quadrants. In order to fully appreciate regions of reduced RNFL thickness, in our study it was necessary to analyse individual quadrants and not only total circumferential RNFL thickness, as some regions of either increased or decreased thickness can be observed in an individual quadrant.
Our findings in regard to RNFL thickness changes in Stargardt disease (a primary outer retinal layer degenerative disease) raises questions regarding the role of trans-synaptic retinal degeneration or direct causal effects from the consequences of outer retinal layer degeneration with possible secondary anatomical and physiological inner retinal layer alternations. A knowledgeable explanation for these changes in the RNFL thickness awaits further future investigations.
A recent study by Lim and associates, by using Fourier-domain OCT in inherited retinal dystrophies, showed that inner retinal layer thickness was mildly thinner than normal in a patient with Stargardt disease.15
Our findings add to the list of inherited retinal dystrophies in which abnormalities in RNFL thickness may be found.9–11,16 In a recent report by Pasadhika and Fishman,11 thinning of the peripapillary RNFL was found in 73% of eight patients with autosomal-recessive cone–rod dystrophy, including those with identified ABCA4 gene mutations. Mutations in the ABCA4 gene are responsible for Stargardt disease,17 but also have been implicated in autosomal recessive forms of cone–rod dystrophy and retinitis pigmentosa.18–20
We were unable to document an association between the stages of severity in retinal fundus changes14 and the presence of RNFL thickness defects. For example, we found that patients with Stargardt disease stage III could show thinner, thicker or normal RNFL quadrant thickness measurements. These data showed that the degrees of thinning were most often similar between both eyes of the same patient (N=9), although asymmetry was also observed (N=4).
One of the current study’s limitations that might have influenced the RNFL thickness calculated by SD-OCTwas a lack of knowledge as to the axial lengths of the eyes.21,22 However, in our patients, we only included patients with a lower degree of refractive error in whom a normal or close to normal axial length might be assumed.
In conclusion, our study demonstrated the presence of defects in the peripapillary RNFL thickness in patients with Stargardt disease by using SD-OCT. Potential treatment strategies, such as gene-directed therapy, targeted at restoring or preserving outer retinal structure, will likely be most useful in patients who have a suitable degree of preservation in their inner retinal layers. Our findings support the conclusion that Stargardt patients considered for various treatment options should also be considered for RNFL thickness measurements.
Acknowledgments
We like to thank JJ McAnany, for his help in the statistical analysis.
Funding Supported by funds from the Foundation Fighting Blindness, Owings Mills, Maryland; Grant Healthcare Foundation, Lake Forest, Illinois; NIH core grant EYO1792; and an unrestricted departmental grant from Research to Prevent Blindness.
Footnotes
Patient consent Obtained.
Ethics approval Ethics approval was provided by an institutional review board at the University of Illinois and the University of Illinois ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Stargardt K. Über familiäre, progressive Degeneration in der Maculagegend des Auges. Albrecht von Graefes Arch Klin Exp Ophthalmol. 1909;71:534–50. [Google Scholar]
- 2.Genead MA, Fishman GA, Stone EM, et al. The natural history of stargardt disease with specific sequence mutation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2009;50:5867–71. doi: 10.1167/iovs.09-3611. [DOI] [PubMed] [Google Scholar]
- 3.Walia S, Fishman GA. Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet. 2009;30:63–8. doi: 10.1080/13816810802695550. [DOI] [PubMed] [Google Scholar]
- 4.Lois N, Holder GE, Bunce C, et al. Phenotypic subtypes of Stargardt macular dystrophy–fundus flavimaculatus. Arch Ophthalmol. 2001;119:359–69. doi: 10.1001/archopht.119.3.359. [DOI] [PubMed] [Google Scholar]
- 5.Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan J, Gerber S, Larget-Piet D, et al. A gene for Stargardt’s disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet. 1993;5:308–11. doi: 10.1038/ng1193-308. [DOI] [PubMed] [Google Scholar]
- 7.Eagle RC, Jr, Lucier AC, Bernardino VB, Jr, et al. Retinal pigment epithelial abnormalities in fundus flavimaculatus: a light and electron microscopic study. Ophthalmology. 1980;87:1189–200. doi: 10.1016/s0161-6420(80)35106-3. [DOI] [PubMed] [Google Scholar]
- 8.Steinmetz RL, Garner A, Maguire JI, et al. Histopathology of incipient fundus flavimaculatus. Ophthalmology. 1991;98:953–6. doi: 10.1016/s0161-6420(91)32197-3. [DOI] [PubMed] [Google Scholar]
- 9.Genead MA, Pasadhika S, Fishman GA. Retinal nerve fiber layer thickness analysis in X-linked retinoschisis using Fourier-domain OCT. Eye. 2009;23:1019–27. doi: 10.1038/eye.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walia S, Fishman GA. Retinal nerve fiber layer analysis in RP patients using Fourier-domain OCT. Invest Ophthalmol Vis Sci. 2008;49:3525–8. doi: 10.1167/iovs.08-1842. [DOI] [PubMed] [Google Scholar]
- 11.Pasadhika S, Fishman GA, Allikmets R, et al. Peripapillary retinal nerve fiber layer thinning in patients with autosomal recessive cone–rod dystrophy. Am J Ophthalmol. 2009;148:260–5. doi: 10.1016/j.ajo.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carpineto P, Ciancaglini M, Zuppardi E, et al. Reliability of nerve fiber layer thickness measurements using optical coherence tomography in normal and glaucomatous eyes. Ophthalmology. 2003;110:190–5. doi: 10.1016/s0161-6420(02)01296-4. [DOI] [PubMed] [Google Scholar]
- 13.Kanamori A, Nakamura M, Escano MF, et al. Evaluation of the glaucomatous damage on retinal nerve fiber layer thickness measured by optical coherence tomography. Am J Ophthalmol. 2003;135:513–20. doi: 10.1016/s0002-9394(02)02003-2. [DOI] [PubMed] [Google Scholar]
- 14.Fishman GA. Fundus flavimaculatus: A clinical classification. Arch Ophthalmol. 1976;94:2061–7. doi: 10.1001/archopht.1976.03910040721003. [DOI] [PubMed] [Google Scholar]
- 15.Lim JI, Tan O, Fawzi AA, et al. A pilot study of Fourier-domain optical coherence tomography of retinal dystrophy patients. Am J Ophthalmol. 2008;146:417–26. doi: 10.1016/j.ajo.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman NM, Stevens RA, Heckenlively JR. Nerve fibre layer loss in diseases of the outer retinal layer. Br J Ophthalmol. 1987;71:21–6. doi: 10.1136/bjo.71.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klevering BJ, Deutman AF, Maugeri A, et al. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005;243:90–100. doi: 10.1007/s00417-004-1079-4. [DOI] [PubMed] [Google Scholar]
- 18.Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone–rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–62. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Mir A, Paloma E, Allikmets R, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet. 1999;18:11–12. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 20.Maugeri A, Klevering BJ, Rohrschneider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone–rod dystrophy. Am J Hum Genet. 2000;67:960–6. doi: 10.1086/303079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Budenz DL, Anderson DR, Varma R, et al. Determinants of normal retinal nerve fiber layer thickness measured by Stratus OCT. Ophthalmology. 2007;114:1046–52. doi: 10.1016/j.ophtha.2006.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bayraktar S, Bayraktar Z, Yilmaz OF. Influence of scan radius correction for ocular magnification and relationship between scan radius with retinal nerve fiber layer thickness measured by optical coherence tomography. J Glaucoma. 2001;10:163–9. doi: 10.1097/00061198-200106000-00004. [DOI] [PubMed] [Google Scholar]