

Abstract
Gammaherpesviruses are ubiquitious pathogens that establish lifelong infection and are associated with several malignancies. All gammaherpesviruses encode a conserved protein kinase that facilitates viral replication and chronic infection and thus represents an attractive therapeutic target. In this study, we identify a novel function of gammaherpesvirus protein kinase as a regulator of class I histone deacetylases (HDAC). Mouse gammaherpesvirus 68 (MHV68)-encoded protein kinase orf36 interacted with HDAC1 and 2 and prevented association of these HDACs with the viral promoter driving expression of RTA, a critical immediate early transcriptional activator. Furthermore, the ability to interact with HDAC1 and 2 was not limited to the MHV68 orf36, as BGLF4, a related viral protein kinase encoded by Epstein-Barr virus, interacted with HDAC1 in vitro. Importantly, targeting of HDAC1 and 2 by orf36 was independent of the kinase's enzymatic activity. Additionally, orf36 expression, but not its enzymatic activity, induced changes in the global deacetylase activity observed in infected primary macrophages. Combined deficiency of HDAC1 and 2 rescued attenuated replication and viral DNA synthesis of the orf36 null MHV68 mutant, indicating that the regulation of HDAC1 and 2 by orf36 was relevant for viral replication. Understanding the mechanism by which orf36 facilitates viral replication, including through HDAC targeting, will facilitate the development of improved therapeutics against gammaherpesvirus kinases.
INTRODUCTION
Histone deacetylases (HDACs) are chromatin-modifying enzymes that mediate the removal of acetyl moieties from proteins, including core histones. Generally, HDACs repress transcription by causing localized chromatin compaction; in contrast, HDAC activity is required for expression of some genes, including beta interferon and interferon-stimulated genes (1, 2). HDACs can also deacetylate nonhistone proteins, including transcriptional factors, providing an additional level of transcriptional regulation (3). The HDAC family comprises four classes that are distinguished by enzymatic mechanism, cell type specificity, cellular localization, and regulation (4, 5). Class I HDACs, containing HDACs 1 to 3 and 8, have wide tissue distribution and are nuclear. HDAC1 and 2 are metalloenzymes that share 83% sequence similarity and are frequently found in the same protein complex that is generally associated with transcriptional repression, such as the NuRD and CoREST. While many functions of HDAC1 and 2 overlap, these HDACs also participate in distinct processes, as deficiency of HDAC1, but not HDAC2, is embryonically lethal (6, 7).
Herpesviruses are nuclear DNA viruses that establish lifelong infection of the host. Class I HDACs frequently suppress immediate early herpesvirus gene expression by deacetylation of histones at the viral gene promoters (8). It is not surprising that herpesviruses encode proteins that target class I HDACs in lytically infected cells. Herpes simplex virus 1 (HSV-1)-encoded ICP0 displaces HDAC1 from the transcriptionally repressive CoREST complex to facilitate viral gene expression (9). HDAC1 and -2 are also phosphorylated by the HSV-1 protein kinase US3, and this phosphorylation coincides with enhanced expression of transgenes driven by the human cytomegalovirus (HCMV) immediate early promoter (10). US3-related varicella-zoster virus (VZV) protein kinase orf66 indirectly induces phosphorylation of HDAC1 and -2. Furthermore, HDAC inhibitors rescue attenuated viral gene expression in orf66 null infections, suggesting that the VZV kinase targets HDACs to facilitate viral gene expression (11, 12). HCMV encodes IE1, IE2, and pUL29/28, which target HDACs to promote viral gene expression during lytic infection (13–15). Paradoxically, Kaposi's sarcoma-associated herpesvirus (KSHV)-encoded K-bZIP has recently been shown to recruit HDAC1 and -2 to KSHV RTA promoters during reactivation (16). While treatment with HDAC inhibitors enhances immediate early mouse gammaherpesvirus-68 (MHV68) gene expression during de novo lytic infection and induces reactivation of Epstein-Barr virus (EBV), KSHV, and MHV68 (17–19), the mechanisms by which gammaherpesviruses counteract HDACs during lytic replication remain poorly understood.
MHV68 is genetically and biologically related to EBV and KSHV (20, 21) and represents a tractable system that allows studies of gammaherpesvirus-host interactions in the context of de novo infection of physiologically relevant primary immune cells. In this study, we identified MHV68 orf36, a conserved gammaherpesvirus protein kinase, as a regulator of HDAC1 and -2 during de novo lytic infection of primary macrophages. Both MHV68 orf36 and a related EBV-encoded kinase, BGLF4, interacted with HDAC1 and -2. Importantly, orf36 prevented association of HDAC1 and -2 with the distal promoter of RTA, an immediate early viral protein essential for gammaherpesvirus lytic replication and reactivation from latency (22–26). In addition to regulation of HDAC1 and -2 at the RTA promoter, orf36 modulated global HDAC enzymatic activity in infected macrophages throughout the course of lytic replication. Intriguingly, orf36 enzymatic activity was not required for interaction with HDAC1 and -2, clearance of these HDACs from the RTA promoter, or regulation of global HDAC activity in infected cells, providing additional evidence for enzymatic-activity-independent functions of gammaherpesvirus kinases (27, 28). Finally, depletion of HDAC1 and -2 from primary macrophages rescued attenuated replication of the orf36 null MHV68 mutant, providing physiological relevance of the orf36-HDAC interactions.
MATERIALS AND METHODS
Animals and primary cell cultures.
C57BL/6J (BL6) mice were obtained from Jackson Laboratories (Bar Harbor, ME). HDAC1/2 flox/flox (HDAC1/2F/F) mice (7) were generously provided by Eric Olson. These mice were crossed to mice expressing modified estrogen receptor (ER)-Cre fusion protein under the control of Rosa promoter ([29], obtained from Jackson Laboratories) to generate HDAC1/2F/F × ER-Cre mice. All mice were housed and bred in a specific-pathogen-free barrier facility in accordance with federal and institutional guidelines. All experimental manipulations of mice were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin. Bone marrow was harvested from mice between 3 and 10 weeks of age. Primary bone marrow-derived macrophages were generated and infected as previously described (30).
OHT treatment of primary macrophages.
Primary bone marrow-derived macrophages were generated as described previously (30) with the following modifications: cells were treated with 1 μM 4-hydroxytamoxifen (OHT; Sigma-Aldrich, St. Louis, MO) on days 4 and 7 of culture. Allele recombination and HDAC1 and -2 protein levels were assayed at day 12 of culture, and cells were infected at day 13 of culture. Hydroxytamoxifen was not present in the medium of infected cultures.
Western blot analysis.
Macrophages were collected into Laemmli buffer and analyzed as previously described (30). Antibodies used were anti-HDAC1 clone 2e10 (1:1,000; Millipore, Billerica, MA), anti-HDAC2 (1:20,000; Sigma-Aldrich, St. Louis, MO), anti-Flag (M2; Sigma-Aldrich, St. Louis, MO), anti-β-actin (1:20,000; Novus Biologicals, Littleton, CO), anti-γH2AX (1:2,000; Bethyl Laboratories, Montgomery, TX), anti-H3 (1:100,000; Abcam Inc., Cambridge, MA), and a secondary goat anti-mouse or anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (1:20,000; Jackson ImmunoResearch, Westgrove, PA).
qRT-PCR quantitation of viral messages.
Total RNA was harvested, DNase treated, reverse transcribed, and analyzed by real-time PCR as described in reference 31.
Assessment of recombination efficiency.
Cellular DNA was isolated from HDAC1/2F/F × ER-Cre-positive or control macrophages as previously described (32). Recombined HDAC1 and -2 alleles were detected by PCR, using primers 5′-GCC-TCT-GCT-TCC-TTA-GTG-TTG-3′ (forward) and 5′-CAA-CTG-ACT-ACA-GAC-TGT-TGG-G-3′ (reverse, R2) for HDAC1 and 5′-GTG-GGA-AGC-ATG-GCA-GCA-TGC-3′ (forward) and 5′-GCC-TTC-TAA-GAA-CCC-CAG-GGA-AC-3′ (reverse) for HDAC2. Levels of intact, nonrecombined HDAC1 allele were quantified by real-time PCR using the same forward HDAC1 primer as above and 5′-GAG-CAA-GGA-AAG-AGC-ACA-AGC-CTG-3′ for the reverse primer (R1), with subsequent normalization to cellular GAPDH gene.
HDAC activity assays.
Infected macrophages were lysed in 1× reporter lysis buffer (Promega, Madison, WI) and freeze-thawed twice. Global HDAC activity was measured in lysates using a fluorescent HDAC activity kit according to the manufacturer's recommendations (Enzo Life Sciences, Farmingdale, NY). Protein content was determined using Bradford reagent (Fermentas, Glen Burnie, MD) and used to normalize HDAC activity. Normalized HDAC activity levels in mock-infected macrophage lysate were set at 1, and values for other conditions were expressed as fold HDAC activity over mock within each experiment. For trichostatin A (TSA)-treated samples, 200 nM TSA was added to the sample prior to HDAC activity analysis.
Immunoprecipitation.
Primary bone marrow macrophages were mock infected or infected at a multiplicity of infection (MOI) of 10 with wild-type or 36-FLAG MHV68 (36F MHV68) (27, 30). At 30 hpi, cells were lysed in 0.2% NP-40 lysis buffer (0.2% NP-40, 50 mM Tris HCl, 300 mM NaCl, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF]) and briefly sonicated (Microson Cell Disruptor; Misonix, Farmingdale, NY). Lysates were precleared with protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h at 4°C and combined with 1 μg α-HDAC1 (clone 2e10; Millipore, Billerica, MA) or 1 μl anti-FLAG M2 monoclonal antibody (Sigma-Aldrich, St. Louis, MO). Lysates and antibodies were incubated while rotating for 16 h at 4°C. Protein A/G beads were added, and rotation continued for 4 h at 4°C. Immunoprecipitated complexes on agarose beads were washed six times with lysis buffer and once with phosphate-buffered saline (PBS) and then eluted with Laemmli buffer.
Immunofluorescence.
Primary macrophages were seeded on coverslips and infected at an MOI of 10 with 36F MHV68. At 16, 30, and 40 hpi, coverslips were subjected to immunofluorescence as previously described (30). Primary antibodies included anti-HDAC1 (mouse monoclonal, 1:500; Millipore, Billerica, MA) and anti-orf6/ssDBP (rabbit polyclonal, 1:500) (32). Alexa Fluor goat anti-rabbit 488 and goat anti-mouse 594 (Invitrogen, Eugene, OR) were used as secondary antibodies. Colocalization of HDAC1 and orf6 was measured using Pearson's correlation coefficient, as calculated by JACoP (33) in ImageJ (34). The significance of correlation coefficients (compared to r2 = 0) was measured using Student's t test with α of 0.05.
Expression constructs.
MHV68 orf36, BGLF4, KSHV orf36, HSV-1 UL13, and HDACs 1 to 3 were amplified with specific primers flanked by BamHI and EcoRI restriction sites (Table 1), cloned into the pT7CFE1-CHis vector (Pierce Biotechnology, Rockford, IL), and sequenced to verify the insertion. Plasmids containing HDAC1 and -2 cDNA were a kind gift from Eric Olson. U69 from HHV-7B was amplified from viral genomic DNA provided by Amy Hudson. FLAG-tagged Vaccinia I3 was expressed from the pTM1 vector, courtesy of Paula Traktman. Cellular Fas (14-3-3 zeta/delta) was subcloned from pET13b vector, provided by Joseph Barbieri. Viral kinases were amplified with primers including FLAG tags at the N terminus; cellular proteins, including HDACs, were cloned using primers encoding the hemagglutinin (HA) epitope on the N terminus. The MHV68 RTA construct in a pFLAG-CMV-2 vector was generously contributed by Ren Sun. Orf36 and Orf36KN in the pFLAG-CMV-2 vector were previously described (30).
Table 1.
Primer sequences for cloning
| Insert | Sensea | Restriction enzyme | Sequence (5′–3′) |
|---|---|---|---|
| FLAG-tagged proteinb | F | BamHI | GAT AAT GGA TCC GAC TAC AAA GAC GAT GAC GAC AAG |
| MHV68 Orf36/Orf36KN | R | XhoI | CAG GGA CTC GAG TCA AAA AAA TCC AGA ATA ATC |
| EBV BGLF4 | R | XhoI | CAG GGA CTC GAG ACA GTT GAA GAA GTA GCT CTC |
| KSHV Orf36 | R | XhoI | CAG GGA CTC GAG TCA GAA AAC AAG TCC GCG GG |
| HSV-1 UL13 | F | BamHI | GAT AAT GGA TCC GAT TAC AAG GAT GAC GAT GAC AAG ATG GAT GAG TCC CGC AGA CAG |
| R | PstI | CAG GGA CTG CAG TCA ACG ACA GCG CGT GCC GCG | |
| HHV-7b U69 | F | EcoRI | GAT AAT GAA TTC GAT TAC AAG GAT GAC GAT GAC AAG ATG GAG CAG CTT AAG ACA CC |
| R | PstI | CAG GGA CTG CAGTCA TTA CAC CCG AAA TAC AGA ATA TG | |
| HDAC1 | F | BamHI | GAT AAT GGA TCC TAC CCA TAC GAC GTC CCA GAC TAC GCT ATG GCG CAG ACT CAG GGC ACC |
| R | XhoI | CAG GGA CTC GAG GGC CAA CTT GAC CTC TTC | |
| HDAC2 | F | BamHI | GAT AAT GGA TCC TAC CCA TAC GAC GTC CCA GAC TAC GCT ATG GCG TAC AGT CAA GGA G |
| R | XhoI | CAG GGA CTC GAG AGG GTT GCT GAG TTG TTC | |
| HDAC3 | F | BamHI | GAT AAT GGA TCC TAC CCA TAC GAC GTC CCA GAC TAC GCT ATG GCC AAG ACC GTG GCG TAT |
| R | XhoI | GAT AAT CTC GAG CTA AAT CTC CAC ATC ACT TTC | |
| Fas | F | BamHI | GAT AAT GGA TCC TAC CCA TAC GAC GTC CCA GAC TAC GCT ATG GAT AAA AAC GAG CTG GT |
| R | PstI | CAG GGA CTG CAG TCA TTA ATT TTC CCC TCC TTC |
F, forward; R, reverse.
Primer used for subcloning sequences containing FLAG at the N terminus (MHV68 Orf36/Orf36KD, BGLF4, KSHV Orf36).
In vitro transcription-translation assays.
Plasmid DNA (500 ng) was combined with rabbit reticulocyte lysate (Promega, Madison, WI) and 10 μCi [35S]methionine. Reaction mixtures were incubated at 30°C for 90 min. Translated protein products (10% of total reaction volume) were then combined as designated below (see Fig. 2) in 0.4% NP-40 buffer (50 mM Tris-HCl [pH 8.0], 300 mM NaCl, 5 mM EDTA, 0.4% [wt/vol] NP-40, 1 mM PMSF). Complexes were immunoprecipitated overnight with anti-HDAC1, -HDAC2, -FLAG, and -HA antibodies and Prot A/G agarose beads, washed five times with 0.4% NP-40 buffer, resuspended in Laemmli buffer, and analyzed by SDS-PAGE and autoradiography.
Fig 2.

Orf36 inhibits association of HDAC1 and 2 with the distal RTA promoter. (A, B) Primary macrophages were infected as indicated, and chromatin was collected at 16 hpi and subjected to ChIP using HDAC1-specific (A) or HDAC2-specific (B) antibodies or a nonspecific IgG control. Enrichment of RTA core and distal promoter sequences was calculated using the ΔΔCT method. Results are presented as fold enrichment over IgG. (C) Schematic of the core and distal RTA promoters. Arrows above the sequence indicate the locations of PCR primers used in experiments depicted in panels A and B. (D, E) Primary macrophages were infected, and chromatin was harvested as described for panels A and B. ChIP enrichment of orilyt and orf57 promoter sequences immunoprecipitated with anti-HDAC1 were calculated as above. (F) ChIP was performed using chromatin from primary macrophages infected as indicated for 5 h at an MOI of 1. (G) Primary macrophages were infected with wild-type (WT) MHV68 or the N36S mutant at an MOI of 10. Chromatin was harvested at 16 h postinfection and subjected to ChIP as described above. All ChIP data in this figure were pooled from 3 to 5 independent experiments; error bars represent the standard errors of the means. *, P < 0.05.
Nucleofection of primary bone marrow macrophages.
Macrophages were cultured as described previously (30), and cells were lifted on day 10 of culture, combined with plasmid DNA, and resuspended in Buffer T (Lonza, Basel, Switzerland). Electroporation was performed using program T-20 on Amaxa Nucleofector (Lonza, Basel, Switzerland). At 6 h posttransfection, cells were collected in 1× rapid lysis buffer (Promega, Madison, WI) and subjected to HDAC activity assays with subsequent normalization against protein content as measured by the Bradford assay (Fermentas, Glen Burnie, MD).
ChIP.
For chromatin immunoprecipitation (ChIP) assays, primary macrophages were infected at an MOI of 1, and chromatin was collected at 16 hpi and prepared as described previously (32, 35). Chromatin was immunoprecipitated with anti-HDAC1 (clone 2e10; Millipore, Billerica, MA), anti-HDAC2 (Sigma-Aldrich, St. Louis, MO), or rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Coimmunoprecipitated DNA was purified and analyzed by RT-PCR using primers described previously (32). Right orilyt was amplified using 5′-GTG-TGG-CCT-TTG-TGT-GCC-TGT-AAA-3′ (forward) and 5′-AAA-TCG-GTT-TGC-GGT-TAG-ACC-AGG-3′ (reverse) primers, and the orf57 promoter was amplified using 5′-AGA-ACA-GCT-TCG-TGC-TGA-CAA-ACC-3′ (forward) and 5′-TTT-GGT-AAG-CTG-GCC-ACA-GTC-TTG-3′ (reverse) primers.
Viral stock preparation and infections.
Wild-type MHV68 and orf36 mutant virus stocks (27, 30) were prepared and measured on NIH 3T12 cells. Bone marrow-derived macrophages were infected at the indicated MOI as previously described (30). 36F MHV68 encoding a Flag-tagged orf36 was kindly provided by Ren Sun (27). For TSA experiments, viral inoculum was removed and cells were replenished with medium containing 40 nM TSA (Sigma-Aldrich, St. Louis, MO).
Statistical analysis.
Student's t test (Graphpad Prism, La Jolla, CA) was used to measure statistical significance with an α value of 0.05.
RESULTS
orf36 prevents association of HDAC1 and -2 with the distal RTA promoter.
orf36 expression and enzymatic activity are required for optimal expression of RTA in primary macrophages under conditions of low but not high MOI (32). The MOI-dependent phenotype of orf36 is reminiscent of herpesvirus HDAC regulators, such as HSV ICP0, that stimulate viral gene expression under low but not high MOI conditions (9). To determine if attenuated RTA expression in the absence of orf36 could be rescued by HDAC inhibition, primary bone marrow-derived macrophages were infected at an MOI of 1 with wild-type MHV68, an N36S mutant that encodes a translational stop codon in the orf36 sequence, or a 36KN mutant that encodes a catalytically null orf36 (27, 30). Immediately following absorption, macrophages were treated with 40 nM TSA, a broad HDAC inhibitor (5), and RTA expression was measured at 16 h postinfection by qRT-PCR. As expected, RTA transcript levels were decreased in macrophages infected with the orf36 MHV68 mutants compared to wild-type MHV68 (Fig. 1A). Consistent with previous reports (17), TSA treatment increased RTA expression in wild-type MHV68-infected macrophages, indicating that the RTA promoter is sensitive to HDAC inhibition. Interestingly, TSA treatment rescued RTA expression in N36S- and 36KN-infected macrophages (Fig. 1A), suggesting that orf36 may counteract HDACs to facilitate RTA expression. Orf36 was expressed at 16 h postinfection (Fig. 1B), consistent with its contribution to RTA transcription.
Fig 1.
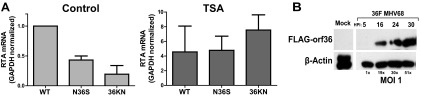
Global inhibition of HDACs rescues RTA transcription in the absence of functional orf36. (A) Primary bone marrow-derived BL6 macrophages were infected at a multiplicity of infection (MOI) of 1 PFU/cell with wild-type (WT) or indicated orf36 mutant MHV68 viruses and treated with 40 nM TSA immediately after virus absorption. Total RNA was collected at 16 hpi, and RTA mRNA levels were measured by qRT-PCR. Data were pooled from three independent experiments. (B) Primary macrophages were mock infected or infected at an MOI of 1 with the MHV68 virus expressing Flag-tagged orf36 (36F). Orf36 and β-actin protein levels were measured at the indicated times postinfection.
HDAC1 and 2 are ubiquitously expressed, frequently localize to gene promoters to mediate transcriptional repression, and are targeted by alpha- and betaherpesvirus proteins. Thus, HDAC1 and -2 could suppress transcription of RTA by localizing to the two known RTA promoters (core and distal [Fig. 2C]) (22, 36). To determine if HDAC1 and -2 directly regulated RTA promoters, primary macrophages were infected with wild-type MHV68 or orf36 mutant viruses at an MOI of 1. The association of HDAC 1 and 2 with RTA promoters and proximal sequences (primers indicated by arrows in Fig. 2C) was measured by chromatin immunoprecipitation (ChIP). Interestingly, HDAC1 and -2 were significantly enriched at the distal RTA promoter in N36S-infected cells at 16 h postinfection (Fig. 2A and B), coincident with attenuated RTA expression (Fig. 1A). Association of HDAC1 and -2 with the distal RTA promoter was minimal in wild-type virus- and 36KN mutant-infected cells (up to 2-fold above background levels [Fig, 2A and B]), suggesting that orf36 enzymatic activity was not required to prevent association of HDAC 1 and 2 with the distal RTA promoter. In contrast to the distal promoter, low levels of HDAC 1 and 2 associated with the core RTA promoter, and this association was not regulated by orf36 (Fig. 2A and B).
In the absence of orf36 expression, enrichment of HDAC1 and -2 was specific to the distal RTA promoter, as evidenced by decreased association of these HDACs with sequences 5′ and 3′ of the distal promoter (Fig. 2A and B) and with the orf57 promoter and right orilyt (Fig. 2D and E). Association of HDAC1 and 2 with the distal RTA promoter was also controlled temporally, as no enrichment of either HDAC was observed at the RTA promoters at 5 h postinfection (Fig. 2F). Finally, differential association of HDAC1 and -2 with the distal RTA promoter was specific to the low MOI conditions. When macrophages were infected at an MOI of 10, HDAC1 was not detected at either distal or core promoter in N36S- or 36KN-infected macrophages (Fig. 2G and data not shown), suggesting that another viral HDAC regulatory mechanism compensated for the absence of orf36 at a high MOI. Similar HDAC1 and -2 protein levels were detected in wild-type-, N36S-, and 36KN-infected cells at 5 and 16 h postinfection (see Fig. 5), suggesting that the observed differences in HDAC1 and -2 association with the distal RTA promoter were not due to differential expression of HDAC1 and -2. In summary, orf36, in an enzymatically independent manner, regulated association of HDAC1 and -2 with the distal RTA promoter, and HDAC inhibition rescued RTA expression in orf36-mutant-infected cells.
Fig 5.

orf36 modulates global HDAC activity throughout lytic infection of primary macrophages. (A to C) Primary macrophages were mock infected or infected at an MOI of 1 or 10 with wild-type or orf36 mutant MHV68 viruses. HDAC enzymatic activity, total protein concentration, and HDAC1, HDAC2, and β-actin protein levels were measured in whole-cell lysates collected at the indicated times postinfection. HDAC activity was normalized to total protein content and, subsequently, to mock-infected lysates. In parallel, HDAC activity was measured in cell lysates spiked with TSA. HDAC activity data were pooled from 3 to 6 independent experiments; each condition was assessed in two replicates within each experiment. Densitometric analysis was used to measure HDAC1 and -2 protein content, normalized to β-actin. Normalized values are listed below the respective lanes. (D) Primary macrophages were nucleofected with the indicated plasmids, and HDAC activity and protein levels were measured as above at 6 h postnucleofection. (E) Primary macrophages were mock infected or infected with 36F MHV68 at an MOI of 10. orf36 and β-actin protein levels were measured at the indicated times postinfection. All error bars represent one standard error of the mean. *, P < 0.05; ***, P < 0.001.
orf36 interacts with HDAC1 and 2.
The finding that orf36 prevented association of HDAC1 and -2 with the distal RTA promoter suggested that orf36 may directly interact with these two HDACs. To determine if orf36 and HDAC1 were present in the same protein complex during lytic infection, primary macrophages were mock infected or infected with wild-type or 36F MHV68, which encodes a Flag-tagged orf36 (27). Lysates were collected at 30 h postinfection and immunoprecipitated with anti-Flag or anti-HDAC1. Immunoprecipitated HDAC1 ran at a higher molecular weight than did HDAC1 in cell lysates, likely due to the proximity of the heavy chain (Fig. 3A). HDAC1 coimmunoprecipitated with anti-Flag in lysates collected from 36F MHV68-infected macrophages. However, HDAC1 was not detected in anti-Flag immunoprecipitates from wild-type- or mock-infected cells (Fig. 3A). Thus, orf36 and HDAC1 were present in the same protein complex in infected macrophages.
Fig 3.

orf36 and BGLF4 interact with HDAC1 and -2. (A) Primary macrophages were infected at an MOI of 10 with wild-type or 36F MHV68 virus for 30 h, and lysates were immunoprecipitated with anti-HDAC1 or anti-Flag antibodies. Immunoprecipitates were probed for HDAC1. (B to D) Radioactively labeled proteins were generated using the in vitro transcription/translation system (TNT). TNT-generated proteins were combined and immunoprecipitated using indicated antibodies (against HDAC1, HDAC2, Flag, or HA tag). Immunoprecipitated complexes were analyzed by SDS-PAGE and autoradiography. Images are representative of at least three independent experiments. Signal in lanes 1 to 5 represents input.
To determine if orf36 could directly interact with HDAC1 and -2 in the absence of infection, radioactively labeled proteins were generated using in vitro transcription/translation system, allowed to interact, and subjected to immunoprecipitation using anti-HDAC1, anti-Flag, or anti-HA antibodies. When orf36 and HDAC1 were coincubated, both proteins were pulled down with either antibody, suggesting that the HDAC1-orf36 association is likely to be direct (Fig. 3B, lanes 6 and 7). This association was not dependent on the enzymatic activity of orf36 (Fig. 3B, lanes 8 and 9) and was specific, as neither HDAC1 nor orf36 bound G protein beads (Fig. 3B, lanes 14 and 15), vaccinia virus-encoded I3 failed to coimmunoprecipitate with HDAC1 (Fig. 3B, lanes 10 and 11), and orf36 failed to interact with an irrelevant cellular protein (Fig. 3B, lanes 12 and 13). Similarly, both wild-type orf36 and the catalytically null 36KN mutant specifically interacted with HDAC2 (Fig. 3C). Thus, orf36 interacted with HDAC1 and -2 in vitro, and this interaction was independent of catalytic activity of the kinase.
EBV BGLF4 interacts with HDAC1 and -2.
To determine the conservation of the viral kinase-HDAC1 interaction, members of the UL13 family of herpesvirus kinases, including U69, encoded by human herpesvirus 7 (HHV-7), BGLF4, encoded by EBV, and orf36, encoded by KSHV, were tested for interaction with HDAC1 in vitro. When the viral kinases and HDAC1 were coincubated and immunoprecipitated with an antibody against HDAC1, both MHV68 orf36 and EBV BGLF4 coimmunoprecipitated with HDAC1 (Fig. 3D, lanes 6 and 7) and HDAC2 (data not shown), suggesting that the interaction between gammaherpesvirus protein kinases and HDAC1 and -2 is not limited to the MHV68 system.
HDAC1 colocalizes with MHV68 single-stranded DNA binding protein (ssDBP) during infection.
HSV-1 protein kinase (US3) relocalizes HDAC1 to the cytoplasm (10). In contrast, HDAC2 colocalizes with nuclear prereplication foci and viral replication compartments in HCMV-infected cells (14). To define the effect of orf36 on HDAC1 nuclear localization, primary macrophages were infected with wild-type or orf36 mutant MHV68 viruses, and subcellular localization of HDAC1 was examined throughout the replication cycle. Similar to that observed in mock-infected macrophages, HDAC1 remained nuclear at all times postinfection (Fig. 4 and data not shown). To determine if HDAC1 was enriched in viral replication compartments, cells were costained with the antibody recognizing MHV68 ssDBP (32). At 16 h postinfection, ssDBP-positive nuclear structures were present in macrophages infected with wild-type or orf36 mutant MHV68 viruses but not in mock-infected controls (Fig. 4). Over the course of the replication cycle, the ssDBP staining progressed from a punctate pattern to large globular domains. While HDAC1 did not exclusively colocalize with ssDBP in the nucleus (r2 = 0.6, P < 0.01), an enrichment of HDAC1 in replication compartments was detected in all infected cells, independent of orf36 expression or catalytic activity (Fig. 4). The pattern of HDAC1 nuclear staining was similar in mock- and MHV68-infected macrophages prior to ssDBP expression (data not shown). Thus, HDAC1 was recruited to MHV68 nuclear replication compartments in an orf36-independent manner.
Fig 4.

HDAC1 colocalizes with viral replication compartments. Primary macrophages were infected at an MOI of 10 with wild-type or orf36 mutant MHV68 viruses. At 16 hpi, cells were fixed and stained with DAPI (4′,6-diamidino-2-phenylindole) and antibodies directed against MHV68 ssDBP and HDAC1. Images were merged, with HDAC1 in green, ssDBP in red, and DAPI in blue. Colocalization was tested using Pearson's correlation coefficient.
orf36 modulates global HDAC activity during lytic infection of primary macrophages.
In spite of a significant number of reports dissecting regulation of individual HDACs within alpha- and betaherpesvirus systems, there is a paucity of information regarding global HDAC activity in infected cells. Of the four defined classes of HDACs, class I and class III (sirtuins) HDACs are thought to be responsible for the majority of global deacetylase activity, whereas class IIa HDACs have lower deacetylase activity and are thought to repress gene expression primarily via interactions with other transcriptional repressors (37). Importantly, the relative contribution of individual HDACs to the overall deacetylase activity in primary macrophages has not been defined. To measure changes in global HDAC activity during gammaherpesvirus lytic replication, primary macrophages were infected with wild-type and orf36 mutant MHV68 viruses at MOIs of 1 and 10, and HDAC activity of total cell lysates was measured at 5, 16, and 30 h postinfection using a commercially available fluorimetric assay. At 5 h postinfection, a time prior to expression of immediate early MHV68 genes (35), global HDAC activity was elevated in N36S-infected compared to wild-type- and 36KN-infected macrophages (P < 0.05; Fig. 5A). These differences in global HDAC enzymatic activity were not a result of increased expression of HDAC1 and -2 (Fig. 5A). Furthermore, measured HDAC activity was susceptible to TSA inhibition, suggesting that TSA-insensitive class III HDACs (sirtuins) were not the major contributors to the overall HDAC enzymatic activity observed in primary macrophages (Fig. 5A).
By 16 h postinfection, concurrent with expression of immediate early and early viral genes and viral DNA synthesis (35), global HDAC activity was similar in all infected conditions and appeared to be slightly elevated above mock-infected levels, although the difference did not reach statistical significance (Fig. 5B). Surprisingly, at 30 h postinfection global HDAC activity remained significantly elevated in wild-type- and 36KN-infected macrophages but not N36S-infected cells (Fig. 5C). The elevated global HDAC activity during later stages of MHV8 replication is consistent with a previous report showing elevated global HDAC activity in HCMV-infected fibroblasts during late stages of lytic replication (14). orf36 was detected in high-MOI-infected macrophages as early as 5 h postinfection (Fig. 5E), consistent with the orf36-dependent effects on total HDAC activity during early and late stages of infection. Thus, orf36, independent of its enzymatic function, regulated overall HDAC activity in infected macrophages.
orf36 overexpression induces global HDAC activity in an enzymatically dependent manner.
To determine if orf36 was sufficient to regulate global HDAC activity, HDAC activity was measured in primary macrophages transiently expressing wild-type orf36 or a catalytically defective orf36 mutant 1 (orf36KN) that has a single amino acid substitution of the same ATP-positioning lysine as the 36KN virus (30). As expected, expression of wild-type, but not catalytically null orf36 increased γH2AX levels in primary macrophages (Fig. 5D). Surprisingly, HDAC activity was found to be elevated in cells expressing wild-type orf36, but not catalytically null 36KN, in spite of similar levels of expression (Fig. 5D; P < 0.05). Furthermore, global HDAC activity was not altered in cells expressing MHV68 RTA, and increased global HDAC activity observed in orf36-expressing macrophages was not due to increased expression of HDAC1 and -2 (Fig. 5D and data not shown). Thus, orf36 expression and catalytic activity were sufficient to increase cellular HDAC activity in primary macrophages.
Depletion of HDAC1 and -2 rescues MHV68 replication in the absence of orf36.
We observed that while global HDAC inhibition with TSA rescued attenuated RTA expression in the absence of catalytically active orf36 (36KN; Fig. 1A), catalytic activity of orf36 was not required to prevent association of HDAC1 and -2 with the distal RTA promoter (Fig. 2A and B), raising the possibility that several HDACs may suppress RTA transcription in the absence of orf36. Because treatment with HDAC inhibitors is associated with inadequate specificity and potential off-target effects, a genetic approach was used to address the specific role of HDACs 1 and 2 in RTA transcription. We took advantage of a previously described mouse system in which HDAC1 and -2 alleles are rendered conditional via the insertion of exon-flanking loxP sites (7) (see schematic in Fig. 6A). These HDAC1/2 conditional mice (HDAC1/2F/F) were crossed to mice with ubiquitous expression of the modified estrogen receptor fused to Cre recombinase (ER-Cre) (29). Because ER-Cre-mediated recombination is induced by 4-hydroxytamoxifen (OHT), this system offers a tractable approach that allows depletion of HDAC1 and 2 in primary cells. Primary macrophages were derived from bone marrow cultures of HDAC1/2F/F × ER-Cre-positive and Cre-negative mice and were treated with OHT on day 4 of in vitro differentiation. Recombined HDAC1 and -2 alleles were detected 8 days after initial OHT treatment (day 12 of culture; Fig. 6B). On average, approximately 84% of genomic HDAC1 alleles were recombined following OHT treatment of Cre-positive compared to OHT treatment of Cre-negative HDAC1/2F/F macrophages (Fig. 6C). Consistent with genomic recombination, on average, OHT treatment reduced HDAC1 and -2 protein levels by 85% and 70%, respectively, in Cre-positive compared to Cre-negative HDAC1/2F/F cells (8 days following OHT treatment; combined data are shown in Fig. 6F, and data from a representative experiment are shown in Fig. 6D and E).
Fig 6.

Conditional depletion of HDAC1 and -2 in primary bone marrow-derived macrophages. (A) Schematic of the ER-Cre-mediated recombination of the conditional HDAC1 and HDAC2 alleles. Numbered boxes represent exons, and circles represent loxP sites for Cre-mediated recombination. Primers are indicated using arrows above the approximate annealing loci. Recombination products after 4-hydroxytamoxifen treatment are depicted below the genomic loci. (B to E) Bone marrow isolated from ER-Cre-positive or -negative mice with conditional HDAC1 and -2 alleles (F/F) was treated with 1 μM 4-hydroxytamoxifen (OHT) or carrier solution at 4 and 7 days of in vitro macrophage differentiation. (B, C) Total DNA was isolated on day 12 of culture, recombined HDAC1 and -2 alleles were detected by PCR (B), and the intact HDAC1 allele was quantified by real-time PCR using primers F and R1 with data pooled from 2 independent experiments (C). (D to F) Cell lysates were harvested on day 12 of culture, and protein levels of HDAC1, HDAC2, and histone H3 were measured by Western analysis (D and E); HDAC1 and -2 protein content was analyzed by densitometry and normalized to H3 (F). Data were pooled from 3 independent experiments.
To identify the effect of combined HDAC1 and 2 depletion on RTA transcription, OHT- or mock-treated HDAC1/2F/F macrophages that were ER-Cre positive or negative were infected at an MOI of 1 with wild-type, N36S, or 36KN MHV68. At 16 h postinfection, RTA expression was measured via qRT-PCR. As expected, macrophages infected with either the N36S or the 36KN mutant exhibited reduced RTA expression in all control infections (Fig. 7A). RTA transcription in infected, HDAC1/2-depleted (ER-Cre-positive, OHT-treated) macrophages was similar to that observed in control cultures, including attenuated RTA expression in the absence of a catalytically active orf36 (Fig. 7A).
Fig 7.

Effects of HDAC1 and -2 depletion on MHV68 replication. (A) Primary macrophages generated and treated as described for Fig. 6 were infected at an MOI of 1 (in the absence of OHT) with wild-type MHV68 and orf36 mutant viruses on day 13 of in vitro culture. RNA was collected at 16 h postinfection, and RTA expression was measured via qRT-PCR and normalized to corresponding GAPDH mRNA levels using the ΔCT method. Data were pooled from 2 to 4 experiments. (B) Primary, OHT-treated macrophages with indicated Cre genotypes were infected at an MOI of 1 with wild-type or N36S MHV68 as described for panel A, and viral yield was measured at the indicated times postinfection. (C) Primary, OHT-treated macrophages with the indicated Cre genotypes were infected at an MOI of 10, and viral DNA accumulation was measured at the indicated time points by real-time PCR. *, P < 0.05. Data are representative of three independent experiments. Error bars represent a standard error of the mean.
Having observed similar RTA expression in HDAC1- and HDAC2-depleted and control macrophages, we wished to investigate the extent to which HDAC1 and -2 affect MHV68 replication, as orf36 expression and activity are required for efficient MHV68 replication in primary macrophages (30, 32). Primary macrophages were derived from HDAC1/2F/F ER-Cre-positive or -negative littermates and treated with OHT to induce recombination. Subsequently, macrophages were infected at an MOI of 1, and virus yield was assessed at 72 h postinfection. As expected, growth of the N36S virus was attenuated in Cre-negative macrophages (Fig. 7B). Intriguingly, the attenuated replication of the N36S virus mutant was rescued in HDAC1- and HDAC2-depleted macrophages (Fig. 7B). A similar phenotype was observed at a high MOI (MOI of 10; data not shown). Thus, HDAC1 and -2 depletion rescued attenuated MHV68 replication in the absence of orf36, despite having very little effect on RTA transcription at 16 h postinfection.
The N36S virus mutant displays a significant attenuation in viral DNA synthesis at both high and low MOIs (32). Because RTA expression was not rescued in N36S-infected HDAC1- and HDAC2-depleted macrophages (Fig. 7A) in spite of rescued N36S replication, accumulation of viral DNA was assessed in HDAC1- and HDAC2-depleted or control macrophages. As expected (32), viral DNA synthesis was significantly attenuated in control, Cre-negative macrophages infected with the N36S mutant MHV68 (Fig. 7C). Interestingly, accumulation of viral DNA was similar in HDAC1- and HDAC2-depleted macrophages infected with either wild-type or N36S MHV68 (Fig. 7C, Cre+), suggesting that HDAC1 and -2 counteract viral DNA synthesis and orf36 alleviates this inhibitory effect in infected macrophages.
DISCUSSION
In this study, we identify orf36, a conserved gammaherpesvirus protein kinase, as a regulator of HDAC1 and -2 during de novo lytic infection of primary macrophages. orf36 interacted with HDAC1 and -2 and prevented association of these two enzymes with the distal RTA promoter, independent of orf36 enzymatic activity. While attenuated RTA transcription in the absence of a catalytically active orf36 was rescued by TSA, a global HDAC inhibitor, combined depletion of HDAC1 and -2 failed to rescue attenuated RTA transcription in macrophages infected with orf36 mutant viruses (Fig. 7A). These and other data demonstrating catalytic activity-independent regulation of HDAC1 and -2 by orf36 suggest that, in addition to HDAC1 and -2, orf36 is likely to target other HDACs (Fig. 8). Importantly, we found that attenuated replication of the N36S mutant was rescued in HDAC1- and HDAC2-depleted primary macrophages, indicating that the regulation of HDAC1 and -2 by orf36 is critical to ensure optimal MHV68 replication. Interestingly, while HDAC1 and -2 depletion failed to rescue RTA expression in N36S infections, attenuated viral DNA synthesis in the absence of orf36 was rescued in HDAC1- and HDAC2-depleted macrophages (Fig. 7C). This suggests that, in the context of de novo lytic infection, orf36 interaction with HDAC1 and 2 has evolved to counteract inhibitory effects of these HDACs on viral DNA replication.
Fig 8.

Working model. orf36 targets HDAC1 and 2 to facilitate MHV68 replication. At the core RTA promoter, orf36, in an enzymatic activity-dependent manner, induces phosphorylation of H2AX. At the distal promoter, orf36 inhibits HDAC1 and -2 association with this RTA promoter independent of the viral kinase enzymatic activity. However, orf36 enzymatic activity is likely necessary to also target as-yet-unidentified HDAC in order to enhance RTA promoter activity. In addition to directly regulating RTA promoters, orf36 counteracts HDAC1 and -2 to promote viral DNA synthesis and MHV68 replication in primary macrophages.
Gammaherpesvirus kinases as regulators of lytic infection.
All gammaherpesviruses encode a protein kinase that supports lytic gene expression, DNA replication, nuclear egress, and reactivation from latency (28, 32, 38–41). However, the role of gammaherpesvirus kinase in the context of de novo lytic infection has not been fully appreciated, in spite of the fact that gammaherpesvirus kinases are virion components (42). In this study, orf36 was found to regulate HDACs, a role shared with other herpesvirus virion-associated regulators of HDAC-containing complexes, such as HCMV pp71 and pUL29/28 (15, 43). The conserved nature of HDAC regulation by herpesvirus kinases is further highlighted in an accompanying article by Bigley et al. (44). Thus, we propose the following model based on published and current results (Fig. 8). orf36 regulates early events in de novo lytic infection by differentially targeting the two known RTA promoters. In doing so, the kinase taps into two major signaling networks of the host. At the core promoter, orf36 enzymatic activity induces γH2AX, a marker of the DNA damage response (32). At the distal RTA promoter, orf36, independent of its enzymatic activity, prevents association of HDAC1 and -2, two important transcriptional repressors. Regulation of HDACs by orf36 is not limited to HDAC1 and -2, based on the phenotype of the catalytically null orf36 mutant and the TSA phenotype (Fig. 1 and 2), suggesting that orf36-mediated phosphorylation of an unknown HDAC also supports RTA expression. Thus, orf36 is poised as a master HDAC regulator, due to its potential role in counteracting multiple HDACs at the RTA promoters, a role to be explored in future studies.
While effects of orf36 on RTA transcription are MOI dependent, viral DNA synthesis remains attenuated in the absence of orf36, regardless of the MOI (32). We have found that while HDAC1 and -2 depletion did not rescue attenuated RTA expression, both viral replication and viral DNA synthesis were rescued in the absence of orf36 when HDAC1 and 2 expression was decreased (Fig. 7C and 8). This suggests that, in spite of many viral processes that may be regulated by gammaherpesvirus protein kinases, the critical bottleneck for virus replication is the inability to efficiently synthesize viral DNA in the absence of orf36. However, it is also possible that the ability of orf36 to stimulate RTA expression becomes paramount in situations other than de novo infection (i.e., during virus reactivation), a possibility to be explored in future studies.
In addition to its regulation of HDAC1 and -2, the MHV68 kinase usurps two other important cellular systems. orf36 is responsible for induction of γH2AX, a marker of an activated DNA damage response in infected cells, and attenuation of beta interferon transcription via interaction with interferon regulatory factor 3 (IRF-3) (27, 30). Importantly, the ability of MHV68 orf36 to interact with HDAC1 and -2, induce γH2AX, and target IRF-3 is shared with EBV-encoded BGLF4 (references 30 and 45 and this study), emphasizing the conserved nature of gammaherpesvirus protein kinase biology that transcends across species. An interesting puzzle that remains to be solved is the interplay between the orf36-targeted cellular networks in infected cells and the effect of these interactions on RTA expression and viral replication.
The abilities of orf36 to repress beta interferon transcription and regulate HDACs are independent of orf36 enzymatic activity, highlighting the multifunctional nature of this viral protein. Because of important roles in viral replication and latency, herpesvirus protein kinases are attractive therapeutic targets. Maribavir, an inhibitor of HCMV-encoded protein kinase, is a prototype drug developed to control HCMV infections in immunocompromised patients (46). Unfortunately, maribavir failed to meet its efficacy endpoints in the initial phase 3 clinical trial (47). Numerous factors could have contributed to the lack of maribavir efficacy, including the design of the clinical protocol. However, it is also clear that there is a need for better herpesvirus kinase inhibitors that specifically target both enzymatic and nonenzymatic functions of herpesvirus kinases.
Gammaherpesviruses and histone deacetylases.
HDAC1 and -2 are classically considered to repress transcription and are thus targeted by several alpha- and betaherpesvirus proteins (9, 10, 14, 15, 48, 49). The identity of a gammaherpesvirus protein that regulates HDACs in lytically infected cells has remained elusive until recently, when KSHV-encoded K-bZIP was shown to regulate RTA expression via targeting HDAC 1 and -2 (16). Paradoxically, K-bZIP promotes recruitment of HDAC1 to the RTA promoter. Intriguingly, KSHV protein kinase orf36 associates with K-bZIP and regulates its sumoylation (50). However, it is not known if KSHV protein kinase also interferes with the recruitment of HDACs to the KSHV RTA promoters. In the MHV68 system, HDACs 3 and 4 associate with the core RTA promoter and repress transcription in a retinoic acid-dependent manner (17). Intriguingly, the same study showed that small interfering RNA (siRNA) directed against HDAC1 or -2 had no effect on wild-type MHV68 replication, consistent with the role of orf36 as a regulator of HDAC1 and -2.
It is intriguing that, in spite of clearance of HDAC1 and -2 from the distal RTA promoters in 36KN-infected cells (Fig. 2A and B), RTA transcription remains attenuated and can be rescued by global inhibition of HDACs (Fig. 1A), but not HDAC1 and -2 depletion (Fig. 7A). A trivial explanation could attribute this phenotype to incomplete depletion of HDAC1 and -2 or to off-target effects of TSA. However, a more intriguing possibility is that orf36 regulates another cellular HDAC and this regulation is dependent on the orf36 enzymatic activity. Herpesviruses target several HDACs and their complexes, including class II HDACs. HSV-1-encoded ICP0, in addition to its role in antagonizing HDAC1-containing CoREST complexes (9), interacts with class II HDACs 4, 5, and 7 to relieve myocyte enhancer factor 2D (MEF2D)-mediated repression of viral promoters (51). Additionally, a complex containing HDACs 4 and 5 is recruited to the BZLF1 promoter of EBV (52). Intriguingly, phosphorylation is responsible for negative regulation of HDACs 4 and 5 (53). Thus, targeting of additional HDACs by orf36 is an important area of future studies.
Aside from orf36's potential role as a master HDAC regulator, this study establishes the physiological relevance of HDAC1/2-orf36 interaction, as replication of the orf36 null MHV68 mutant (N36S) was rescued in macrophages depleted of HDAC1 and -2 (Fig. 7B). This rescue occurred in spite of attenuated RTA expression in N36S- and 36KN-infected macrophages (Fig. 7A). Intriguingly, we found that HDAC1 and -2 depletion rescued viral DNA synthesis in the absence of orf36, suggesting that HDAC1 and -2 counteract viral DNA replication and orf36 inhibits such effects. HDAC1 and -2 are well-known regulators of gene expression; thus, they could inhibit viral DNA synthesis indirectly, by regulating host gene expression. However, we have also observed that HDAC1 and -2 localize to the MHV68 replication compartments (Fig. 4 and data not shown), suggesting an intriguing possibility that these HDACs may directly inhibit viral DNA synthesis by deacetylating key viral or cellular proteins. These two nonexclusive mechanisms are currently being explored.
This study demonstrates that HDAC1 and -2 are physiologically relevant targets of MHV68 protein kinase orf36. While global HDAC inhibitors induce gammaherpesvirus reactivation from tumor cell lines and in vivo (17–19), the role of individual HDACs in the regulation of chronic gammaherpesvirus infection is not defined. Pan-HDAC inhibition has emerged as a promising therapeutic for lymphomas and solid tumors, and HDAC inhibitors with increased specificity against individual HDACs are currently undergoing clinical trials as cancer therapy agents (54). Because of the high seroprevalence of gammaherpesvirus infection in humans, defining the regulation of specific HDACs during chronic gammaherpesvirus infection is another important focus of future studies.
ACKNOWLEDGMENTS
Expert technical assistance was provided by Brittani Wood. We are grateful to Eric Olson for his generous gift of HDAC1/2 conditional mice and HDAC cDNA. We thank Amy Hudson's, William Jackson's, and Lisa Cirillo's laboratories for helpful discussions. Reagents were kindly provided by Ren Sun, Paula Traktman, Amy Hudson, and Joseph Barbieri.
This work was supported by Advancing a Healthier Wisconsin and 1R56AI084889 (V.L.T.). The funding sources had no involvement in the studies.
Footnotes
Published ahead of print 24 April 2013
REFERENCES
- 1. Nusinzon I, Horvath CM. 2003. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc. Natl. Acad. Sci. U. S. A. 100:14742–14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sakamoto S, Potla R, Larner AC. 2004. Histone deacetylase activity is required to recruit RNA polymerase II to the promoters of selected interferon-stimulated early response genes. J. Biol. Chem. 279:40362–40367 [DOI] [PubMed] [Google Scholar]
- 3. Peng L, Seto E. 2011. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb. Exp. Pharmacol. 206:39–56 [DOI] [PubMed] [Google Scholar]
- 4. Haigis MC, Sinclair DA. 2010. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5:253–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Witt O, Deubzer HE, Milde T, Oehme I. 2009. HDAC family: what are the cancer relevant targets? Cancer Lett. 277:8–21 [DOI] [PubMed] [Google Scholar]
- 6. Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. 2009. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459:55–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. 2007. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21:1790–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van ON, Favoreel H, Van de Walle GR. 2012. Histone modifications in herpesvirus infections. Biol. Cell 104:139–164 [DOI] [PubMed] [Google Scholar]
- 9. Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A. 104:17134–17139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poon AP, Gu H, Roizman B. 2006. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. U. S. A. 103:9993–9998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walters MS, Erazo A, Kinchington PR, Silverstein S. 2009. Histone deacetylases 1 and 2 are phosphorylated at novel sites during varicella-zoster virus infection. J. Virol. 83:11502–11513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walters MS, Kinchington PR, Banfield BW, Silverstein S. 2010. Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J. Virol. 84:9666–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nevels M, Paulus C, Shenk T. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. U. S. A. 101:17234–17239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park JJ, Kim YE, Pham HT, Kim ET, Chung YH, Ahn JH. 2007. Functional interaction of the human cytomegalovirus IE2 protein with histone deacetylase 2 in infected human fibroblasts. J. Gen. Virol. 88:3214–3223 [DOI] [PubMed] [Google Scholar]
- 15. Terhune SS, Moorman NJ, Cristea IM, Savaryn JP, Cuevas-Bennett C, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog. 6:e1000965. 10.1371/journal.ppat.1000965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martinez FP, Tang Q. 2012. Leucine zipper domain is required for Kaposi sarcoma-associated herpesvirus (KSHV) K-bZIP protein to interact with histone deacetylase and is important for KSHV replication. J. Biol. Chem. 287:15622–15634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goodwin MM, Molleston JM, Canny S, Abou EH, Willert EK, Bremner R, Virgin HW. 2010. Histone deacetylases and the nuclear receptor corepressor regulate lytic-latent switch gene 50 in murine gammaherpesvirus 68-infected macrophages. J. Virol. 84:12039–12047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luka J, Kallin B, Klein G. 1979. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 94:228–231 [DOI] [PubMed] [Google Scholar]
- 19. Yang Z, Tang H, Huang H, Deng H. 2009. RTA promoter demethylation and histone acetylation regulation of murine gammaherpesvirus 68 reactivation. PLoS One 4:e4556. 10.1371/journal.pone.0004556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD, Gompels UA. 1990. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J. Gen. Virol. 71:1365–1372 [DOI] [PubMed] [Google Scholar]
- 21. Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu S, Pavlova IV, Virgin HW, Speck SH. 2000. Characterization of gammaherpesvirus 68 gene 50 transcription. J. Virol. 74:2029–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312 [DOI] [PubMed] [Google Scholar]
- 24. Sun R, Lin SF, Gradoville L, Yuan Y, Zhu FX, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu TT, Tong L, Rickabaugh T, Speck S, Sun R. 2001. Function of Rta is essential for lytic replication of murine gammaherpesvirus 68. J. Virol. 75:9262–9273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu TT, Usherwood EJ, Stewart JP, Nash AA, Sun R. 2000. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J. Virol. 74:3659–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hwang S, Kim KS, Flano E, Wu TT, Tong LM, Park AN, Song MJ, Sanchez DJ, O'Connell RM, Cheng G, Sun R. 2009. Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe 5:166–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tarakanova VL, Stanitsa E, Leonardo SM, Bigley TM, Gauld SB. 2010. Conserved gammaherpesvirus kinase and histone variant H2AX facilitate gammaherpesvirus latency in vivo. Virology 405:50–61 [DOI] [PubMed] [Google Scholar]
- 29. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. 2007. Restoration of p53 function leads to tumour regression in vivo. Nature 445:661–665 [DOI] [PubMed] [Google Scholar]
- 30. Tarakanova VL, Leung-Pineda V, Hwang S, Yang C-W, Matatall K, Basson M, Sun R, Piwnica-Worms H, Sleckman BP, Virgin HW. 2007. Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1:275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tarakanova VL, Molleston JM, Goodwin M, Virgin HW, IV 2010. MHV68 complement regulatory protein facilitates MHV68 replication in primary macrophages in a complement independent manner. Virology 306:323–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mounce BC, Tsan FC, Droit L, Kohler S, Cirillo LA, Tarakanova VL. 2011. Gammaherpesvirus gene expression and DNA synthesis are facilitated by viral protein kinase and histone variant H2AX. Virology 420:73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224:213–232. 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 34. Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mounce BC, Tsan FC, Kohler S, Cirillo LA, Tarakanova VL. 2011. Dynamic association of gammaherpesvirus DNA with core histone during de novo lytic infection of primary cells. Virology 421:167–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gray KS, Allen RD, Farrell ML, Forrest JC, Speck SH. 2009. Alternatively initiated gene 50/RTA transcripts expressed during murine and human gammaherpesvirus reactivation from latency. J. Virol. 83:314–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haberland M, Montgomery RL, Olson EN. 2009. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10:32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang PC, Fitzgerald LD, Van Geelen A, Izumiya Y, Ellison TJ, Wang DH, Ann DK, Luciw PA, Kung HJ. 2009. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi's sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 69:5681–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gershburg E, Raffa S, Torrisi MR, Pagano JS. 2007. Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J. Virol. 81:5407–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee CP, Huang YH, Lin SF, Chang Y, Chang YH, Takada K, Chen MR. 2008. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 82:11913–11926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meng Q, Hagemeier SR, Kuny CV, Kalejta RF, Kenney SC. 2010. Simian virus 40 T/t antigens and lamin A/C small interfering RNA rescue the phenotype of an Epstein-Barr virus protein kinase (BGLF4) mutant. J. Virol. 84:4524–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A. 101:16286–16291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bigley TM, Reitsma JM, Mirza SP, Terhune SS. 2013. Human cytomegalovirus pUL97 regulates the viral major immediate early promoter by phosphorylation-mediated disruption of histone deacetylase 1 binding. J. Virol. 87:7393–7408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang JT, Doong SL, Teng SC, Lee CP, Tsai CH, Chen MR. 2009. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 83:1856–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prichard MN. 2009. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev. Med. Virol. 19:215–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, Young JA, Rodriguez T, Maertens J, Schmitt M, Einsele H, Ferrant A, Lipton JH, Villano SA, Chen H, Boeckh M. 2011. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial. Lancet Infect. Dis. 11:284–292 [DOI] [PubMed] [Google Scholar]
- 48. Du T, Zhou G, Khan S, Gu H, Roizman B. 2010. Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. Proc. Natl. Acad. Sci. U. S. A. 107:15904–15909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou G, Te D, Roizman B. 2011. The CoREST/REST repressor is both necessary and inimical for expression of herpes simplex virus genes. mBio 2:e00313–10. 10.1128/mBio.00313-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Izumiya Y, Izumiya C, Van Geelen A, Wang DH, Lam KS, Luciw PA, Kung HJ. 2007. Kaposi's sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J. Virol. 81:1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lomonte P, Thomas J, Texier P, Caron C, Khochbin S, Epstein AL. 2004. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J. Virol. 78:6744–6757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gruffat H, Manet E, Sergeant A. 2002. MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 3:141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martin M, Kettmann R, Dequiedt F. 2007. Class IIa histone deacetylases: regulating the regulators. Oncogene 26:5450–5467 [DOI] [PubMed] [Google Scholar]
- 54. Venugopal B, Evans TR. 2011. Developing histone deacetylase inhibitors as anti-cancer therapeutics. Curr. Med. Chem. 18:1658–1671 [DOI] [PubMed] [Google Scholar]