

The structure of the cellulose-binding module of the C. thermocellum cellulosome-integrating protein A scaffoldin with portions of the C- and N-terminal flanking linker regions has been determined at 1.98 Å resolution.
Keywords: family 3 cellulose-binding module, CipA, Clostridium thermocellum, cellulosome
Abstract
The cellulosome of the cellulolytic bacterium Clostridium thermocellum has a structural multi-modular protein called CipA (cellulosome-integrating protein A) that includes nine enzyme-binding cohesin modules and a family 3 cellulose-binding module (CBM3a). In the CipA protein, the CBM3a module is located between the second and third cohesin modules and is connected to them via proline/threonine-rich linkers. The structure of CBM3a with portions of the C- and N-terminal flanking linker regions, CBM3a-L, has been determined to a resolution of 1.98 Å. The structure is a β-sandwich with a structural Ca2+ ion. The structure is consistent with the previously determined CipA CBM structure; however, the structured linker regions provide a deeper insight into the overall cellulosome structure and assembly.
1. Introduction
Cellulose, the major structural component in plants and the most abundant carbohydrate on the planet, is insoluble and resistant to hydrolysis. In nature, cellulolytic organisms have developed a number of strategies to efficiently degrade cellulose, one of which is a complex extracellular multi-enzyme system of cellulosomes (Lamed & Bayer, 1988 ▶; Bayer et al., 2008 ▶). The cellulosome of the anaerobic thermophilic bacterium Clostridium thermocellum has a structural multi-domain protein called CipA (cellulosome-integrating protein A), a scaffoldin that includes nine enzyme-binding cohesin modules and a family 3 cellulose-binding module (CBM3a). The first step in the cellulose-degradation process is binding of the cellulosome enzyme complex to the substrate cellulose: this is the role of CBM3a.
The CBM3a of CipA from C. thermocellum is located between cohesin 2 and cohesin 3 and is separated from these domains by linker regions rich in threonine and proline residues (Figs. 1 ▶ and 2 ▶). In accordance with the current annotation of the CipA gene (Swiss-Prot Q06851.2), the full-length linker from Coh2 to CBM3a is 46 residues in length and that from CBM3a to Coh3 is 39 residues in length. The known CBM3a protein structure (Tormo et al., 1996 ▶) is based upon the current annotation and does not include any of the residues designated as flanking linker regions (Fig. 2 ▶). The cloning of the latter protein from which the structure was determined was originally performed before any structural information on family 3 CBMs was known and was chosen on the basis of sequence alignments with CBMs from other bacteria and on the basis of predicted secondary structure. In order to gain deeper insight into the entire CipA structure, we have expressed the CipA CBM with extended linker regions (CBM3a-L) and determined its structure. In the present communication, we describe the crystallization and X-ray structure analysis of C. thermocellum CBM3a-L.
Figure 1.

Schematic representation of the multi-domain structural protein CipA from the cellulosome of C. thermocellum. There are nine highly homologous cohesin modules; the CBM3a is between the second and third cohesins, flanked by linker regions.
Figure 2.

The entire linker sequences from the Coh2 module to CBM3a and from CBM3a to the Coh3 module are shown in lower case. The linker residues expressed for this study are shown in grey. The linker residues and CBM3a observed in the CBM3a-L structure are shown in bold.
2. Materials and methods
2.1. Protein production
A DNA fragment encoding CBM3a, corresponding to residues 323–540 of the full-length CipA cellulosomal scaffoldin (GenBank accession No. CCV01464.1), was amplified by PCR from C. thermocellum ATCC 27405 genomic DNA isolated as described by Murray & Thompson (1980 ▶) using the primers 5′-GAGCTGGATCCATGGGGAATGCAACACCGACCAAG-3′ and 5′-GTACGCGGATCCTGTTGTTCGAGGTGGTGT-3′. The PCR product was inserted into pET-28a(+) (Novagen, Madison, Wisconsin, USA) via NcoI and BamHI (restriction sites are shown in bold in the primer sequences) to generate the expression plasmid.
The plasmid encoding recombinant CBM3a-L (molecular mass 23 268.4 Da) was cloned into Escherichia coli BL21 (DE3) cells using T7 polymerase high-expression vector. The cell culture was grown under aerobic conditions at 310 K to an A 600 of 0.6. Isopropyl β-d-1-thiogalactoside (IPTG) was added to a final concentration of 5 mM to induce gene expression, and cultivation was continued for an additional 16 h.
After sonication, the expressed polypeptide was purified by taking advantage of its inherent affinity for cellulose. The supernatant fluid was incubated with cellulose (microcrystalline cellulose, type 50; Sigma, St Louis, Missouri, USA) for 1 h with gentle stirring at 277 K. The cellulose pellet was recovered by centrifugation and was washed three times with 1 M sodium bicarbonate and three times with 200 mM sodium bicarbonate. The resulting cellulose pellet was resuspended in 1%(w/v) aqueous triethylamine solution to elute the CBM protein, and the cellulose powder was removed by centrifugation. The eluate was neutralized to pH 7.5 using 1 M Tris–HCl. Protein purity was evaluated by SDS–PAGE.
2.2. Crystallization, data collection and processing
The protein was crystallized at 291 K by hanging-drop vapour diffusion using Linbro 24-well plates. The drops consisted of 2 µl protein solution mixed with 2 µl reservoir solution. A single crystal of CipA CBM with long linkers (CBM3a-L) was obtained using the reservoir composition 1.275 M ammonium sulfate, 0.085 M Tris pH 8.5, 25.2%(v/v) glycerol. The cube-shaped crystal was discovered on a final examination before the crystallization plate was to be discarded, more than a year after the drops were set up. The crystal only grew after regular inspection of the plates had been abandoned approximately four months after initial seeding and reached a size of 0.15 mm in all directions. A thick skin had formed over the drop, which is most likely to be the reason why the drop had not dried out completely by this time. The crystal was transferred to Hampton Research Paratone oil prior to being flash-cooled in a 100 K nitrogen cold stream using an Oxford Crysostream apparatus in situ on the beamline.
X-ray diffraction data were collected on beamline ID14-4 at the ESRF, Grenoble using an ADSC Q315 area detector. Data were collected as 0.5° frames with 2 s exposure at a crystal-to-detector distance of 301.02 mm. The beam of 0.1 mm cross-section was attenuated by a factor of three. Data processing with DENZO and SCALEPACK as implemented in HKL-2000 (v. 0.98.704o; Otwinowski & Minor, 1997 ▶) revealed that the crystal was cubic, with space group P4132 and unit-cell parameters a = b = c = 108.81 Å. Data-rejection criterion were not applied with the exception of internal default decisions implemented in SCALEPACK. It was estimated using unit-cell parameters and the amino-acid sequence (Kantardjieff & Rupp, 2003 ▶) that there was one molecule of CBM3a-L in the asymmetric unit, corresponding to a Matthews coefficient of 3.1 Å3 Da−1 (Matthews, 1968 ▶) and a solvent content of 60.35%. A decision on the resolution cutoff was made utilizing CC1/2 and CC* statistics (0.523 and 0.829, respectively, for the outer shell; Karplus & Diederichs, 2012 ▶). 5% of the reflections were marked for further use in R free calculations. Crystal parameters and data-collection statistics are given in Table 1 ▶.
Table 1. X-ray data-collection and structure-refinement statistics for CipA CBM3a-L.
Values in parentheses are for the outer resolution shell.
| Resolution range (Å) | 29.13–1.98 (2.01–1.98) |
| Space group | P4132 |
| Unit-cell parameters (Å) | a = b = c = 108.81 |
| Total reflections | 371850 |
| Unique reflections | 16030 (771) |
| Multiplicity | 23.2 (23.1) |
| Completeness (%) | 100.0 (100.0) |
| Mean I/σ(I) | 34.40 (1.12) |
| Mosaicity range (°) | 0.35–0.63 |
| Wilson B factor (Å2) | 28.07 |
| R merge † | 0.116 (0.582) |
| Reflections for refinement | 15806 |
| Reflections in test set | 790 |
| R work | 0.1720 (0.2470) |
| R free | 0.2150 (0.3452) |
| No. of atoms | |
| Total | 2721 |
| Protein | 1328 |
| Ligands | 32 |
| Water | 92 |
| No. of protein residues | 170 |
| R.m.s.d., bonds (Å) | 0.012 |
| R.m.s.d., angles (°) | 1.17 |
| Ramachandran favoured (%) | 98.0 |
| Ramachandran outliers (%) | 0.0 |
| Clashscore | 0.74 |
| Average B factors (Å2) | |
| Overall | 33.40 |
| Macromolecules | 32.40 |
| Ligands | 61.60 |
| Solvent | 37.10 |
R
merge = 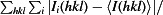
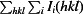 , where
, where 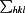 denotes the sum over all reflections and
denotes the sum over all reflections and 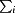 denotes the sum over all equivalent and symmetry-related reflections (Stout & Jensen, 1968 ▶).
denotes the sum over all equivalent and symmetry-related reflections (Stout & Jensen, 1968 ▶).
2.3. Structure determination and refinement
A molecular-replacement search using the program MOLREP (Vagin & Teplyakov, 2010 ▶) with the structure of CipA CBM (PDB entry 1nbc; Tormo et al., 1996 ▶) as a search model was completed, yielding a clear solution with one molecule per asymmetric unit and a CC of 0.621. The structure was initially refined using REFMAC (v.5.7.0032; Murshudov et al., 2011 ▶) and final cycles of refinement against intensities were performed with PHENIX (v.1.8.2-1316; Adams et al., 2011 ▶). One set of TLS parameters was refined for the entire molecule. H atoms were refined in riding mode. The inclusion of TLS parameters and H atoms in the refinement was supported by an improvement in R work and R free. Model building was performed using Coot (v.0.7.1; Emsley et al., 2010 ▶). Although no Ca2+ ions were added to either the purification or the crystallization, as in the case of Tormo and coworkers a large peak corresponding to a Ca2+ ion was clearly visible in the electron-density maps. The final refinement statistics are given in Table 1 ▶.
3. Results and discussion
The structure of CBM3a-L was determined by molecular replacement using CipA CBM3a (PDB entry 1nbc) as a search model. Overall, CBM3a-L forms an antiparallel β-sandwich of nine strands arranged in two sheets with a Ca2+ ion stabilizing the fold. The two sheets are designated ‘top’ for strands 5–6–3–8–9 and ‘bottom’ for strands 1–2–7–4. Cellulose binding takes place via interactions with the ‘bottom’ sheet (Tormo et al., 1996 ▶). In the final structure, three Cl atoms, two sulfate ions and one glycerol molecule (originating from the crystallization solution) were detected and refined. One molecule interpreted as an imidazole of unknown origin was found on a twofold axis and was refined.
The current molecule and the search model only deviate significantly in the region of the C- and N-termini (Figs. 3 ▶ and 4 ▶) and superimpose with an r.m.s.d. of 0.358 Å calculated on 147 Cα-atom positions. Electron density is visible for six additional residues in the N-terminal portion and nine additional residues in the C-terminal portion of the linker residues. Of the total cloned sequence, the first 39 residues and the last nine residues are not seen in the electron-density maps (Fig. 2 ▶).
Figure 3.

Stereoview of the superposition of the Cα traces of CBM3a-L in red and CBM3a in blue (PDB entry 1nbc). The C- and N-termini of both molecules are labelled. The Ca2+ ion is shown as a sphere. The cellulose-binding amino-acid residues of the planar strip are shown as sticks.
Figure 4.

Stereoview of the superposition of the Cα traces of CBM3a-L in red and CBM3a in blue showing the C- and N-termini forming the beginning of a stem-like structure distal to the cellulose-binding strip of amino-acid residues. The Ca2+ ion is shown as a sphere. The cellulose-binding amino-acid residues of the planar strip are shown as sticks (C atoms coloured grey). The view is rotated approximately 90° about the vertical axis from the view in Fig. 3 ▶.
The previous structure of CBM3a without linkers ended at Pro155 (Tormo et al., 1996 ▶). This residue corresponds to Pro200 in the present structure. The van der Waals interactions that these proline residues make with Thr85 and Thr130, respectively, stabilize the C-termini in the case of PDB entry 1nbc (Tormo et al., 1996 ▶) and cause a turn in the C-terminal extension in the current structure. The last C- and N-terminal residues of the previous structure are positioned at exactly the place where the CBM3a-L Cα backbone changes direction. The C- and N-termini of the previous CBM3a are 21.7 Å apart and exit the main protein module heading in opposite directions (Fig. 3 ▶). However, in the current structure the two ends of CBM3a-L are close in space and exit the module as a stem-like structure (Fig. 4 ▶). There are neighbouring symmetry-related molecules in this region; however, we can rule out an influence of crystal-packing contacts on stem formation (see Supplementary Material1).
In CBM3a-L a segment of the additional C-terminal linker extends the β-sandwich fold (Figs. 3 ▶ and 4 ▶ and Supplementary Material). The six residues from Lys198 to Ser203 form a loop that is stabilized by hydrogen bonds from the main-chain O atom of Gln199 to the main-chain N atom of Gly202 and from the main-chain N atom of Gln199 to the main-chain O atom of Ser203 (Fig. 5 ▶). The C-terminal linker sequence begins to diverge from the main protein fold at residue Val205, and the last four visible C-terminal residues approach the N-terminus. The C- and N-termini of the present structure are approximately 7 Å apart; the two ends interact with each other in an antiparallel manner via van der Waals interactions.
Figure 5.

View of the C-termini-stabilizing interactions in CBM3a-L. Hydrogen bonds are shown as dashed lines. The Cα backbone changes direction at Pro200 and makes van der Waals interactions with Thr130.
As discussed above, the C- and N-termini in the current structure approach each other in space and form a stem-like structure. At the C-terminus, the main chain changes direction towards the N-terminus after Pro200 and folds back onto β-strand 9 (see the topography diagram in the Supplementary Material) owing to its interactions with residue Thr130 and the loop between β-strands 5 and 6 and not owing to any contact at the crystal-packing interface. The change in direction at the N-terminus takes place at residue Asn46; this residue is close to a crystal-packing interface and makes a hydrogen bond to Thr97 from a symmetry-related molecule. While it is conceivable that the direction of the N-terminus could be affected by the proximity of a symmetry-related molecule, superposition with related CBMs (PDB entries 1g43 and 2xbt; Shimon et al., 2000 ▶; Yaniv et al., 2011 ▶) shows that the trace of the main chain is consistent with these other structures (Supplementary Fig. S1). We are able to rule out an influence of crystal-packing contacts on the conformation of the termini.
4. Summary
The present molecule, with its ordered flanking region, has implications for the structure and organization of the complete CipA scaffoldin. The C- and N-termini are more structured than previously considered, and some of these residues may actually be considered as part of the CBM3a-L molecular fold. Outside the molecular fold, 44 linker residues remain between the Coh2 and CBM modules and 34 remain between the CBM and Coh3 modules. These linkers are rich in proline and threonine residues, the latter of which are known to be heavily glycosylated (Gerwig et al., 1993 ▶). The presence of glycosylation is known to limit the flexibility of proline/threonine-rich linker regions (Poon et al., 2007 ▶). Moreover, glycosylated linkers have been shown to adopt more extended or elongated geometries. From the location and geometry of the C- and N-terminal stem linkers as they exit the molecule (distal to the cellulose-binding region) and assuming an extended conformation arising from glycosylation, the maximum distance between cohesin module 2 (Shimon, Bayer et al., 1997 ▶; Shimon, Frolow et al., 1997 ▶) and cohesin module 3, and the cellulose-binding strip of CBM3a-L is estimated to be approximately 70 Å.
The structure of CBM3a-L thus provides novel and significant insight into the overall architecture of the C. thermocellum cellulosome. Most importantly, the current structure suggests that CBM3a does not interact laterally with the cohesin modules but rather that the modules are structurally independent (Fig. 6 ▶). The cohesin modules of the CipA molecule are tethered outside an autonomous CBM-stem structure, allowing the cellulosome more freedom to interact in space with the cellulose substrate.
Figure 6.
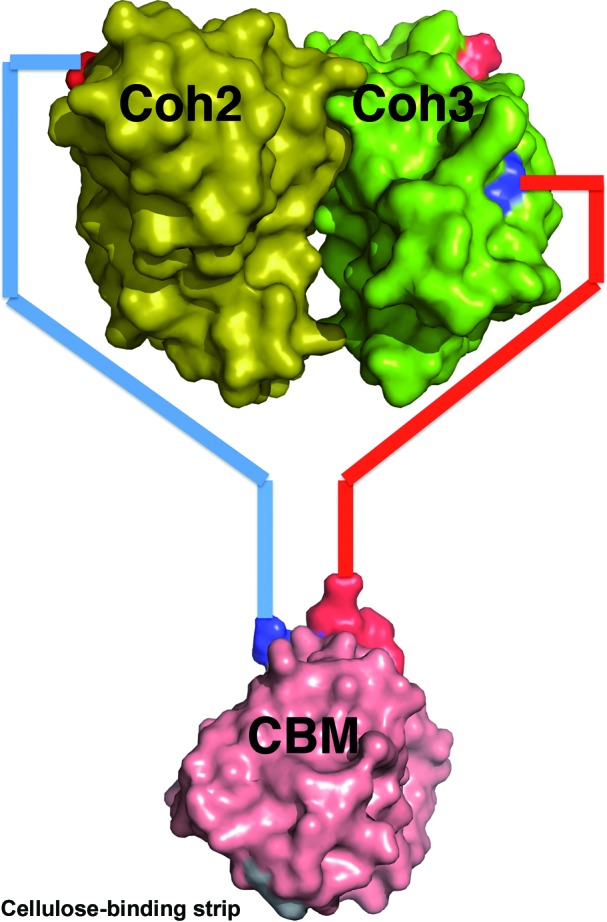
Schematic view of the possible relative positions and connectivity of Coh2–CBM–Coh3. CBM3a-L is in pink, the Coh2 module is in yellow and the Coh3 module is in green. The N- and C-termini of all modules are shown in blue and red, respectively. The cohesin dimer model is taken from the crystallographic dimer observed in the C. thermocellum Coh2 structure (PDB entry 1anu; Shimon, Bayer et al., 1997 ▶). The asymmetric linker lengths (44 and 34) allow the difference in distance of the N- and C-termini of the cohesin modules to be accommodated.
Supplementary Material
PDB reference: carbohydrate-binding module from the cellulosomal scaffoldin CipA, 4jo5
Supplementary material file. DOI: 10.1107/S174430911301614X/hv5233sup1.pdf
Acknowledgments
We thank the ESRF for synchrotron beam time and the staff scientists of the ID14-4 beamline for their assistance. This work was supported by regular and energy grants from the United States–Israel Binational Science Foundation (BSF), Jerusalem, Israel and by the Israel Science Foundation (grants 159/07, 291/08, 966/09 and 715/12). EAB is the incumbent of The Maynard I. and Elaine Wishner Chair of Bio-organic Chemistry.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: HV5233).
References
- Adams, P. D. et al. (2011). Methods, 55, 94–106.
- Bayer, E. A., Lamed, R., White, B. A. & Flint, H. J. (2008). Chem. Rec. 8, 364–377. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Gerwig, G. J., Kamerling, J. P., Vliegenthart, J. F. G., Morag, E., Lamed, R. & Bayer, E. A. (1993). J. Biol. Chem. 268, 26956–26960. [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- Lamed, R. & Bayer, E. A. (1988). Adv. Appl. Microbiol. 33, 1–46.
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Murray, M. G. & Thompson, W. F. (1980). Nucleic Acids Res. 8, 4321–4325. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Poon, D. K., Withers, S. G. & McIntosh, L. P. (2007). J. Biol. Chem. 282, 2091–2100. [DOI] [PubMed]
- Shimon, L. J. W., Bayer, E. A., Morag, E., Lamed, R., Yaron, S., Shoham, Y. & Frolow, F. (1997). Structure, 5, 381–390. [DOI] [PubMed]
- Shimon, L. J. W., Frolow, F., Yaron, S., Bayer, E. A., Lamed, R., Morag, E. & Shohan, Y. (1997). Acta Cryst. D53, 114–115. [DOI] [PubMed]
- Shimon, L. J. W., Pagès, S., Belaich, A., Belaich, J.-P., Bayer, E. A., Lamed, R., Shoham, Y. & Frolow, F. (2000). Acta Cryst. D56, 1560–1568. [DOI] [PubMed]
- Stout, G. H. & Jensen, L. H. (1968). X-ray Structure Determination. A Practical Guide. London: McMillan.
- Tormo, J., Lamed, R., Chirino, A. J., Morag, E., Bayer, E. A., Shoham, Y. & Steitz, T. A. (1996). EMBO J. 15, 5739–5751. [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Yaniv, O., Shimon, L. J. W., Bayer, E. A., Lamed, R. & Frolow, F. (2011). Acta Cryst. D67, 506–515. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: carbohydrate-binding module from the cellulosomal scaffoldin CipA, 4jo5
Supplementary material file. DOI: 10.1107/S174430911301614X/hv5233sup1.pdf