

Abstract
Purpose
Pre-clinical data suggest that combining imatinib with traditional cytotoxic chemotherapy may improve imatinib efficacy. We conducted a Phase I study of imatinib in combination with paclitaxel in patients with advanced or metastatic solid tumors.
Methods
Patients were accrued to the study in a standard 3 + 3 design. Patients were restaged every two cycles, and those with stable disease (SD), or better, continued study treatment without interruption. Maximally tolerated doses (MTDs) and pharmacokinetic profiles of combination imatinib and paclitaxel were assessed.
Results
Fifty-eight patients were enrolled, including 40 in the Phase I dose escalation portion. Alternating dose escalation of imatinib and paclitaxel on a 28-day cycle resulted in MTDs of 800 mg imatinib daily, on days 1–4, 8–11, 15–18, and 22–25, and 100 mg/m2 paclitaxel weekly, on days 3, 10, and 17. Two expansion cohorts, comprising 10 breast cancer patients and 8 patients with soft-tissue sarcomas, were enrolled at the MTDs. The most common adverse events were flu-like symptoms (64 %) and nausea/vomiting (71 %). The most common Grade 3/4 toxicities were neutropenia (26 %), flu-like symptoms (12 %), and pain (12 %). There were no relevant differences in the pharmacokinetic profiles of either drug when given in combination compared with alone. Thirty-eight subjects were evaluable for response, 18 (47.4 %) of whom experienced clinical benefit. Five patients (13.2 %) had a partial response (PR) and 13 patients (34.2 %) had SD; the average time to progression in those with clinical benefit was 17 weeks (range: 7–28 weeks).
Conclusions
This combination of imatinib and paclitaxel was reasonably safe and tolerable, and demonstrated evidence of anti-tumor activity. Further exploration in disease-specific Phase II trials is warranted.
Keywords: Imatinib, Paclitaxel, Phase I
Introduction
Imatinib is a well-described protein tyrosine kinase inhibitor that has potent activity against the oncogene fusion protein, BCR-ABL, the platelet-derived growth factor receptor (PDGFR), and the growth factor receptor of the tyrosine kinase subclass III family, C-Kit (or CD117) [1–4]. The clinical activity of imatinib was first established in the treatment of chronic myelogenous leukemia (CML), a disease defined by the overexpression of BCR-ABL [5]. When administered to patients with CML, the response rate (RR) to imatinib was over 90 %, with most patients experiencing long-term disease control [6]. Imatinib is also indicated for the treatment of gastrointestinal stromal tumor (GIST) in which C-Kit is typically overexpressed, resulting in a disease control rate of over 80 % [7]. Finally, recent data from a limited number of patients have suggested that imatinib can induce a positive response in at least 90 % of patients with dermatofibrosarcoma protuberans (DFSP). Most of these cases involved a t(17;22) chromosomal translocation that results in overexpression of functional platelet-derived growth factor (PDGF)-β, a ligand for the PDGFR-β also expressed by DFSP tumor cells [8]. However, imatinib has limited efficacy as a single agent in conditions where overexpression of its tyrosine kinase target has not been well defined. For example, imatinib has been ineffective as a single agent when used in unselected patients with soft-tissue sarcomas [9, 10]. The emergence of imatinib resistance, as well as the incomplete response seen in a number of patients receiving this therapy, has led to a search for combination therapies that could potentially enhance the efficacy of imatinib [11].
Pre-clinical data suggest that combining imatinib with traditional cytotoxic or more molecularly targeted chemotherapies may improve imatinib efficacy. Imatinib has demonstrated additive or synergistic activity in pre-clinical models when combined with a number of chemotherapeutic agents including carboplatin, paclitaxel, docetaxel, estramustine, gemcitabine, cisplatin, doxorubicin, and rapamycin [12–24]. Furthermore, several Phase I/II studies provide promising data supporting the benefits of combination chemotherapy with imatinib. For example, a Phase II study of imatinib plus multi-agent chemotherapy in patients with newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia (ALL) demonstrated a complete response (CR) rate of 96 % with an overall survival that was superior to historical controls of chemotherapy alone [25]. A Phase I study of imatinib and gemcitabine demonstrated that the addition of intermittently dosed imatinib, to full or reduced doses of gemcitabine, was associated with greater than expected broad anti-tumor activity [26]. In a separate study, 23 patients with advanced platinum resistant ovarian cancer and primary peritoneal carcinomatosis, both of which expressed PDGFRα and/or C-Kit, were treated with imatinib in combination with docetaxel, resulting in an RR of 22 %, including 1 CR [27]. Mathew et al. [28] conducted a Phase I study using imatinib and docetaxel in patients with prostate cancer and, of the 21 participants, 8 (38 %) had a prostate specific antigen (PSA) level decline of greater than 50 % and 6 (29 %) had a PSA decline of less than 50 %. In fact, one patient experienced an apparent reversal of docetaxel resistance with imatinib after previous disease progression on docetaxel alone.
There have been several mechanisms proposed to explain the synergistic effects of imatinib in combination with other chemotherapeutic agents. For example, the efficacy of chemotherapy may be enhanced by an imatinib-mediated anti-angiogenic effect [29, 30]. Additionally, imatinib has been shown to enhance cytotoxicity through increased apoptosis in cell lines [14]. Finally, imatinib may enhance drug delivery via modulation of adenosine triphosphate-dependent transporter proteins responsible for regulating uptake and efflux of agents at the blood brain barrier and at tumor cell membranes [31]. However, the most compelling explanation for the synergistic effects of imatinib in combination with cytotoxic chemotherapy has been that imatinib enhances the efficacy of chemotherapy primarily by reducing tumor interstitial fluid pressure (IFP), resulting in an increase in transcapillary transport, and thus in an improved delivery of chemotherapeutic agent [32–34]. Pietras et al. demonstrated that the interstitial pressure of a tumor cell microenvironment is elevated, primarily due to the local overexpression of PDGF [32]. In another study by the same group, imatinib was administered in combination with paclitaxel to immunodeficient (SCID) mice with subcutaneous KAT-4 tumors; long-term (21-day) treatment with PDGFR inhibitors was found to lower tumor IFP and subsequently enhance the effect of paclitaxel on apoptosis and proliferation, in vivo [33].
Based on this scientific rationale, we conducted a Phase I study of imatinib in combination with the broad acting taxane, paclitaxel, in patients with advanced or metastatic solid tumors. Pharmacokinetic profiles were characterized, and the dose-limiting toxicities and maximum tolerated doses of each agent in the combined regimen were determined.
Patients and methods
Patient eligibility
Patients who were 18 years or older with a histologically confirmed advanced or metastatic solid tumor and no standard treatment options were eligible for enrollment. Inclusion criteria included an Eastern Cooperative Oncology Group (ECOG) performance status of ≤2 and adequate hematologic, hepatic, and renal function. Patients must have recovered from any previous therapeutic side effects prior to initiating protocol treatment. Exclusion criteria included significant baseline neuropathies from previous chemotherapy, poorly controlled diabetes, a history of prior myocardial infarction within the previous 6 months, and poorly controlled congestive heart failure. Additionally, patients with inadequate hematologic function (WBC count <3,000/mm3, absolute neutrophil count ≤1,500/mm3, or platelet count ≤100,000/mm3); inadequate hepatic function (PT/PTT ≥ 1.5 × the upper limit of normal [ULN], total bilirubin ≥1.5 × the ULN, or AST/ALT ≥ 2.5 × the ULN); or inadequate renal function (creatinine ≥1.5 mg/dL or creatinine clearance ≤50 mL/min) were also excluded from the study. Pregnant or lactating women were disallowed, as were patients with the presence of symptomatic or unstable central nervous system metastases or carcinomatous meningitis, or any other severe concurrent medical or psychiatric disease that could interfere with informed consent and follow-up.
A breast-cancer extension cohort consisted of 10 breast cancer patients who, in addition to fitting the criteria above, had measurable and progressive disease on prior taxane therapy. Likewise, the sarcoma extension cohort consisted of eight patients with sarcomas (soft tissue or bone), with measurable disease that was refractory to standard therapy.
Study design and treatment schedule
The primary endpoint of this Phase I study was to determine the MTDs of imatinib and paclitaxel when given as combination therapy. The secondary endpoints were pharmacokinetic analysis and RR. Patients were accrued in a standard 3 + 3 manner. The original protocol involved the continuous daily oral administration of imatinib with weekly paclitaxel infusions. However, this regimen resulted in several patients developing febrile neutropenia, and the protocol was amended such that patients received only intermittent dosing of imatinib (Fig. 1). Thus, an abbreviated schedule of imatinib therapy was used, involving oral administration of the drug for four consecutive days (D1–4) every 7 days for four consecutive weeks. Paclitaxel was given IV on days 3, 10, and 17 of each 28-day cycle. Patients were pre-treated with dexamethasone, diphenhydramine, and famotidine prior to each paclitaxel infusion. Patients who underwent two treatment cycles were restaged by radiographic imaging by CT scan unless additional scans, such as PET or MRI, were indicated in order to evaluate for disease response.
Fig. 1.

Patient entry and evaluability
Pharmacokinetic analysis
Peripheral blood samples were collected in heparinized tubes prior to and at 10 min, 1, 1.5, 3, 4, 7, and 24 h after imatinib administration/paclitaxel infusion initiation on Cycle 1, day 1 (imatinib alone), Cycle 1, day 3 (imatinib and paclitaxel), and Cycle 1, day 10 (imatinib and paclitaxel). Each blood sample was centrifuged for 10 min at approximately 1,000×g, and plasma was stored at −70 °C or colder until analysis. Plasma concentrations of paclitaxel, imatinib, and the active imatinib metabolite, CGP74588, were determined using validated LC–MS assays [35, 36].
Imatinib and paclitaxel pharmacokinetic analyses were carried out using drug plasma concentration–time data and model-independent (non-compartmental) methodology, employing PK Solutions 2.0™ (Summit Research Services, Montrose, CO, USA).
Because of imatinib’s long plasma half-life, this agent would not have reached steady-state plasma concentrations prior to dosing of patients on day 3 or day 10. Thus, exposure (AUC0-inf) to imatinib on those days was estimated using 0–24 h plasma concentration–time data for all QD dose levels, and 0–7 h data for BID dose levels, universally corrected for exposure to imatinib at 0 h by subtracting Cpre/kel (the pre-dose [0 h] plasma concentration divided by the elimination rate constant) in each case.
To assess any delayed effect of imatinib on paclitaxel pharmacokinetics, we chose day 3 and day 10 to study the effect of the full range of imatinib dosing on the maximum plasma concentration (Cmax) and clearance (Cl) of paclitaxel, at each paclitaxel IV dose level (Table 3).
Table 3.
Paclitaxel plasma pharmacokinetic parameters
| DL | N (d3/d10) | Ima (mg) | Pac (mg/m2) | Cmax d3 (SD) (μg/mL) | Cmax d10 (SD) (μg/mL) | Tmax d3 (SD) (h) | Tmax d10 (SD) (h) | T½ d3 (SD) (h) | T½ d10 (SD) (h) | Cl d3 (SD) (L/h/m2) | Cl d10 (SD) (L/h/m2) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6/6 | 400 QD | 60 | 1.71 (0.71) | 3.80 (2.76) | 1.0 (0) | 0.86 (0.34) | 12.6 (2.9) | 11.9 (2.8) | 16.3 (8.0) | 11.4 (6.1) |
| −1 | 7/5 | 300 QD | 60 | 0.96 (0.27) | 1.29 (0.55) | 0.88 (0.31) | 1.0 (0) | 16.0 (5.5) | 11.0 (1.8) | 23.9 (8.0) | 26.7 (10.9) |
| 1A | 3/3 | 400 QD | 60 | 1.86 (0.95) | 1.64 (0.13) | 1.0 (0) | 0.72 (0.48) | 16.6 (8.2) | 11.5 (4.6) | 20.4 (7.0) | 21.0 (2.7) |
| 2A | 3/3 | 400 QD | 80 | 2.11 (0.44) | 1.77 (0.48) | 1.0 (0) | 1.0 (0) | 11.3 (3.5) | 10.6 (0.8) | 18.2 (2.1) | 19.3 (4.1) |
| 3A | 3/3 | 300 BID | 80 | 1.65 (0.39) | 1.02 (0.46) | 1.0 (0) | 1.2 (0.3) | 10.8 (4.4) | 12.7 (2.3) | 21.9 (2.6) | 29.6 (10.4) |
| 4A | 4/4 | 300 BID | 100 | 6.37 (1.80) | 6.78 (3.13) | 1.0 (0) | 1.0 (0) | 9.59 (1.67) | 10.7 (2.8) | 9.4 (1.8) | 9.6 (2.9) |
| 5A | 8/8 | 400 BID | 100 | 6.97 (3.33) | 6.32 (3.26) | 1.1 (0.2) | 1.1 (0.2) | 8.21 (1.27) | 10.8 (3.5) | 7.4 (2.1) | 8.4 (1.9) |
| P value | (N = 31) | 0.688 | 0.500 | 0.812 | 0.783 | ||||||
Differences between paclitaxel parameters on day 3 and day 10 were tested with a two-tailed Wilcoxon exact rank test, whereby a P value<0.05 was considered significant
DL dose level
To assess any effect of paclitaxel on imatinib pharmacokinetics, we compared the maximum imatinib plasma concentration (Cmax), time to Cmax (Tmax), apparent clearance (Cl/F), and half-life (t½) on day 1 (imatinib alone) with Cmax, Tmax, Cl/F, and t½ on day 3 (the first day of combination therapy).
Statistical methods
This Phase I trial was designed to enter enough patients to complete up to five dose cohorts for escalation by threes to establish the maximum tolerated dose (MTD), with two additional supplemental cohorts of 10 patients, each with breast cancer and sarcoma. Toxicities were tabulated according to the total number of toxicities that occurred within each adverse event category as well as by Grade 3 or higher. Only toxicities that were at least possibly related to the study drug were included. The highest toxicity grade a subject experienced within a category was used for the tabulation. Each subject was counted once within each adverse event, regardless of the number of treatment cycles they received. Subjects were considered for toxicity data as long as they had at least one study drug administered. Patients were evaluable for dose-limiting toxicities (DLT) if they either experienced a DLT or completed a full cycle of therapy without a DLT. Patients not evaluable for DLT were replaced. Best response was tabulated using the RE-CIST best response criteria. Subjects were considered evaluable for response if they had response data either through imaging or clinical evaluation. For subjects who were evaluable for response, time on study was determined as the number of weeks from study entry to the official date off study.
Statistical analyses for pharmacokinetic parameters and concentration values were performed using SPSS 17.0 for Windows (SPSS Inc., Chicago, IL). Cmax, Cl, and t½ values were compared using a two-tailed Mann–Whitney signed rank test. A P <0.05 was considered significant.
Results
Patient characteristics
Between February 19, 2003 and December 8, 2008, 58 patients were enrolled, 40 in the main Phase I portion, 10 in the breast-cancer extension cohort, and 8 in the sarcoma extension cohort. Fifty-six patients received treatment (two patients were ineligible on day 1 prior to starting therapy). Patient characteristics are listed in Table 1. Prior therapies included chemotherapy (n = 42), surgery (n = 44), hormonal therapy (n = 6), and radiation therapy (n = 26). Patients who went off the study did so for a variety of reasons, the most prevalent of which was disease progression (66 %). Eight patients, who were taken off the study before the completion of Cycle 1 for reasons other than dose-limiting toxicities, were replaced.
Table 1.
Patient demographics and disease characteristics
| Characteristic | N (%) |
|---|---|
| All patients | 58 (100) |
| Age, years | |
| Median (range) | 53.2 (21–80) |
| Sex | |
| Male | 23 (40) |
| Female | 35 (60) |
| Race | |
| White | 40 (69) |
| Black | 10 (17) |
| Asian | 5 (9) |
| Hispanic | 3 (5) |
| Performance status | |
| Missing | 10 (17) |
| 0 | 14 (24) |
| 1 | 31 (53) |
| 2 | 3 (5) |
| Tumor type | |
| Breast-female | 12 (21) |
| Colorectal | 10 (17) |
| Soft-tissue sarcoma | 8 (14) |
| Osteo- or chondrosarcoma | 5 (9) |
| Lung | 4 (7) |
| Pancreas | 4 (7) |
| Esophagus | 4 (7) |
| Cervix | 2 (3) |
| Ovary | 2 (3) |
| Thyroid | 1 (2) |
| Lip, oral cavity, and pharynx | 1 (2) |
| Kidney | 1 (2) |
| Small intestine | 1 (2) |
| Other | 3 (5) |
| Prior therapy | |
| Chemotherapy | 42 (72) |
| Surgery | 44 (76) |
| Hormonal therapy | 6 (10) |
| Radiation | 26 (45) |
Figure 1 details the dose escalation plan; the trial was originally designed to use continuous daily dosing of oral imatinib with weekly paclitaxel infusions (Cohort 1 = imatinib, 400 mg daily (D1–28), plus paclitaxel, 60 mg/m2 on days 1, 8, and 15). However, four out of eight patients enrolled in the first cohort developed Grade 3 neutropenia. Thus, the continuous oral dose of imatinib was de-escalated to 300 mg daily on days 1–28 (Cohort-1). When yet another patient in Cohort-1, developed febrile neutropenia, the treatment regimen was amended such that patients received only intermittent dosing of imatinib, initially concurrent with the paclitaxel (Cohort−1A), but, due to neutropenia, ultimately offset from the paclitaxel dosing (Cohorts +1A to +5A and the extension cohorts). Thus, the majority of patients were treated using the intermittent abbreviated dosing schedule of imatinib on days 1–4, 8–11, 15–18, and 22–25, together with IV paclitaxel on days 3, 10, and 17. Alternating dose escalation of imatinib and paclitaxel proceeded until the MTDs of imatinib and paclitaxel were reached: 800 mg of imatinib, orally administered daily on days 1–4, 8–11, 15–18, and 22–25, combined with 100 mg/m2 paclitaxel, administered IV on days 3, 10, and 17, of each 28-day cycle.
Upon nearing completion of the Phase I study, a potential benefit was seen in patients with breast cancer and patients with sarcomas. Thus, two extension cohorts were added at the MTD level (+5A, final cohort, Phase I dose). One extension cohort was for breast cancer patients, in which 10 patients were enrolled, and the other was for patients with sarcomas, in which eight patients were enrolled.
Suspected drug-related adverse events
All 58 subjects were evaluable for adverse events. Table 2 provides the number of adverse events by category and cohort for those categories that had ≥10 % of evaluable subjects experiencing the adverse event. Both the total number of adverse events and those with a Grade 3 or higher are provided. There were a number of common toxicities typically seen with imatinib and/or paclitaxel as single agents, such as nausea/vomiting, diarrhea, anemia, neutropenia, peripheral edema, and sensory neuropathy. The most common adverse events experienced in the current study were flu-like symptoms and nausea/vomiting (seen in 64 and 71 % of subjects, respectively). The most commonly reported Grade 3 or 4 toxicities related to therapy were neutropenia (26 %), flu-like symptoms (12 %), and pain (12 %). Even at the MTDs, treatment with paclitaxel had to be held periodically due to neutropenia, most often on day 17 of a cycle.
Table 2.
Summary of clinically relevant* adverse events
| Imatinib (mg) Paclitaxel (mg/m2) Adverse event | 400 Daily | 300 Daily | 400 | 400 | 600 | 600 | 800 | Total AEs N (%) | Grade 3/4 AEs N (%) |
|---|---|---|---|---|---|---|---|---|---|
| 60 | 60 | 60 | 80 | 80 | 100 | 100 | |||
| No. of patients (no. of patients with Grade 3 or 4 AEs)
|
|||||||||
| N = 8 | N = 7 | N = 3 | N = 3 | N = 4 | N = 7 | N = 26 | N = 58 (100) | N = 58 (100) | |
| Alopecia | 3 (0) | 0 | 0 | 0 | 1 (0) | 2 (0) | 10 (0) | 16 (28) | 0 (0) |
| Constitutional | |||||||||
| Fever | 2 (0) | 3 (1) | 2 (0) | 0 | 0 | 0 | 3 (2) | 10 (17) | 3 (5) |
| Flu-like symptoms | 4 (0) | 4 (1) | 3 (0) | 3 (1) | 3 (1) | 3 (1) | 17 (3) | 37 (64) | 7 (12) |
| Myalgia/arthralgia | 2 (0) | 1 (0) | 0 | 0 | 0 | 1 (0) | 2 (0) | 6 (10) | 0 (0) |
| Weight loss | 0 | 1 (0) | 0 | 0 | 1 (0) | 2(0) | 2 (0) | 6 (10) | 0 (0) |
| Neuro | |||||||||
| Headache | 2 (0) | 3 (0) | 2 (0) | 0 | 0 | 0 | 2 (0) | 9 (16) | 0 (0) |
| Sensory | 2 (0) | 1 (0) | 0 | 0 | 1 (0) | 0 | 10 (2) | 14 (24) | 2 (3) |
| Other | 2 (0) | 2 (0) | 2 (0) | 0 | 1 (0) | 0 | 3 (0) | 10 (17) | 0 (0) |
| Motor | 2 (0) | 0 | 1 (0) | 1 (0) | 0 | 2 (0) | 6 (2) | 12 (21) | 2 (3) |
| Pain | 4 (1) | 3 (1) | 1 (0) | 1 (1) | 2 (1) | 0 | 5 (3) | 16 (28) | 7 (12) |
| Pulmonary | |||||||||
| Dyspnea | 0 | 0 | 1 (0) | 0 | 0 | 0 | 7 (1) | 8 (14) | 1 (2) |
| Cough | 0 | 2 (0) | 0 | 1 (0) | 0 | 1 (0) | 5 (0) | 9 (16) | 0 (0) |
| Other | 1 (0) | 1 (0) | 1 (0) | 1 (0) | 0 | 1 (0) | 1 (1) | 6 (10) | 1 (2) |
| Gastrointestinal | |||||||||
| Anorexia | 0 | 1 (0) | 0 | 2 (0) | 1 (0) | 2 (0) | 2 (0) | 8 (14) | 0 (0) |
| Nausea/vomiting | 5 (0) | 6 (1) | 3 (0) | 3 (0) | 3 (1) | 5 (0) | 16 (1) | 41 (71) | 3 (5) |
| Diarrhea | 4 (0) | 2 (0) | 2 (0) | 1 (0) | 1 (0) | 0 | 12 (0) | 22 (38) | 0 (0) |
| LFT abnormalities | 1 (0) | 0 | 0 | 0 | 1 (0) | 1 (0) | 4 (1) | 7 (12) | 1 (2) |
| Vomiting | 1 (0) | 4 (2) | 0 | 2 (0) | 3(0) | 2 (0) | 10 (0) | 22 (38) | 2 (3) |
| Other | 4 (0) | 2 (0) | 1 (0) | 3 (0) | 1 (0) | 1 (0) | 8 (1) | 20 (34) | 1 (2) |
| Hematologic | |||||||||
| Neutropenia | 6 (4) | 1 (1) | 0 | 2 (2) | 1 (0) | 1 (0) | 14 (8) | 25 (43) | 15 (26) |
| Anemia | 3 (0) | 2 (0) | 3 (0) | 1 (0) | 2 (0) | 3 (0) | 10 (2) | 24 (41) | 2 (3) |
| Skin | 4 (0) | 3 (0) | 0 | 1 (0) | 0 | 4 (0) | 8 (0) | 20 (34) | 0 (0) |
| Infection | 2 (0) | 2 (0) | 1 (0) | 1 (0) | 0 | 0 | 2 (1) | 8 (14) | 1 (2) |
| Metabolic | |||||||||
| Hyperglycemia | 0 | 0 | 0 | 0 | 1 (0) | 1 (0) | 4 (0) | 6 (10) | 0 (0) |
| Hypocalcemia | 0 | 0 | 0 | 0 | 0 | 0 | 6 (0) | 6 (10) | 0 (0) |
| Hyponatremia | 1 (0) | 1 (0) | 0 | 0 | 1 (0) | 1 (0) | 4 (0) | 8 (14) | 0 (0) |
| Other | 1 (0) | 1 (0) | 0 | 1 (0) | 2 (1) | 1 (0) | 8 (2) | 14 (24) | 3 (5) |
| Edema | |||||||||
| Unspecified | 3(0) | 2 (0) | 1 (0) | 2(0) | 2(1) | 1 (0) | 12(0) | 23 (40) | 1 (2) |
Breast cancer and sarcoma extension patients are included in the last cohort
Adverse events are included if they occurred in at least 10 % of the patients.
Pharmacokinetics
Of the 58 patients enrolled, 37 had paclitaxel pharmacokinetic data. Of these 37 patients, 31 had sufficient data to estimate AUC values on both day 3 and day 10. None of the tested pharmacokinetic parameters differed between day 3 and 10 (Table 3). Although data is limited, paclitaxel Cmax appeared to display a greater than proportional increase at higher paclitaxel doses, as expected, which correlated with a greater than proportional decrease in clearance [37, 38]. Thus, when the dose of paclitaxel was increased to 100 mg/m2, paclitaxel pharmacokinetics appeared to display non-linearity. Imatinib did not appear to have a delayed effect on paclitaxel pharmacokinetics (day 10 vs. day 3) (Table 3).
Thirty-nine of the patients enrolled in this study had sufficient imatinib plasma concentration–time data to obtain pharmacokinetic parameter estimates. Of these 39 patients, 30 had sufficient data to estimate AUC values on both day 1 and day 3, while only 21 patients had sufficient data to estimate pharmacokinetic parameters on all 3 days (Cycle 1: day 1, day 3, and day 10). There was no apparent difference in Cmax, Tmax, Cl/F, and t½ between day 3 and day 10, but no formal statistical tests were performed for day 10 parameters so these data were excluded. No significant difference was observed between day 1 (imatinib alone) and day 3 (imatinib + paclitaxel) for Cmax or Cl/F. However, we did observe a significant difference between day 1 and day 3 with respect to Tmax and t½ (Table 4).
Table 4.
Imatinib plasma pharmacokinetic parameters
| DL | N (d1/d3) | Ima (mg) | Pac (mg/m2) | Cmax d1 (SD) (μg/mL) | Cmax d3 (SD) (μg/mL) | Tmax d1 (SD) (h) | Tmax d3 (SD) (h) | T½ d1 (SD) (h) | T½ d3 (SD) (h) | Cl/F d1 (SD) (L/h) | Cl/F d3 (SD) (L/h) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7/6 | 400 QD | 60 | 3.00 (1.55) | 2.77 (1.18) | 2.6 (1.0) | 4.1 (1.4) | 12.7 (4.1) | 23.8 (14.0) | 8.93 (5.59) | 7.40 (3.33) |
| −1 | 7/7 | 300 QD | 60 | 2.45 (1.61) | 1.84 (1.18) | 2.6 (0.7) | 6.1 (1.5) | 11.7 (2.1) | 18.3 (11.7) | 9.98 (5.40) | 11.2 (5.7) |
| 1A | 3/3 | 400 QD | 60 | 3.76 (2.43) | 3.83 (2.27) | 3.7 (0.6) | 5.7 (2.3) | 14.6 (5.3) | 12.5 (2.1) | 6.24 (2.47) | 8.22 (2.48) |
| 2A | 3/3 | 400 QD | 80 | 3.15 (0.96) | 2.73 (2.01) | 3.7 (0.6) | 3.3 (0.6) | 11.0 (4.4) | 12.9 (4.8) | 8.40 (4.46) | 24.1 (31.3) |
| 3A | 2/3 | 300 BID | 80 | 4.06 (2.50) | 4.91 (2.28) | 2.8 (1.8) | 3.0 (0) | 5.9 (0.1) | 35.4 (32.7) | 8.75 (5.45) | 5.13 (3.34) |
| 4A | 6/3 | 300 BID | 100 | 2.15 (0.83) | 2.24 (0.47) | 2.9 (0.8) | 3.7 (0.6) | 3.5 (16.8) | 22.5 (3.2) | 10.6 (9.30) | 14.7 (4.7) |
| 5A | 8/6 | 400 BID | 100 | 5.15 (2.13) | 6.65 (2.47) | 3.0 (1.0) | 3.7 (0.5) | 10.2 (3.7) | 19.5 (5.8) | 5.72 (2.38) | 9.13 (3.3) |
| P value | (N = 30) | 0.256 | 8.0 E-6 | 1.4 E-6 | 0.136 |
Differences between imatinib parameters on day 1 and day 3 were tested with a two-tailed Wilcoxon exact rank test, whereby a P value <0.05 was considered significant
DL dose level
Clinical responses
Of the 58 patients enrolled in the study, 38 were evaluable for response. Twenty patients were taken off the study prior to first restaging analysis. The reasons for withdrawal were rapid clinical deterioration due to intercurrent illness (12 patients), unexpected and unacceptable abnormalities in liver function tests on day 1 (two patients), and withdrawal of consent prior to first restaging analysis (six patients). Among the remaining 38 patients, there were five partial responses (PRs) with two RECIST confirmed PRs. Of the five patients, there was one patient each with esophageal cancer, soft-tissue sarcoma, carcinoma of unknown primary, ovarian cancer, and colon cancer. The overall RR was 13.2 % (Table 5). Additionally, 13 patients (34.2 %) had stable disease (SD), 10 of whom had SD for >12 weeks (range 14–32 weeks). Four of the 10 patients had SD >24 weeks (Fig. 2). The median time on study for all patients with PR or SD was 13.8 weeks. The overall clinical benefit rate (PR + SD) was 47.4 %. Interestingly, examination of best response and time on study as a function of dose level revealed no significant differences, and certainly no incremental increase with increasing doses of imatinib and paclitaxel (Table 5; Fig. 2).
Table 5.
Best response and median time to progression by dose level and overall (evaluable patients only)
| Cohort | Imatinib (mg) | Paclitaxel (mg/m2) | Na | Best response
|
|
|---|---|---|---|---|---|
| Partial response N (%) | Stable disease N (%) | ||||
| 1 | 400 Daily (D1–28) | 60 (D1, 8, 15) | 6 | 1 (16.7) | 1 (16.7) |
| −1 | 300 Daily (D1–28) | 60 (D1, 8, 15) | 4 | 0 | 2 (50.0) |
| +1A | 400 (D1–4) | 60 (D3, 10, 17) | 3 | 0 | 1 (33.3) |
| +2A | 400 (D1–4) | 80 (D3, 10, 17) | 3 | 0 | 2 (66.7) |
| +3A | 600 (D1–4) | 80 (D3, 10, 17) | 3 | 1 (33.3) | 1 (33.3) |
| +4A | 600 (D1–4) | 100 (D3, 10, 17) | 3 | 0 | 1 (33.3) |
| +5A | 800 (D1–4) | 100 (D3, 10, 17) | 4 | 3 (75.0) | 0 |
| Breast extension | 800 (D1–4) | 100 (D3, 10, 17) | 5 | 0 | 0 |
| Sarcoma extension | 800 (D1–4) | 100 (D3, 10, 17) | 7 | 0 | 5 (62.5) |
| Total | 38 | 5 (13.2) | 13 (34.2) | ||
N represents the number of subjects who were response evaluable
Fig. 2.
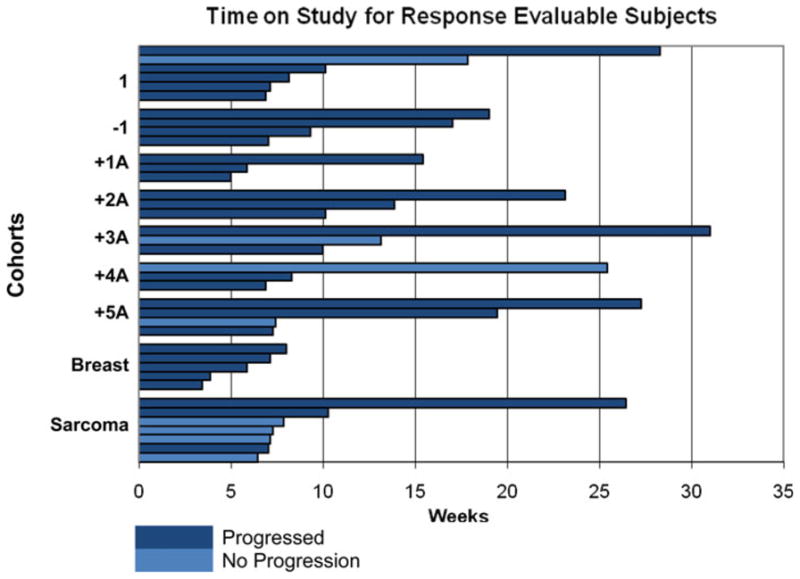
Study duration by progression status
Discussion
The combination of intermittently dosed imatinib and paclitaxel is reasonably well tolerated at the MTDs and schedules of 800 mg imatinib, orally administered daily on days 1–4, 8–11, 15–18, and 22–25, combined with 100 mg/m2 paclitaxel, administered IV on days 3, 10, and 17, of a 28-day cycle. In this heavily pre-treated population, the combination of paclitaxel and imatinib resulted in a clinically meaningful RR of 13.2 %, with an additional 34.2 % experiencing stable disease (the majority of whom had clinically meaningful stable disease of at least ≥12 weeks). Continuous daily dosing of imatinib resulted in unacceptable toxicity, primarily severe neutropenia.
The increased dosing of, and thus the increased exposure to, imatinib explains (in part) the observed efficacy and toxicity of the drug regimen, as opposed to the combined pharmacodynamic effect of imatinib and paclitaxel. In the current study, paclitaxel pharmacokinetic parameter values, estimated following administration of 60 and 80 mg/m2 paclitaxel to patients, were in agreement with parameter values reported in the literature, following paclitaxel administration as a 1-h infusion at doses of 80 and 100 mg/m2 [39]. However, in the current study, following administration of 100 mg/m2 of paclitaxel, Cmax values were higher than expected and the apparent clearance decreased dramatically. The pharmacokinetic profile of paclitaxel is known to have a non-linear component, even at 3-h and 24-h infusion durations. A mechanistic explanation for this non-linear behavior has been under debate for some time, and proposed mechanisms include one or more of the following: saturable tissue transport; saturable tissue binding; non-linear elimination; non-linear binding to plasma proteins and blood cells; and binding influenced by Cremophor EL, part of the vehicle for paclitaxel [37, 38]. It could be expected that if paclitaxel were infused over a shorter duration, as was the case for our current study, the higher paclitaxel input rate would result in higher initial concentrations of both paclitaxel and its vehicle, thus augmenting the non-linearity of the pharmacokinetic profile. In addition, both imatinib and paclitaxel are predominantly metabolized by cytochrome P450 3A4 (CYP3A4) liver enzymes, and could potentially interfere with one another’s clearance [40, 41]. However, any effect of imatinib on the dose-dependent clearance of paclitaxel is likely to be overshadowed by: (1) the intrinsic variability of paclitaxel pharmacokinetics observed within patient cohorts and (2) the increase in non-linearity of paclitaxel pharmacokinetics with increasing paclitaxel dose.
Imatinib apparent clearance values observed were in the range of 5.1–25.7 L/h, which agrees with apparent clearance values of 12.5 ± 7.2 L/h reported in the literature [40]. Imatinib pharmacokinetics did not change in the presence of paclitaxel. Cmax values on day 3 (following both imatinib and paclitaxel administration) were not statistically different from those on day 1 (following imatinib treatment alone). This could be due to the short interval between the two sampling times (the second sample was taken only 48 h after the first and this may not be enough time for any induction or inhibition effects on drug metabolism to take effect). The long half-life of imatinib could not be accurately estimated using plasma concentration–time data taken over 24 h only; hence, the observed statistically significant difference in t½ between day 1 and day 3 is likely due to imatinib being closer to steady-state levels on day 3. The difference in Tmax in some cohorts is more difficult to explain, but at a constant clearance rate, neither Tmax nor t½ differences between day 1 and day 3 are likely to be clinically relevant.
At the time of conception of our currently reported trial, it was anticipated that chemotherapy would enhance the anti-tumor efficacy of imatinib. However, since that time, imatinib has been shown to be ineffective as a single agent against solid tumors; particularly where there is no evidence of overexpression of its tyrosine kinase target(s) [9, 10]. Nevertheless, the combination of imatinib and cytotoxic chemotherapy clearly results in additive, if not synergistic anti-tumor activity [12–28]. In fact, the prevailing hypothesis is that imatinib enhances the anti-tumor efficacy of chemotherapy by decreasing tumor IFP and enhancing cytotoxic drug delivery to cancer cells.
Tumor stromal cells and local blood vessels typically exhibit high PDGFR expression, and signaling through these receptors seems to enhance tumor IFP [32]. Inhibition of PDGFR signaling decreases tumor IFP and enhances drug delivery to tumors [32–34]. This finding may explain the clinical benefit seen in our very heavily pre-treated patient population, in which an overall disease control rate of 42 % following treatment with the imatinib plus paclitaxel combination does suggest enhanced anti-tumor activity of this drug combination. Furthermore, our dosing schedule is consistent with pre-clinical evidence that enhanced delivery of cytotoxic chemotherapy by imatinib is seen when imatinib is given before and after chemotherapy, on a “bracketed” schedule [34]. Finally, because the enhanced efficacy of the imatinib plus paclitaxel combination is probably a result of the effects of imatinib on tumor IFP, there is likely no dependence on tumor BCR-ABL, or C-kit, making this a drug combination with potentially broad applicability.
There is a clear evidence (particularly in the dermis) that PDGFR signaling also regulates IFP in the non-tumor microenvironment, which likely accounts for the increased toxicity observed with the imatinib plus paclitaxel combination [32, 42]. Thus, it is possible that imatinib decreases IFP in normal tissues. If this decrease was to occur in the bone marrow microenvironment, the delivery of paclitaxel would be enhanced and this would result in increased paclitaxel-related toxicities. The degree of neutropenia, anemia, nausea, vomiting, diarrhea, sensory neuropathy, and peripheral edema observed in this study was consistent with the side effects typically seen upon patient treatment with single agent paclitaxel. However, the rate and the severity of side effects were much greater than expected, particularly given that nearly half of the patients received a lower than usual weekly dose of paclitaxel. Clearly, imatinib enhanced paclitaxel activity, despite the fact that there was no evidence of any pharmacokinetic interaction between imatinib and paclitaxel.
These results are consistent with the outcome of similar Phase I trials where imatinib is combined with cytotoxic chemotherapy. We compared results from two clinical trials carried out in prostate cancer patients: one pivotal trial involved treatment of patients with docetaxel alone, with the aim of establishing the efficacy of the drug in this group of patients [43] and the other trial involved treatment of patients with a combination of docetaxel plus imatinib [28]. The combination of docetaxel and imatinib resulted in significantly greater rates of Grade 3 toxicities than docetaxel alone, including fatigue (35 vs. 5 %), nausea/vomiting (20 vs. 0 %), and diarrhea (10 vs. 0 %). Interestingly, the rates of Grade 3 neutropenia were identical (2 %) [28, 43]. Similarly, increased toxicities were seen when imatinib was combined with 5-fluorouracil [44], cisplatin + irinotecan [19], and carboplatin + irinotecan [45]. Ali et al. [26] combined imatinib with gemcitabine and found unexpectedly high rates of nausea and vomiting, neutropenia, elevated liver transaminases, and neuropathy, particularly given the relatively low doses of gemcitabine administered to patients. Surprisingly, only our study and the study by Ali et al. demonstrated a greater than expected clinical response benefit upon the combination of imatinib with chemotherapy, although Al-Batran et al. did comment on a possible increase in clinical activity on combination of imatinib with 5-fluorouracil (in all cases, compared with historical data). It is noteworthy that only clinical trials following a treatment schedule in which imatinib “bracketed” chemotherapy (our current study, [26, 44]), rather than being administered every day [19, 28, 45], suggested enhanced anti-tumor activity. These findings suggest that the combination of imatinib and cytotoxic chemotherapy, using a bracketed imatinib schedule, warrants further exploration in disease-specific Phase II trials.
Acknowledgments
We would like to thank Novartis for funding this clinical trial. Jan H. Beumer (JHB) and Merrill J. Egorin (MJE) were supported by grant P30-CA47904 from the National Cancer Institute. JHB is the recipient of a Hillman Fellows for Innovative Cancer Research Award. MJE is the recipient of an American Society of Clinical Oncology Cancer Foundation Translational Research Professorship.
Abbreviations
- PDGF
Platelet-derived growth factor
- PDGF-R
Platelet-derived growth factor receptor
- CML
Chronic myelogenous leukemia
- GIST
Gastrointestinal stromal tumor
- DFSP
Dermatofibrosarcoma protuberans
- RR
Response rate
- CR
Complete response
- IFP
Interstitial fluid pressure
- ULN
Upper limit of normal
- MTD
Maximally tolerated doses
Footnotes
Conflict of interest None.
Contributor Information
Michael J. Pishvaian, Email: pishvaim@georgetown.edu, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA
Rebecca Slack, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA. Department of Biostatistics, University of Texas MD Anderson Cancer Center, Houston, TX 77230, USA.
Eunice Y. Koh, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA
Jan H. Beumer, Molecular Therapeutics/Drug Discovery Program, University of Pittsburgh Cancer Institute, Pittsburgh, PA 15213, USA. Department of Pharmaceutical Sciences, University of Pittsburgh School of Pharmacy, Pittsburgh, PA 15213, USA. Melanoma Program, University of Pittsburgh Cancer Institute, Pittsburgh, PA 15213, USA
Marion L. Hartley, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA
Ion Cotarla, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA.
John Deeken, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA.
Aiwu Ruth He, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA.
Jimmy Hwang, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA.
Shakun Malik, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA.
Kashif Firozvi, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA.
Minetta Liu, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA.
Beth Elston, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA. Center for Population Health and Clinic Epidemiology, Brown University, Providence, RI 02912, USA.
Sandy Strychor, Molecular Therapeutics/Drug Discovery Program, University of Pittsburgh Cancer Institute, Pittsburgh, PA 15213, USA.
Merrill J. Egorin, Division of Hematology/Oncology, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA. Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA
John L. Marshall, Lombardi Comprehensive Cancer Center, Developmental Therapeutics Program, Georgetown University Medical Center, Podium B, 3800 Reservoir Road, NW, Washington, DC 20007, USA
References
- 1.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian HM, Giles F, Quintas-Cardama A, Cortes J. Important therapeutic targets in chronic myelogenous leukemia. Clin Cancer Res. 2007;13:1089–1097. doi: 10.1158/1078-0432.CCR-06-2147. [DOI] [PubMed] [Google Scholar]
- 3.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, Lydon NB. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimi-dine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- 4.Savage DG, Antman KH. Imatinib mesylate—a new oral targeted therapy. N Engl J Med. 2002;346:683–693. doi: 10.1056/NEJMra013339. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian H, Sawyers C, Hochhaus A, Guilhot F, Schiffer C, Gambacorti-Passerini C, Niederwieser D, Resta D, Capdeville R, Zoellner U, Talpaz M, Druker B, Goldman J, O’Brien SG, Russell N, Fischer T, Ottmann O, Cony-Makhoul P, Facon T, Stone R, Miller C, Tallman M, Brown R, Schuster M, Loughran T, Gratwohl A, Mandelli F, Saglio G, Lazzarino M, Russo D, Baccarani M, Morra E. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- 6.Druker BJ. Translation of the Philadelphia chromosome into therapy for CML. Blood. 2008;112:4808–4817. doi: 10.1182/blood-2008-07-077958. [DOI] [PubMed] [Google Scholar]
- 7.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 8.McArthur GA. Dermatofibrosarcoma protuberans: a surgical disease with a molecular savior. Curr Opin Oncol. 2006;18:341–346. doi: 10.1097/01.cco.0000228739.62756.df. [DOI] [PubMed] [Google Scholar]
- 9.Verweij J, van Oosterom A, Blay JY, Judson I, Rodenhuis S, van der Graaf W, Radford J, Le Cesne A, Hogendoorn PC, di Paola ED, Brown M, Nielsen OS. Imatinib mesylate (STI-571 Glivec, Gleevec) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003;39:2006–2011. [PubMed] [Google Scholar]
- 10.Chugh R, Thomas D, Wathen K, Thall PF, Benjamin RS, Maki RS, Samuels BL, Keohan ML, Priebat DA, Baker LH. Imatinib mesylate in soft tissue and bone sarcomas: interim results of a Sarcoma Alliance for Research thru Collaboration (SARC) phase II trial. ASCO annual meeting proceedings (post-meeting edition) J Clin Oncol. 2004;22(14S) [Google Scholar]
- 11.Jabbour E, Cortes J, O’Brien S, Giles F, Kantarjian H. New targeted therapies for chronic myelogenous leukemia: opportunities to overcome imatinib resistance. Semin Hematol. 2007;44:S25–S31. doi: 10.1053/j.seminhematol.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 12.Vlahovic G, Ponce AM, Rabbani Z, Salahuddin FK, Zgonjanin L, Spasojevic I, Vujaskovic Z, Dewhirst MW. Treatment with imatinib improves drug delivery and efficacy in NSCLC xenografts. Br J Cancer. 2007;97:735–740. doi: 10.1038/sj.bjc.6603941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubler HR, van Randenborgh H, Treiber U, Wutzler S, Battistel C, Lehmer A, Wagenpfeil S, Hartung R, Paul R. In vitro cytotoxic effects of imatinib in combination with anticancer drugs in human prostate cancer cell lines. Prostate. 2005;63:385–394. doi: 10.1002/pros.20201. [DOI] [PubMed] [Google Scholar]
- 14.Bertino P, Piccardi F, Porta C, Favoni R, Cilli M, Mutti L, Gaudino G. Imatinib mesylate enhances therapeutic effects of gemcitabine in human malignant mesothelioma xenografts. Clin Cancer Res. 2008;14:541–548. doi: 10.1158/1078-0432.CCR-07-1388. [DOI] [PubMed] [Google Scholar]
- 15.Bertino P, Porta C, Barbone D, Germano S, Busacca S, Pinato S, Tassi G, Favoni R, Gaudino G, Mutti L. Preliminary data suggestive of a novel translational approach to mesothelioma treatment: imatinib mesylate with gemcitabine or pemetrexed. Thorax. 2007;62:690–695. doi: 10.1136/thx.2006.069872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruce IA, Slevin NJ, Homer JJ, McGown AT, Ward TH. Synergistic effects of imatinib (STI 571) in combination with chemotherapeutic drugs in head and neck cancer. Anticancer Drugs. 2005;16:719–726. doi: 10.1097/01.cad.0000168392.04676.bb. [DOI] [PubMed] [Google Scholar]
- 17.Grabellus F, Ebeling P, Worm K, Sheu SY, Antoch G, Frilling A, Schmid KW. Double resistance to imatinib and AMG 706 caused by multiple acquired KIT exon 17 mutations in a gastrointestinal stromal tumour. Gut. 2007;56:1025–1026. doi: 10.1136/gut.2006.115923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coskun HS, Goksu SS, Sahin M, Alanoglu G. Bleomycin, etoposide and cisplatin (BEP) combination with concurrent imatinib mesylate (GLEEVEC) in chronic myeloid leukemia (CML) patient with mesenchymal tumor. Med Oncol. 2008;25:110–112. doi: 10.1007/s12032-007-0053-5. [DOI] [PubMed] [Google Scholar]
- 19.Johnson FM, Krug LM, Tran HT, Shoaf S, Prieto VG, Tamboli P, Peeples B, Patel J, Glisson BS. Phase I studies of imatinib mesylate combined with cisplatin and irinotecan in patients with small cell lung carcinoma. Cancer. 2006;106:366–374. doi: 10.1002/cncr.21640. [DOI] [PubMed] [Google Scholar]
- 20.Yerushalmi R, Nordenberg J, Beery E, Uziel O, Lahav M, Luria D, Fenig E. Combined antiproliferative activity of imatinib mesylate (STI-571) with radiation or cisplatin in vitro. Exp Oncol. 2007;29:126–131. [PubMed] [Google Scholar]
- 21.Majsterek I, Sliwinski T, Poplawski T, Pytel D, Kowalski M, Slupianek A, Skorski T, Blasiak J. Imatinib mesylate (STI571) abrogates the resistance to doxorubicin in human K562 chronic myeloid leukemia cells by inhibition of BCR/ABL kinase-mediated DNA repair. Mutat Res. 2006;603:74–82. doi: 10.1016/j.mrgentox.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Palmberg E, Johnsen JI, Paulsson J, Gleissman H, Wickstrom M, Edgren M, Ostman A, Kogner P, Lindskog M. Metronomic scheduling of imatinib abrogates clonogenicity of neuroblastoma cells and enhances their susceptibility to selected chemotherapeutic drugs in vitro and in vivo. Int J Cancer. 2009;124:1227–1234. doi: 10.1002/ijc.24069. [DOI] [PubMed] [Google Scholar]
- 23.Sleijfer S. With a little help from small friends: enhanced chemotherapeutic effects with imatinib. Eur J Cancer. 2006;42:808–810. doi: 10.1016/j.ejca.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Merimsky O, Gorzalczany Y, Sagi-Eisenberg R. Molecular impacts of rapamycin-based drug combinations: combining rapamycin with gemcitabine or imatinib mesylate (Gleevec) in a human leiomyosarcoma model. Int J Oncol. 2007;31:225–232. [PubMed] [Google Scholar]
- 25.Yanada M, Takeuchi J, Sugiura I, Akiyama H, Usui N, Yagasaki F, Kobayashi T, Ueda Y, Takeuchi M, Miyawaki S, Maruta A, Emi N, Miyazaki Y, Ohtake S, Jinnai I, Matsuo K, Naoe T, Ohno R. High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol. 2006;24:460–466. doi: 10.1200/JCO.2005.03.2177. [DOI] [PubMed] [Google Scholar]
- 26.Ali Y, Lin Y, Gharibo MM, Gounder MK, Stein MN, Lagattuta TF, Egorin MJ, Rubin EH, Poplin EA. Phase I and pharmacokinetic study of imatinib mesylate (Gleevec) and gemcitabine in patients with refractory solid tumors. Clin Cancer Res. 2007;13:5876–5882. doi: 10.1158/1078-0432.CCR-07-0883. [DOI] [PubMed] [Google Scholar]
- 27.Matei D, Emerson RE, Schilder J, Menning N, Baldridge LA, Johnson CS, Breen T, McClean J, Stephens D, Whalen C, Sutton G. Imatinib mesylate in combination with docetaxel for the treatment of patients with advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: a Hoosier Oncology Group trial. Cancer. 2008;113:723–732. doi: 10.1002/cncr.23605. [DOI] [PubMed] [Google Scholar]
- 28.Mathew P, Fidler IJ, Logothetis CJ. Combination docetaxel and platelet-derived growth factor receptor inhibition with imatinib mesylate in prostate cancer. Semin Oncol. 2004;31:24–29. doi: 10.1053/j.seminoncol.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 29.Rezai K, Lokiec F, Grandjean I, Weill S, de Cremoux P, Bordier V, Ekue R, Garcia M, Poupon MF, Decaudin D. Impact of imatinib on the pharmacokinetics and in vivo efficacy of etoposide and/or ifosfamide. BMC Pharmacol. 2007;7:13. doi: 10.1186/1471-2210-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bauman JE, Eaton KD, Martins RG. Antagonism of platelet-derived growth factor receptor in non small cell lung cancer: rationale and investigations. Clin Cancer Res. 2007;13:s4632–s4636. doi: 10.1158/1078-0432.CCR-07-0212. [DOI] [PubMed] [Google Scholar]
- 31.Reardon DA, Egorin MJ, Quinn JA, Rich JN, Gururangan S, Vredenburgh JJ, Desjardins A, Sathornsumetee S, Provenzale JM, Herndon JE, II, Dowell JM, Badruddoja MA, McLendon RE, Lagattuta TF, Kicielinski KP, Dresemann G, Sampson JH, Friedman AH, Salvado AJ, Friedman HS. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005;23:9359–9368. doi: 10.1200/JCO.2005.03.2185. [DOI] [PubMed] [Google Scholar]
- 32.Pietras K, Ostman A, Sjoquist M, Buchdunger E, Reed RK, Heldin CH, Rubin K. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001;61:2929–2934. [PubMed] [Google Scholar]
- 33.Pietras K, Rubin K, Sjoblom T, Buchdunger E, Sjoquist M, Heldin CH, Ostman A. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62:5476–5484. [PubMed] [Google Scholar]
- 34.Pietras K, Stumm M, Hubert M, Buchdunger E, Rubin K, Heldin CH, McSheehy P, Wartmann M, Ostman A. STI571 enhances the therapeutic index of epothilone B by a tumor-selective increase of drug uptake. Clin Cancer Res. 2003;9:3779–3787. [PubMed] [Google Scholar]
- 35.Parise RA, Ramanathan RK, Hayes MJ, Egorin MJ. Liquid chromatographic-mass spectrometric assay for quantitation of i-matinib and its main metabolite (CGP 74588) in plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;791:39–44. doi: 10.1016/s1570-0232(03)00206-x. [DOI] [PubMed] [Google Scholar]
- 36.Parise RA, Ramanathan RK, Zamboni WC, Egorin MJ. Sensitive liquid chromatography-mass spectrometry assay for quantitation of docetaxel and paclitaxel in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;783:231–236. doi: 10.1016/s1570-0232(02)00659-1. [DOI] [PubMed] [Google Scholar]
- 37.Karlsson MO, Molnar V, Freijs A, Nygren P, Bergh J, Larsson R. Pharmacokinetic models for the saturable distribution of paclitaxel. Drug Metab Dispos. 1999;27:1220–1223. [PubMed] [Google Scholar]
- 38.Henningsson A, Karlsson MO, Vigano L, Gianni L, Verweij J, Sparreboom A. Mechanism-based pharmacokinetic model for paclitaxel. J Clin Oncol. 2001;19:4065–4073. doi: 10.1200/JCO.2001.19.20.4065. [DOI] [PubMed] [Google Scholar]
- 39.Smorenburg CH, ten Tije AJ, Verweij J, Bontenbal M, Mross K, van Zomeren DM, Seynaeve C, Sparreboom A. Altered clearance of unbound paclitaxel in elderly patients with meta-static breast cancer. Eur J Cancer. 2003;39:196–202. doi: 10.1016/s0959-8049(02)00611-1. [DOI] [PubMed] [Google Scholar]
- 40.Peng B, Lloyd P, Schran H. Clinical pharmacokinetics of imatinib. Clin Pharmacokinet. 2005;44:879–894. doi: 10.2165/00003088-200544090-00001. [DOI] [PubMed] [Google Scholar]
- 41.Spratlin J, Sawyer MB. Pharmacogenetics of paclitaxel metabolism. Crit Rev Oncol Hematol. 2007;61:222–229. doi: 10.1016/j.critrevonc.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 42.Rodt SA, Ahlen K, Berg A, Rubin K, Reed RK. A novel physiological function for platelet-derived growth factor-BB in rat dermis. J Physiol. 1996;495(Pt 1):193–200. doi: 10.1113/jphysiol.1996.sp021584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 44.Al-Batran SE, Atmaca A, Schleyer E, Pauligk C, Hosius C, Ehninger G, Jager E. Imatinib mesylate for targeting the platelet-derived growth factor beta receptor in combination with fluorouracil and leucovorin in patients with refractory pancreatic, bile duct, colorectal, or gastric cancer–a dose-escalation Phase I trial. Cancer. 2007;109:1897–1904. doi: 10.1002/cncr.22622. [DOI] [PubMed] [Google Scholar]
- 45.Spigel DR, Hainsworth JD, Simons L, Meng C, Burris HA, III, Yardley DA, Grapski R, Schreeder M, Mallidi PV, Greco FA. Irinotecan, carboplatin, and imatinib in untreated extensive-stage small-cell lung cancer: a phase II trial of the Minnie Pearl Cancer Research Network. J Thorac Oncol. 2007;2:854–861. doi: 10.1097/JTO.0b013e31814617b7. [DOI] [PubMed] [Google Scholar]