

Abstract
Objective
The G-protein-coupled receptor (GPCR)-kinase interacting protein-1 (GIT1) is a scaffold protein that is important for phospholipase Cγ (PLCγ) and extracellular signal–regulated kinase (ERK1/2) signaling induced by angiotensin II (AngII) and epidermal growth factor (EGF). Because GIT1 regulates signaling by several vascular smooth muscle cell (VSMC) growth factors, we hypothesized that intima formation would be inhibited by GIT1 depletion.
Approach and Results
Complete carotid ligation was performed on GIT1 wild type (WT) and knockout (KO) mice. We compared changes between GIT1 WT and KO mice in carotid vascular remodeling, VSMC proliferation and apoptosis in vivo and in vitro. Our data demonstrated that GIT1 deficiency significantly decreased intima formation after carotid ligation due to both reduced VSMC proliferation and enhanced apoptosis. To confirm the effects of GIT1 in vitro, we performed proliferation and apoptosis assays in VSMC. In mouse aortic smooth muscle cells (MASM), we found that the growth rate and [3H]-thymidine incorporation of the GIT1 KO MASM were significantly decreased compared to the WT MASM. Cyclin D1, which is a key cell cycle regulator, was significantly decreased in GIT1 KO cells. Serum deprivation of GIT1 KO MASM increased apoptosis 3-fold compared to WT MASM. Treatment of rat aortic smooth muscle cells (RASM) with GIT1 siRNA impaired cell migration. Both PLCγ and ERK1/2 signaling were required for GIT1 dependent VSMC proliferation and migration, whereas only PLCγ was involved in GIT1 mediated VSMC apoptosis.
Conclusions
GIT1 is a novel mediator of vascular remodeling by regulating VSMC proliferation, migration and apoptosis through PLCγ and ERK1/2 signaling pathways.
Keywords: GIT1, VSMC proliferation, VSMC apoptosis, intima formation, phospholipase Cγ, extracellular signal–regulated kinase (ERK) 1/2
Introduction
The proliferation and migration of vascular smooth muscle cells (VSMC) contributes to the development of atherosclerosis and restenosis after angioplasty1, 2. Several growth factors that activate G-protein-coupled receptors (GPCR such as endothelin-1 and angiotensin II (AngII)) and tyrosine kinase receptors (TKRs, such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF)) trigger and control these processes2–4. We and others previously showed a number of shared signal events downstream of these receptors that require tyrosine phosphorylation5, 6 mediated by c-Src. Among these signals, we found that a key molecule was the G-protein-coupled receptor (GPCR)-kinase interacting protein-1 (GIT1). GIT1 is a multi-domain scaffold protein7 that associates with GRK2 to regulate GPCR endocytosis8; GIT1 interacts with MEK1 to activate extracellular signal-regulated kinase (ERK) 1/29; it can also bind to phospholipase Cγ (PLCγ), which is required for PLCγ phosphorylation10. In addition, our laboratory showed that GIT1 is required for ERK1/2 translocation and activation in focal adhesions, which are important for cell spreading and migration9, 11. Based on these data, we propose that GIT1 is essential for VSMC proliferation, apoptosis and migration through PLCγ and ERK1/2 pathway induced by GPCR and TKR, which subsequently affects vascular remodeling.
Results
Intima Formation Was Decreased in GIT1 KO Mice
To determine the physiological role of GIT1 in vascular remodeling, we performed complete carotid artery ligation on GIT1 WT and KO mice (n=8 in each group). At 3 months age, there were no significance differences in the endothelium, media and adventitia in GIT1 WT and KO mice. Four weeks after carotid artery ligation, WT mice exhibited a substantial increase in the intima (Fig. 1A–B & E, 6.37±1.3 ×104 μm2), while GIT1 KO mice had significantly less intima (Fig. 1C–D & E, 3.05±0.72 ×104 μm2, p<0.05) and a smaller intima/media ratio (Fig. 1F, 0.86±0.12 vs 0.45± 0.06, p<0.05). However, there was no significant difference in media or adventitia area between the two genotypes. In addition, we did not observer any differences between sham groups.
Fig. 1. Intima formation was reduced in GIT1 KO mice.
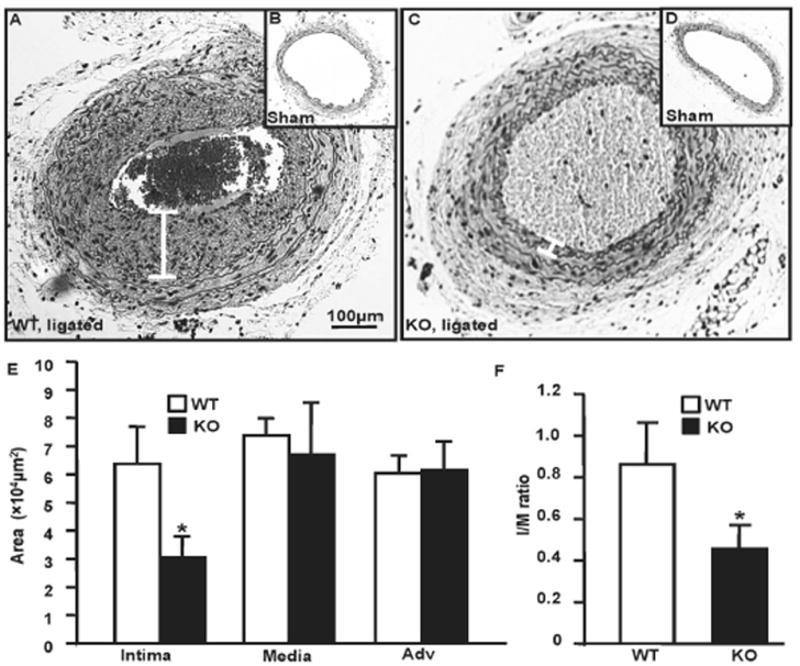
A–D. Morphometry of carotid arteries from sham and ligated mice (28 days after ligation). Representative cross sections of carotid arteries on day 28 (H&E staining). White line indicates intima width. Bar is 100μm. E–F. Quantification of intima, media, adventitial volume and intima/media ratio. There is a significant decrease in intima volume and I/M ratio in GIT1 KO mice (*p<0.05 vs GIT1 WT mice, n=8).
GIT1 Expression Was Enhanced after Complete Ligation
To determine the role of GIT1 in vascular remodeling, we measured the expression of GIT1 in carotid arteries after ligation or sham surgery in GIT1 WT and KO mice. GIT1 was expressed in both VSMC and endothelial cells in GIT1 WT carotid artery (Fig. 2A). After ligation, GIT1 expression increased in intima of WT mice (Fig. 2B). Western blot data demonstrated that GIT1 expression increased by 3.01-fold in intima of WT mice compared to the sham group (Fig. 2E–F). However, there was no GIT1 expression in GIT1 KO mice (Fig. 2C–E). To confirm our finding in vitro, GIT1 expression was detected in RASM treated with various growth factors including AngII, PDGF, EGF and FBS. We found that treatment with AngII, PDGF or FBS for 8h significantly increased GIT1 mRNA expression as measured by standard end-point PCR (Supplemental Fig.I, 2.87, 2.83 and 2.73 fold respectively). Consistently, GIT1 protein expression significantly increased after 24h stimulation with AngII, FBS and PDGF (Fig. 2. G–H). However, administration of EGF had no effect on GIT1 expression (data not shown).
Fig. 2. GIT1 protein expression was highly regulated in vivo and in vitro.
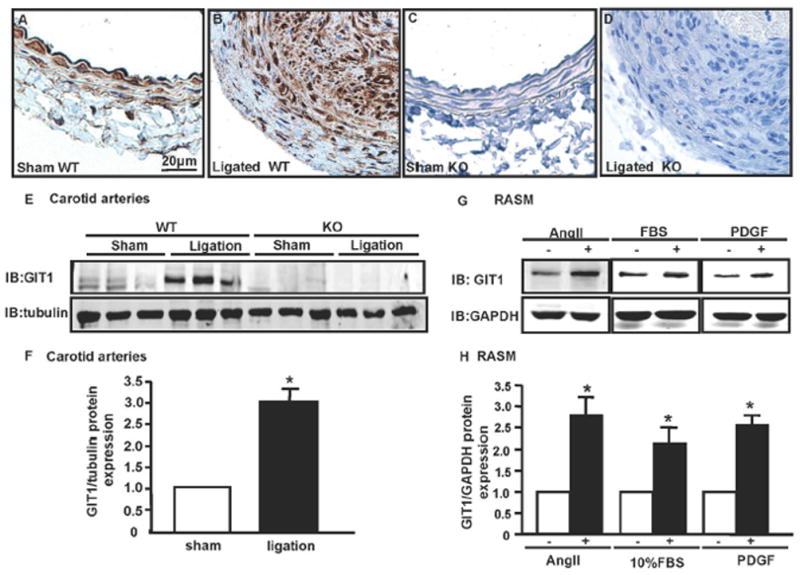
A–D. Immunostaining of GIT1 in carotid arteries of WT and KO mice 14 days after sham or ligation. GIT1 is highly expressed in the intima. Bar=20μm. E. Immunoblot of GIT1 expression in carotid arteries in GIT1 WT and KO mice 7 days after ligation. F. Quantification of GIT1 relative expression normalized to tubulin (Sham = 1.0). *p< 0.05 compared with sham group (mean ±SE; n =3). G. RASM were cultured in serum-free medium for 24h, then treated with and without AngII (10−7 m/L), PDGF (10ng/ml) and 10% FBS for 24h. GIT1 expression was detected by western blot. H. Quantification of GIT1 relative expression normalized to GAPDH (control = 1.0). *p< 0.05 compared with control group (mean ±SE; n =3).
GIT1 Was Required for VSMC Proliferation
To determine the role of GIT1 in VSMC proliferation in vivo, staining for PCNA, a cell proliferation maker, was performed on paraffin sections of carotid arteries (14 days after ligation). PCNA positive cells in the GIT1 KO mice were significantly reduced compared to the GIT1 WT mice (Fig. 3A–C, WT 10.01±0.67% vs KO 1.24±0.72%), which suggests that GIT1 is critical for cell proliferation. To confirm a difference in rates of proliferation, MASM were isolated from GIT1 WT and KO mice. MASM proliferation was measured by cell number counting and [3H]-thymidine incoporation. In response to 5% serum, the growth rate of KO cells was dramatically decreased compared to WT cells (Fig. 3D). [3H]-thymidine incorporation in response to 5% FBS of GIT1 KO cells was significantly decreased compared with that of WT (0.98±0.21 vs 3.18±0.35, P<0.05; n=6 in each group, Fig. 3E). We published previously that GIT1 is critical for PLCγ and ERK1/2 activation9, 10. We presume that GIT1 mediated VSMC proliferation is through PLCγ-ERK1/2 pathway. Thus, we determined the effects of PD98059 (ERK1/2 inhibitor) and U73122 (PLCγ inhibitor) on VSMC proliferation. The growth curve in Figure 3F showed that inhibition of either PLCγ or ERK1/2 significantly reduced the RASM growth induced by FBS, which suggested that GIT1- PLCγ-ERK1/2 pathway is important for VSMC proliferation. In addition, U73122 had stronger inhibitory effect than PD98059, which implied other downstream targets involved in GIT1-PLCγ pathway beyond ERK1/2.
Fig. 3. GIT1 was required for VSMC proliferation in vivo and in vitro.
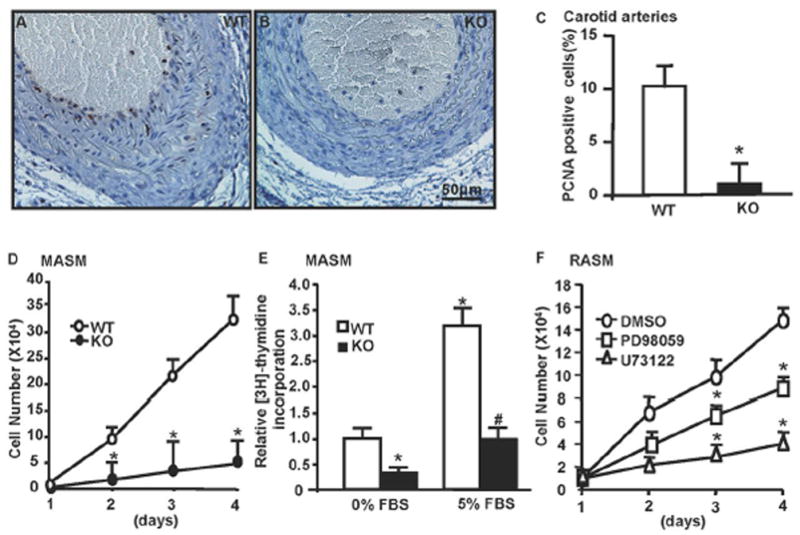
A–B. Immunostaining of PCNA in sham and ligated carotids from WT and KO mice at day 14 after ligation. Brown cells indicate PCNA positive cells. Bar=50μm. C. Quantification of PCNA positive cells in GIT1 WT and KO mice shows a significant decrease in proliferating cells in GIT1 KO (mean ±SE; *p<0.05 vs WT, n =5). D. GIT1 WT and KO MASM were stimulated with 5% serum for the indicated time points. The number of cells was significantly decreased at all times in GIT1 KO MASM. Data represent mean of triplicates (mean±SE), *p<0.05 vs WT group; n=3. E. GIT1 WT and KO MASM were stimulated with 5% serum for 24h and [3H]-thymidine incorporation was assayed. The [3H]-thymidine incorporation was significantly decreased in both FBS free and 5%FBS in GIT1 KO MASM. (*p<0.05 vs WT group without FBS; #p<0.05 vs WT group+ 5% FBS). F. RASM were stimulated with 5% serum in the presence or absence of DMSO, PD98059 (30nm/L) or U73122 (1μm) for the indicated time points. Cells number was significantly decreased in the PD98059 or U73122 treated group. Data represent mean of triplicates (mean±SE), *p<0.05 vs control group (DMSO treated cell), n=4.
GIT1 Was Critical for Cell Cycle Progression
In mammalian cells, cyclin D1 regulates the G1 to S cell cycle transition12. To show further the role of GIT1 in VSMC growth, we measured cyclin D1 expression induced by serum in GIT1 WT and KO MASM. Serum stimulation for 9h increased cyclin D1 expression by 2 fold in GIT1 WT MASM, while in GIT1 KO MASM, both baseline and FBS induced cyclinD1 expression decreased, which is consistent with the finding that GIT1 KO MASM showed significantly decreased cell proliferation both at basal condition and after FBS treatment (Fig. 4A–B, Fig. 3E). Inhibition of PLCγ and ERK1/2 showed similar effects to GIT1 deletion (Fig. 4C–D). We next evaluated the stage of cell cycle most regulated by GIT1 in MASM. As expected, GIT1 deletion dramatically decreased the amount of cells in S phase with a corresponding increase in G0–G1 (Fig. 4E).
Fig. 4. Effects of GIT1 on cell cycle progression.
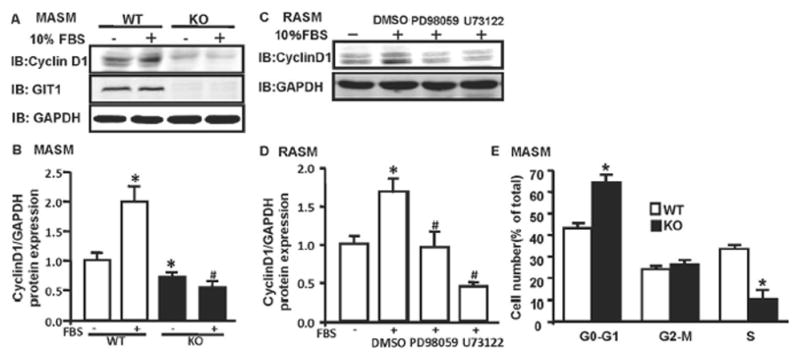
A. MASM from GIT1 WT and KO mice were treated with 10% FBS for 9h and cyclin D1, GIT1 and GAPDH expression was measured by western blot. B. Quantification of the relative expression of cyclinD1 normalized to GAPDH (untreated WT group = 1.0). *p< 0.05 compared with untreated WT group, # p< 0.05 compared with FBS treated WT group (mean ±SE; n =3). C. RASM were pretreated with DMSO, PD98059 (30nm/L) or U73122 (1μm/L) for 30min, then stimulated with 5% serum for 9h. D. Quantification of the relative expression of cyclinD1 normalized to GAPDH (untreated group = 1.0). *p< 0.05 compared with untreated group, # p< 0.05 compared with FBS treated group (mean ±SE; n =3). E. MASM from GIT1 WT and KO mice were growth-arrested by serum starvation for 24h and treated with 10% serum for 24h. Cells were then collected and subjected to flow cytometry (mean ±SE; *p<0.05 vs WT, n =7).
GIT1 Reduced VSMC Apoptosis
It has been shown that VSMC apoptosis plays a key role in vascular remodeling13. To elucidate the role of GIT1 in VSMC apoptosis, we performed TUNEL assays on carotid arteries 1 and 14 days after ligation. We detected apoptotic cells at both the early time point (1 day) and later time point (14 days) after ligation. We did not observe any difference in apoptotic cells at the early time point (data not shown), but at the later time point, the number of apoptotic cells in the GIT1 KO carotid was significantly increased compared to GIT1 WT (4.3±0.61 fold, p<0.05, Fig. 5A–C). To confirm the role of GIT1 in VSMC apoptosis, we studied the effect of serum starvation on MASM isolated from GIT1 WT and KO aortas. Serum starvation significantly increased apoptosis in KO compared to WT cells (23.1±2.1% vs 8.56±2.67%, p<0.05, Fig. 5D–F). These data show that GIT1 is required for VSMC survival. Furthermore, the effects of PLCγ and ERK1/2 on VSMC apoptosis were assayed. Interestingly, we found that inhibition of PLCγ increased RASM apoptosis by 8.63 fold (Fig. 5G–I), whereas inhibition of ERK1/2 had no effect on RASM apoptosis (data not shown). These data suggested that GIT1-PLCγ pathway is crucial for VSMC survival.
Fig. 5. GIT1 was important for cell survival in vivo and in vitro.

A–B. TUNEL assay was performed on carotid arteries of ligated GIT1 WT and KO mice at day 14 after ligation. The red staining indicates TUNEL positive cells. GIT1 KO carotids demonstrated significant an increase in apoptotic cells (arrow). Bar=10μm. C. Quantification of TUNEL positive cells in both groups (mean ±SE; *p<0.05 vs WT, n =5). D–F. MASM from GIT1 WT and KO mice were growth-arrested by serum starvation for 24h. Apoptotic cells were identified by condensed nuclei (arrow) after Hoechst staining. Data are mean ±SE; * p<0.05 vs WT, n=5. G–I. RASM were growth-arrested by serum starvation for 24h in the presence of DMSO or U73122 (1μm). Apoptotic cells were identified by condensed nuclei (arrow) after Hoechst staining. Data are mean ±SE; *p<0.05 vs DMSO treated, n=5.
GIT1 Was Critical for VSMC Migration
VSMC migration is critical for vascular remodeling14. GIT1 has been shown to play an important role in migration of various cell types, including endothelial cell, neuron, fibroblast and A7R5 cell15–17. However, the role of GIT1 in primary cultured VSMC migration is still unknown. Thus, wound scratch assay was performed on RASM treated with control siRNA or GIT1 siRNA (Fig. 6A–E). Consistent with previous findings, GIT1 depleted cells demonstrated impaired migration stimulated by PDGF, although basal migration did not appear to differ at this level of GIT1 depletion11, 15, 17. Moreover, inhibition of PLCγ and ERK1/2 showed similar results as GIT1 depleted cells (Fig. 6F–J). All these data suggests an important role of GIT1-PLCγ and ERK1/2 pathway in VSMC migration.
Fig. 6. GIT1 was important for VSMC migration.

A–D. RASM were transfected with control siRNA or GIT1 siRNA for 48h. Scratch wounds were created in the monolayer using 100mm sterile pipet tips. Cells were treated with PDGF (10ng/ml) for 16h. Images are representative of wound area after 16h treatment. E. GIT1 expression was detected by western blot. Wound area was analyzed by using Image J. * p<0.05 vs untreated control siRNA group, # p<0.05 vs PDGF+control siRNA group; n=3. F–I. RASM were growth-arrested by serum starvation for 24h. Scratch wounds were created and cells were treated with PDGF (10ng/ml) in the presence of DMSO, PD98059 (30nm/L) or U73122 (1μm/L) for 16h. Images are the representative of wound area after 16h treatment. J. Wound area was analyzed by using Image J. *p<0.05 vs untreated control siRNA group, # p<0.05 vs PDGF+DMSO group; n=3.
GIT1 Was Required for ERK1/2 Activation after Carotid Ligation
Both GPCR and TKR signaling are essential for vascular remodeling. We have published previously that GIT1 is required for PLCγ and ERK1/2 activation induced by AngII and EGF9, 10. To explore the role of GIT1 in PLCγ and ERK1/2 activation in vivo during vascular remodeling, we measured PLCγ and ERK1/2 phosphorylation in carotid arteries after 7 and 14 days ligation by immunohistochemistry and western blot. Due to the limitation of the phospho-PLCγ antibody, we couldn’t detect the expression of phospho-PLCγ at any time point (data not shown). There was minimal ERK1/2 activation at 14 days (data not shown). However, ERK1/2 activation was dramatically increased in VSMC in the GIT1 WT carotid at 7 days, whereas there was no change in the GIT1 KO carotid and sham groups (Supplemental Fig. II A–D). Western blot analysis showed that there was 2.47± 0.27 fold increase in ERK1/2 activation in GIT1 WT ligated carotid compared to WT sham (Supplemental Fig. II E–F). In contrast, there was no obvious increase in ERK1/2 activation in KO carotid.
Discussion
The major finding of this study is that GIT1 is an important regulator of vascular remodeling. Specifically, we found that GIT1 depletion inhibited intima formation after carotid ligation by 50%. In vivo and in vitro analysis showed a key role for GIT1 in VSMC proliferation, migration and apoptosis during vascular remodeling (Supplemental Fig. III). Furthermore, GIT1 is required for VSMC proliferation through PLCγ and ERK1/2 by regulating the expression of cell cycle related proteins, such as cyclin D1. GIT1 is also essential for cell survival by regulating VSMC apoptosis through PLCγ and cell migration through PLCγ and ERK1/2.
VSMC proliferation and migration are key components in vascular remodeling1, 2. The present study shows that GIT1 expression is highly regulated by AngII and PDGF in vitro and during vascular remodeling in vivo. The role of GIT1 in mediating VSMC proliferation was demonstrated by several assays including in vitro cell count, [3H]-thymidine incorporation and cell cycle analysis. Several groups, including our laboratory, showed that GIT1 can regulate cytoskeleton dynamics during cell spreading and migration18–20. Our previous data demonstrated that GIT1-Y392 phosphorylation is critical for PDBU-induced podosome formation in A7R5 cell migration15. Recently, myosin 18A (MYO18A, a member of the myosin super family) was shown to interact with the PAK2/βPIX/GIT1 complex to modulate epithelial cell migration21. Our data demonstrate that GIT1 depletion in RASM impaired migration, which further confirmed previous findings specifically in primary cultured VSMC.
Previously, we showed that GIT1 was a scaffold protein for PLCγ and MEK1 and was required for the sustained activation of PLCγ and ERK1/2 by AngII and EGF9, 10. As a surrogate for VSMC growth signaling, we measured the effect of GIT1 on both PLCγ and ERK1/2 activation in vivo. Consistent with these observations, we found that GIT1 was required for ERK1/2 activation during vascular remodeling, whereas PLCγ activation was not detected due to the limitation of functional antibody. However, using specific PLCγ and ERK1/2 inhibitor, we confirmed the critical role of PLCγ and ERK1/2 in both VSMC proliferation and migration. Moreover, we observed a stronger inhibitory effect of U73122 on VSMC proliferation, which implies additional downstream target of PLCγ other than ERK1/2.
VSMC apoptosis is another critical process during vascular remodeling22. Several studies show that there are two waves of VSMC apoptosis23, 24. The first wave occurs in the media within hours of injury, which leads to decreased VSMC in the vessel wall. The second wave of apoptosis occurs at later time (several days or weeks) at lower frequencies, which is important for intima formation23, 24. Our data demonstrated that GIT1 reduced VSMC apoptosis in vivo and in vitro. Importantly, we found that GIT1 only affected the second wave of apoptosis in vivo and this effect is through PLCγ. The exact downstream target of PLCγ in VSMC apoptosis needs to be determined in future studies.
In the current study, we use both RASM and MASM for the experiments. There are three reasons for this. 1. RASM are a well-established tool for VSMC functional study. 2. GIT1 KO cells are susceptible to undergo apoptosis and grow very slowly. Therefore, it is difficult to perform cell migration assay (wound scratch experiment). 3. MASM easily transform to polyploid cells after passage 5, therefore we can only use cells between passages 3–5.
In conclusion, as a key scaffold protein in multiple growth factor signaling pathways, GIT1 can regulate VSMC proliferation, migration and apoptosis during vascular remodeling. These data suggest that GIT1 is a potential therapeutic target to reduce vascular remodeling.
Supplementary Material
Significance.
GIT1 is important for vascular remodeling by promoting cell proliferation, migration and preventing cell apoptosis.
Acknowledgments
Sources of Funding
This work was supported by a grant from NIH, HL63462 to BCB. This work was also supported by Scientist Development Grant from American Heart Association to Jinjiang Pang (0835626D).
Footnotes
Disclosures
None
References
- 1.Ross R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 2.Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 3.Murakoshi N, Miyauchi T, Kakinuma Y, Ohuchi T, Goto K, Yanagisawa M, Yamaguchi I. Vascular endothelin-B receptor system in vivo plays a favorable inhibitory role in vascular remodeling after injury revealed by endothelin-B receptor-knockout mice. Circulation. 2002;106:1991–1998. doi: 10.1161/01.cir.0000032004.56585.2a. [DOI] [PubMed] [Google Scholar]
- 4.Kirchengast M, Munter K. Endothelin and restenosis. Cardiovasc Res. 1998;39:550–555. doi: 10.1016/s0008-6363(98)00143-6. [DOI] [PubMed] [Google Scholar]
- 5.Berk BC, Alexander RW, Brock TA, Gimbrone MA, Jr, Webb RC. Vasoconstriction: a new activity for platelet-derived growth factor. Science. 1986;232:87–90. doi: 10.1126/science.3485309. [DOI] [PubMed] [Google Scholar]
- 6.Watling D, Guschin D, Müller M, Silvennoinen O, Witthuhn BA, Quelle FW, Rogers NC, Schindler C, Stark GR, Ihle JN, Kerr LM. Complementation by the protein tyrosine kinase JAK2 of a mutant cell line defective in the interferon-gamma signal transduction pathway. Nature. 1993;366:166–170. doi: 10.1038/366166a0. [DOI] [PubMed] [Google Scholar]
- 7.Hoefen RJ, Berk BC. The multifunctional GIT family of proteins. J Cell Sci. 2006;119:1469–1475. doi: 10.1242/jcs.02925. [DOI] [PubMed] [Google Scholar]
- 8.Premont RT, Vitale N. Purification and characterization of GIT family of ADP-ribosylation factor (ARF) GTPase-activating proteins. Methods Enzymol. 2001;329:335–343. doi: 10.1016/s0076-6879(01)29095-8. [DOI] [PubMed] [Google Scholar]
- 9.Yin G, Haendeler J, Yan C, Berk BC. GIT1 functions as a scaffold for MEK1-extracellular signal-regulated kinase 1 and 2 activation by angiotensin II and epidermal growth factor. Mol Cell Biol. 2004;24:875–885. doi: 10.1128/MCB.24.2.875-885.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haendeler J, Yin G, Hojo Y, Saito Y, Melaragno M, Yan C, Sharma VK, Heller M, Aebersold R, Berk BC. GIT1 mediates Src-dependent activation of phospholipase Cgamma by angiotensin II and epidermal growth factor. J Biol Chem. 2003;278:49936–49944. doi: 10.1074/jbc.M307317200. [DOI] [PubMed] [Google Scholar]
- 11.Yin G, Zheng Q, Yan C, Berk BC. GIT1 Is a Scaffold for ERK1/2 Activation in Focal Adhesions. J Biol Chem. 2005;280:27705–27712. doi: 10.1074/jbc.M502271200. [DOI] [PubMed] [Google Scholar]
- 12.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 13.Pollman MJ, Hall JL, Mann MJ, Zhang L, Gibbons GH. Inhibition of neointimal cell bcl-x expression induces apoptosis and regression of vascular disease. Nat Med. 1998;4:222–227. doi: 10.1038/nm0298-222. [DOI] [PubMed] [Google Scholar]
- 14.Ferns GA, Sprugel KH, Seifert RA, Bowen PDF, Kelly JD, Murray M, Raines EW, Ross R. Relative platelet-derived growth factor receptor subunit expression determines cell migration to different dimeric forms of PDGF. Growth Factors. 1990;3:315–324. doi: 10.3109/08977199009003674. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Yin G, Menon P, Pang J, Smolock EM, Yan C, Berk BC. Phosphorylation of G protein-coupled receptor kinase 2-interacting protein 1 tyrosine 392 is required for phospholipase C-gamma activation and podosome formation in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2010;30:1976–1982. doi: 10.1161/ATVBAHA.110.212415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webb DJ, Kovalenko M, Whitmore L, Horwitz AF. Phosphorylation of serine 709 in GIT1 regulates protrusive activity in cells. Biochem Biophys Res Commun. 2006;346:1284–1288. doi: 10.1016/j.bbrc.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Taba Y, Pang J, Yin G, Yan C, Berk BC. GIT1 mediates VEGF-induced podosome formation in endothelial cells: critical role for PLCgamma. Arterioscler Thromb Vasc Biol. 2009;29:202–208. doi: 10.1161/ATVBAHA.108.174391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manabe R, Kovalenko M, Webb DJ, Horwitz AR. GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J Cell Sci. 2002;115:1497–1510. doi: 10.1242/jcs.115.7.1497. [DOI] [PubMed] [Google Scholar]
- 19.Bagrodia S, Bailey D, Lenard Z, Hart M, Guan JL, Premont RT, Taylor SJ, Cerione RA. A tyrosine-phosphorylated protein that binds to an important regulatory region on the Cool family of p21-activated kinase-binding proteins. J Biol Chem. 1999;274:22393–22400. doi: 10.1074/jbc.274.32.22393. [DOI] [PubMed] [Google Scholar]
- 20.Lamorte L, Rodrigues S, Sangwan V, Turner CE, Park M. Crk Associates with a Multimolecular Paxillin/GIT2/{beta}-PIX Complex and Promotes Rac-dependent Relocalization of Paxillin to Focal Contacts. Mol Biol Cell. 2003;14:2818–2831. doi: 10.1091/mbc.E02-08-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu RM, Tsai MH, Hsieh YJ, Lyu PC, Yu JS. Identification of MYO18A as a novel interacting partner of the PAK2/betaPIX/GIT1 complex and its potential function in modulating epithelial cell migration. Mol Biol Cell. 2010;21:287–301. doi: 10.1091/mbc.E09-03-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han DK, Haudenschild CC, Hong MK, Tinkle BT, Leon MB, Liau G. Evidence for apoptosis in human atherogenesis and in a rat vascular injury model. Am J Pathol. 1995;147:267–277. [PMC free article] [PubMed] [Google Scholar]
- 23.Perlman H, Maillard L, Krasinski K, Walsh K. Evidence for the rapid onset of apoptosis in medial smooth muscle cells after balloon injury. Circulation. 1997;95:981–987. doi: 10.1161/01.cir.95.4.981. [DOI] [PubMed] [Google Scholar]
- 24.Walsh K, Smith RC, Kim HS. Vascular cell apoptosis in remodeling, restenosis, and plaque rupture. Circ Res. 2000;87:184–188. doi: 10.1161/01.res.87.3.184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.