

Abstract
Oxidative and nitrative stress responses resulting from inflammation exacerbate liver injury associated with nonalcoholic steatohepatitis (NASH) by inducing lipid peroxidation and protein nitration. The objective of this study was to investigate whether the anti-inflammatory properties of green tea extract (GTE) would protect against NASH by suppressing oxidative and nitrative damage mediated by proinflammatory enzymes. Obese mice (ob/ob) and their 5-week-old C57BL6 lean littermates were fed 0%, 0.5% or 1% GTE for 6 weeks (n=12–13 mice/group). In obese mice, hepatic lipid accumulation, inflammatory infiltrates and serum alanine aminotransferase activity were markedly increased, whereas these markers of hepatic steatosis, inflammation and injury were significantly reduced among obese mice fed GTE. GTE also normalized hepatic 4-hydroxynonenal and 3-nitro-tyrosine (N-Tyr) concentrations to those observed in lean controls. These oxidative and nitrative damage markers were correlated with alanine aminotransferase (P<.05; r=0.410–0.471). Improvements in oxidative and nitrative damage by GTE were also associated with lower hepatic nicotinamide adenine dinucleotide phosphate oxidase activity. Likewise, GTE reduced protein expression levels of hepatic myeloperoxidase and inducible nitric oxide synthase and decreased the concentrations of nitric oxide metabolites. Correlative relationships between nicotinamide adenine dinucleotide phosphate oxidase and hepatic 4-hydroxynonenal (r=0.364) as well as nitric oxide metabolites and N-Tyr (r=0.598) suggest that GTE mitigates lipid peroxidation and protein nitration by suppressing the generation of reactive oxygen and nitrogen species. Further study is warranted to determine whether GTE can be recommended as an effective dietary strategy to reduce the risk of obesity-triggered NASH.
Keywords: Green tea, NADPH oxidase, Myeloperoxidase, Lipid peroxidation, Protein nitration, Nonalcoholic steatohepatitis
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is a constellation of pathologic changes beginning with relatively benign steatosis and progressing to nonalcoholic steatohepatitis (NASH), fibrosis and cirrhosis [1]. The incidence of NAFLD, which is estimated to afflict ~40 million Americans [2], has risen concurrently with the obesity epidemic that now afflicts two thirds of adults [3]. No validated treatments of NAFLD exist beyond weight loss or comorbidity management. Weight loss interventions have a poor success rate [4], thereby emphasizing the need to validate alternative dietary strategies that effectively mitigate the progression to NASH.
NASH increases the risk for liver-related morbidity and mortality [5] and is characterized by histologic evidence of excess hepatic lipid accumulation, lobular inflammation, hepatocyte ballooning and necrosis [2]. The mechanisms leading to NASH remain unclear, but the “two-hit” mechanism is often used as a simple description for its etiology [6]. The “first-hit” resulting in liver steatosis is caused by obesity, insulin resistance and excess lipid accumulation. Then, reactive oxygen/nitrogen species (ROS/RNS)-mediated “second-hits” result in oxidative and nitrative modification to lipids and proteins, exacerbated liver injury and NASH [7,8].
Reactive species implicated in the second-hit of NASH include those originating from mitochondria, cytochrome P-450 induction [9] and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase following inflammatory cell activation [10]. Greater hepatic NADPH oxidase activity increased liver inflammation, injury, lipid peroxidation (LPO) and fibrogenesis in a diet-induced obese model of NAFLD [11]. Other proinflammatory enzymes, including myeloperoxidase (MPO) and inducible nitric oxide synthase (iNOS), also cause hepatocyte injury in NASH. In NASH patients, greater accumulation of MPO-positive Kupffer cells was accompanied by liver injury induced by the MPO–H2O2 system, as evidenced by greater 3-nitro-tyrosine (N-Tyr) levels [12]. Likewise, greater hepatic iNOS in rodents with NASH was paralleled by higher RNS and N-Tyr in association with liver injury, inflammation and fibrosis [8]. However, it has yet to be determined whether regulating these inflammatory events and the resulting oxidative stress responses with novel dietary strategies could attenuate liver injury leading to the development of NASH.
Epidemiologic studies suggest that green tea consumption reduces serum aminotransferases [13], commonly used markers of liver injury. The hepatoprotective activities of green tea are attributed to its catechins that scavenge ROS/RNS in vitro [14–16]. Green tea also protects against oxidative stress indirectly through its hypolipidemic activities that reduce steatosis and by improving antioxidant defenses [17,18]. Epigallocatechin gallate (EGCG), the major green tea catechin, reduces iNOS expression and RNS release from macrophages [19]. It also inhibits NADPH oxidase translocation in mast cells [20]. However, whether green tea reduces these inflammatory responses in livers of obese (ob/ob) mice has yet to be determined. Although no experimental model completely mimics the natural etiology of NASH in humans [21,22], ob/ob mice are commonly used to study obesity-induced NASH because they become obese and develop severe steatosis and have greater inflammation [17,18]. Thus, this model recapitulates many characteristics of patients with NASH [2], thereby making these mice well suited for studies examining whether dietary interventions mitigate liver injury associated with NASH.
Our laboratory demonstrated that green tea extract (GTE) protects against NASH in ob/ob mice through its lipid-lowering and antioxidant activities [18]. GTE reduced liver steatosis by decreasing expression of adipose lipogenic genes and serum-free fatty acids. It also increased enzymatic antioxidant activities and decreased tumor necrosis factor α in association with reduced liver injury. These data strongly support that GTE may reduce the risk for NASH by down-regulating proinflammatory events that otherwise cause oxidative and nitrative damage and exacerbate liver injury. Thus, this study aimed to determine whether GTE mitigates liver injury by suppressing the activities of several inflammatory cell-derived enzymes known to generate ROS/RNS during the development of NASH. Our data reported herein provide the first evidence that GTE attenuates NADPH oxidase activity and the expression of MPO and iNOS in ob/ob mice, thereby suppressing oxidative and nitrative modifications that otherwise lead to liver injury and NASH.
2. Materials and methods
2.1. Materials
The following chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA): diphenyleneiodonium chloride, N-(1-naphthyl)ethylenediamine dihydrochloride, ethylenediamine-tetraacetic acid, ethylene glycol-bis-N, N, N′, N′-tetraacetic acid, flavin adenine dinucleotide, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), magnesium chloride, nitrate reductase from Aspergillus, β-NADPH, potassium chloride, protease inhibitor cocktail, sodium deoxycholate, sodium dodecyl sulfate, sulfanilamide and trizma hydrochloride. Nonidet-P40 was purchased from US Biological (Swampscott, MA, USA). Calcium chloride, D-glucose, magnesium sulfate, phosphate-buffered saline (PBS), phosphoric acid, potassium phosphate, sodium bicarbonate, sodium chloride, sodium nitrite, sodium phosphate and trichloroacetic acid were purchased from Fisher Scientific (Fair Lawn, NJ, USA).
2.2. Animals and study design
The protocol for the care and use of animals was approved by the Institutional Care and Use Committee at the University of Connecticut. Male leptin-deficient (ob/ob) mice and their C57BL6/J lean littermates (5 weeks old) were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and housed individually in a temperature- and humidity-controlled room with a 12-h light–dark cycle. After a 1-week acclimation, obese mice (ob/ob; n=12–13/group) were fed a modified AIN-93G diet containing GTE at 0%, 0.5% or 1% (wt/wt), and a group of lean mice (n=12) were fed the basal diet having no GTE. The modified AIN-93G diet was purchased from Dyets (Bethlehem, PA, USA) and contained egg white substituted for casein. GTE was provided by Unilever BestFoods (Englewood, NJ, USA) and contained 30% total catechins (wt/wt), as verified by high-performance liquid chromatography [17]. After a 6-week intervention, mice were starved overnight (10–12 h). Blood was collected from the retro-orbital sinus into anticoagulant-free collection tubes under isoflurane anesthesia. Following blood collection, mice were killed by cervical dislocation. Liver was excised, rinsed in ice-cold PBS, blotted, snap frozen in liquid nitrogen and stored at −80°C until analysis. A small portion of the same lobe from each mouse was processed for histologic examination. Liver and body mass, food intake and serum alanine aminotransferase (ALT) activity were determined, as described previously [18].
2.3. Liver histology
From formalin-fixed liver tissues embedded in paraffin, sections of 4–5 μm were stained with hematoxylin and eosin to examine general liver morphology and the extent of NAFLD. Images (200×) were captured using an Olympus IX70 microscope (Center Valley, PA, USA) and examined in a blinded manner.
2.4. Oxidative and nitrative damage
Liver 4-hydroxynonenal-histidine (HNE) protein adducts, a marker of LPO, were measured to assess hepatic oxidative damage. In brief, liver was homogenized in PBS containing protease inhibitor cocktail and centrifuged (13,000×g, 4°C, 10 min), and the protein concentration of the supernatant was determined using a Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA). The supernatant was diluted to 20 μg/ml, and HNE was measured from 100 μl of sample using a commercially available enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s instructions (catalog #STA-334; Cell Biolabs; San Diego, CA, USA). In brief, the kit uses a monoclonal anti-HNE–histidine (HIS) mouse immunoglobulin G (IgG) as the primary antibody. The antibody does not cross-react other reactive aldehydes generated during the decomposition of LPO products such as 4-hydroxyhexenaldehyde or malondialdehyde. The kit has a linear range of quantification from 1 to 10 μg/ml and a lower limit of detection of 1 μg/ml, and its intra- and interassay coefficient of variation was <7%.
Hepatic N-Tyr was measured to define the extent to which obesity and GTE affect protein nitration. Liver was homogenized in ice-cold radio immunoprecipitation assay (RIPA) buffer and centrifuged (16,000×g, 4°C, 30 min) as described [23]. The supernatant was diluted 1:300 and used to measure N-Tyr using a commercially available ELISA in accordance with the manufacturer’s instructions (catalog #21055; Oxis Research, Portland, OR, USA). In brief, sample N-Tyr binds with a monoclonal antibody that was generated from a nitrated keyhole limpet hemocyanin raised in mouse. Specificity of the antibody was confirmed against nitrated bovine serum albumin (N-BSA), and its binding affinity to N-BSA is unaffected by tyrosine, chloro-tyrosine and phenylalanine, whereas preincubation with purified N-Tyr inhibits antibody binding to N-BSA [24]. The kit has a limit of detection of 2 nmol/l and a range of quantification from 2.1 to 1500 nmol/l, and its intra- and interassay coefficient of variation was <8%.
2.5. Assessment of reactive oxygen and nitrogen species
NADPH oxidase activity was assessed because of its significant role in generating superoxide following inflammatory cell activation [10]. In brief, its activity was measured by lucigenin-enhanced chemiluminescence as described [25]. Liver homogenates prepared in ice-cold Krebs-4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer were centrifuged (1000×g, 10 min). The supernatant containing 100 μg of protein was incubated (10 min, 37°C) with 5 μM of lucigenin, and then hepatic NADPH oxidase activity was determined by recording luminescence (GloMax 20/20; Sunnyvale, CA, USA) at 10-s intervals for 10 min in the presence or absence of NADPH (100 μM). Control experiments were performed in parallel using diphenyle-neiodonium chloride (100 μM), an inhibitor of NADPH oxidase activity, to confirm that the measured activity was attributed to NADPH oxidase. Data are expressed as relative luminescence units per second per milligram of protein as described [26].
Hepatic total nitrate and nitrite (NOx), end-products of nitric oxide degradation, were measured as an index of RNS using the Griess reaction as described [27]. In brief, liver was homogenized in PBS containing protease inhibitor, centrifuged (13,000×g, 4°C, 10 min), and then 100 μl of diluted supernatant (1:100) was mixed with 5 U/ml nitrate reductase, 12.5 mM β-NADPH and 0.5 mM flavin adenine dinucleotide. The mixture was incubated (37°C, 90 min), then mixed with 62.5% ethanol and Griess reagent containing equal volumes of 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride prepared in water and 1% sulfanilamide in 6% phosphoric acid. After a 30-min incubation, proteins were precipitated by adding 3% trichloroacetic acid (wt/vol; final concentration), the sample was centrifuged (10,000×g, 2 min) and the absorbance of the supernatant was determined at 540 nm. NOx concentrations were determined from a standard curve prepared in parallel using sodium nitrite and normalized to hepatic protein.
2.6. Proinflammatory protein expression
Western blotting was performed to measure protein expression levels of MPO and iNOS, as described previously [28]. In brief, liver was homogenized in 5 vol of ice-cold RIPA buffer containing protease inhibitor cocktail. Following centrifugation (16,000×g, 4°C, 30 min), the supernatant was collected, and 20 μg (iNOS) or 50 μg (MPO) of protein was resolved using an 8% (MPO) or 10% (iNOS) sodium dodecyl sulfate polyacrylamide gel. Separated proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane. Membranes were incubated with rabbit polyclonal antibodies against iNOS (1:2000 dilution) or MPO (1:1000 dilution; Abcam, Cambridge, MA, USA), followed by horseradish peroxidase-conjugated antirabbit IgG (Sigma-Aldrich). Protein loading was verified by probing blots with antibodies against β-actin (1:5000 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). iNOS (131 kd), MPO (84 kd) and β-actin (43 kd) were detected using enhanced chemiluminescence substrate (Pierce Biotechnology, Rockford, IL, USA) and quantified using NIH Image J.
Immunohistochemistry was also performed to visualize the localization of hepatic iNOS. Formalin-fixed, paraffin-embedded liver sections (4–5 μm) were incubated overnight with rabbit polyclonal antibodies against iNOS, followed by HRP-conjugated goat antirabbit IgG (Vector Laboratories, Burlingame, CA, USA). Sections were stained with 3,3′-diaminobenzidine (DAB) substrate to visualize the reaction and counter-stained with hematoxylin, and then images (400×) were examined using an Olympus BX41 microscope (Center Valley, PA).
2.7. Statistical analysis
Data were analyzed using GraphPad Prism (version 5; GraphPad Software, La Jolla, CA, USA). One-way analysis of variance with Newman–Keuls posttest, as appropriate, was performed to evaluate group mean differences. Linear regression analysis was performed to evaluate correlations between study variables. All data are expressed as means±S.E. Statistical significance was set at an α level of P<.05 for all analyses.
3. Results
3.1. GTE attenuates obesity-induced liver steatosis and inflammation
Details of the GTE-mediated effects on antioxidant defenses and biochemical markers of liver steatosis and injury in these mice have been reported previously [18]. In brief, obese mice (ob/ob; 29.4±1.0 g) at 5 week of age weighed 41% more than lean controls (20.9±0.5 g) consistent with their phenotypic propensity to become obese. Following the 6-week intervention, obese controls fed no GTE had 37% greater body mass than lean littermates (42.9±0.7 vs. 31.4±0.6 g). GTE at 0.5% did not affect liver or body mass, but GTE at 1% decreased these parameters by 16% and 46%, respectively, without affecting daily food intake [18]. The ratio of liver mass to body mass was unaffected by GTE. In addition, obese mice had 7.3 times higher serum ALT, which decreased by 19%–25% with GTE. In the present study, histologic analysis of liver sections clearly showed that obese (ob/ob) mice, in agreement with others [17,29], had severe macrovesicular steatosis and moderate inflammation, as evidenced by greater inflammatory cell infiltrates (Fig. 1). In contrast, little or no visual evidence of steatosis or inflammation was apparent in lean mice. Obese mice fed GTE at either level had reduced severity of liver inflammation compared with obese controls fed no GTE (Fig. 1C–D). However, GTE at 1% reduced liver steatosis (Fig. 1D) consistent with our earlier findings where GTE at ≥1% decreases hepatic lipid accumulation [17,18].
Fig. 1.
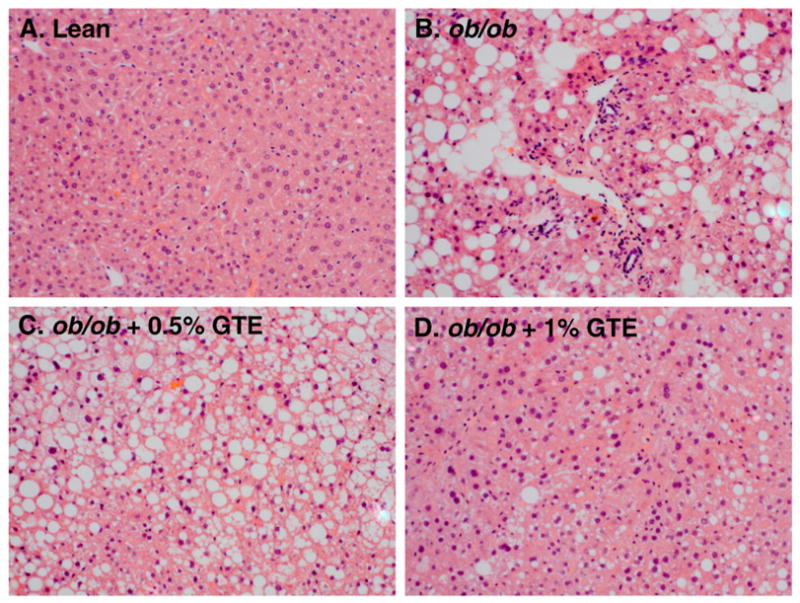
Representative hematoxylin and eosin-stained liver sections from a lean mouse (A) and ob/ob mice fed no GTE (B), 0.5% GTE (C), or 1% GTE (D) for 6 weeks. Hepatic steatosis and inflammatory cell infiltration were apparent in obese mice fed no GTE compared with lean mice. Obese mice fed GTE at 1% markedly reduced obesity-induced steatosis and inflammation (original magnification 200×).
3.2. GTE inhibits hepatic LPO by decreasing ROS generation
ROS-mediated LPO exacerbates hepatocyte injury by inducing cell death as well as by forming adducts between hepatic proteins and aldehyde end-products of LPO [7]. Consistent with obesity-induced oxidative stress, hepatic HNE levels were 22% greater (P<.05) in obese controls fed no GTE compared with the lean littermate mice. GTE at both levels decreased HNE by 17%–19% (Fig. 2A). Regression analysis indicated that liver HNE and serum ALT activity were correlated (r=0.410; P<.05), suggesting that GTE mitigated obesity-induced liver injury in part by reducing LPO.
Fig. 2.
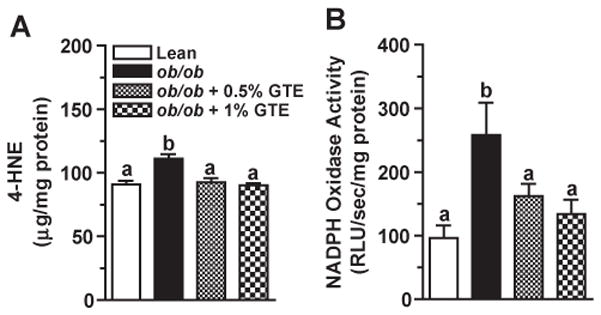
(A) Hepatic 4-HNE concentrations in lean and obese (ob/ob) mice fed 0%, 0.5% or 1% GTE for 6 weeks (n=12–13 mice/group; means±S.E.). Liver homogenates were prepared in RIPA buffer, and HNE was measured by ELISA. (B) Hepatic NADPH oxidase activity in lean and obese (ob/ob) mice fed GTE at 0%, 0.5% and 1% for 6 weeks (n=12–13 mice/group; means±S.E.). NADPH oxidase from liver homogenates was evaluated by lucigenin-enhanced chemiluminescence following incubation with lucigenin and NADPH. Means not sharing a common superscript are significantly different, P<.05.
Following inflammatory cell activation, NADPH oxidase rapidly generates superoxide [10] and increases oxidative stress during the development of NASH [7]. Obese controls fed no GTE had 2.7 times greater hepatic NADPH oxidase activity compared with the lean mice (Fig. 2B) consistent with their greater accumulation of inflammatory infiltrates (Fig. 1B). GTE decreased NADPH oxidase activity by 37%–48% and normalized it to that observed in lean controls. NADPH oxidase activity was also correlated (P<.05) with HNE (r=0.364) and serum ALT (r=0.444).
3.3. GTE reduces RNS-mediated protein nitration
RNS-mediated hepatic protein nitration causes protein dysfunction and provokes hepatic inflammation during the progression to NASH [8]. Obese mice fed no GTE had 54% greater hepatic N-Tyr than lean controls. GTE dose-dependently decreased N-Tyr up to 40% (P<.05; Fig. 3A). GTE at 1% reduced protein nitration to the level observed in the lean controls. To better define GTE-mediated improvements in nitrative damage, we also measured NOx as an index of RNS generation. Consistent with greater N-Tyr levels, obese controls had 4.9 times greater hepatic NOx concentrations (Fig. 3B). Obese mice fed GTE had 23%–34% lower hepatic NOx compared with obese controls fed no GTE. Hepatic N-Tyr was correlated (P<.05) with NOx (r=0.598) and serum ALT (r=0.471).
Fig. 3.
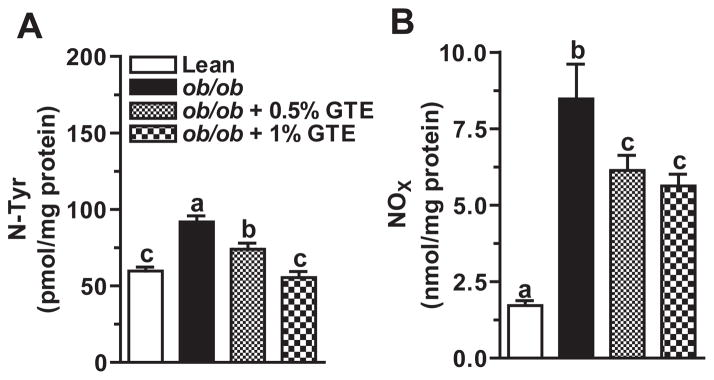
(A) Hepatic N-Tyr in lean and obese (ob/ob) mice fed 0%, 0.5% and 1% GTE for 6 weeks (n=12–13 mice/group; means±S.E.). N-Tyr was measured by ELISA from liver homogenates prepared in PBS. (B) Hepatic total nitrate/nitrite (NOx) in lean and obese (ob/ob) mice fed GTE at 0%, 0.5% and 1% for 6 weeks (n=12–13 mice/group; means ±S.E.). NOx was measured by the Griess reaction from liver homogenates prepared in PBS. Means not sharing a common superscript are significantly different, P<.05.
3.4. GTE decreases the expression of proinflammatory enzymes that induce oxidative and nitrative damage
To further define the mechanisms by which GTE reduces LPO and protein nitration associated with NASH, we examined protein expression levels of hepatic MPO and iNOS. Western blotting studies indicated that the expression of MPO from obese mice fed no GTE was 73% greater than that in the lean littermates (P<.05; Fig. 4). When compared with obese controls, GTE at 1% reduced MPO expression by 27% (P<.05) and decreased it to the levels in lean controls, providing evidence that GTE reduces oxidative and nitrative damage mediated by the MPO–H2O2 system, such as the formation of HNE and N-Tyr.
Fig. 4.

Protein expression of hepatic MPO in lean and obese (ob/ob) mice fed GTE at 0%, 0.5% and 1% for 6 weeks (n=12–13 mice/group; means±S.E.). MPO was measured by Western blot analysis from liver homogenates prepared in RIPA buffer. Proteins (50 μg/lane) was electrophoresed through an 8% polyacrylamide gel, transferred onto PVDF membranes and probed with antibody against MPO (1:1000, top) or β-actin (1:5000, bottom). Data are relative density normalized to β-actin expression. Means not sharing a common superscript are significantly different, P<.05.
Data also indicated that obese controls fed no GTE had greater iNOS expression compared with the lean littermates (P<.05; Fig. 5A), consistent with their greater nitrative stress and damage. GTE at 1% reduced iNOS expression to that of lean mice. Immunohistochemical data (Fig. 5B) further showed that positive staining toward iNOS was more intense in obese controls with liver steatosis. This was particularly evident around the central and portal veins, which is consistent with the greater accumulation of inflammatory infiltrates in those regions (Fig. 1B). Consistent with our Western blot data, iNOS staining intensity was reduced by GTE, particularly at 1%.
Fig. 5.
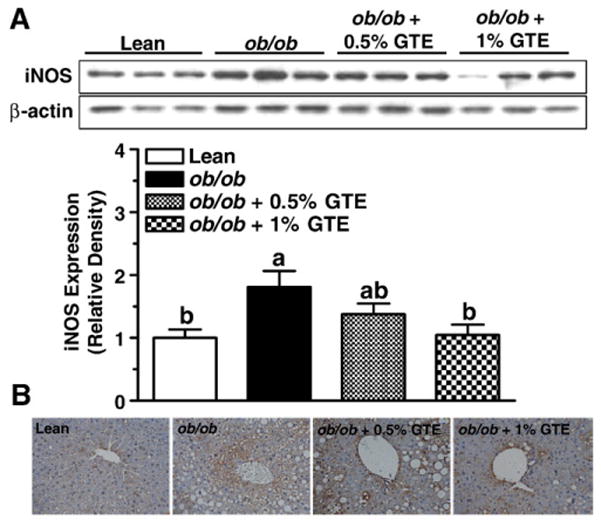
Protein expression and localization of hepatic iNOS in lean and obese (ob/ob) mice fed GTE at 0%, 0.5% and 1% for 6 weeks (n=12–13 mice/group; means±S.E.). (A) iNOS was measured by Western blot analysis from liver homogenates prepared in RIPA buffer. Proteins (20 μg/lane) were electrophoresed through a 10% polyacrylamide gel, transferred onto a PVDF membrane and probed with antibodies against iNOS (1:2000, top) or β-actin (1:5000, bottom). Data are relative densities normalized to β-actin expression. Means not sharing a common superscript are significantly different, P<.05. (B). Localization of iNOS was determined by immunohistochemistry using formalin-fixed and paraffin-embedded sections. Liver sections were incubated with antibody against iNOS, followed by HRP-conjugated antirabbit IgG, and visualized with DAB substrate after counterstaining with hematoxylin. Representative images are shown at 400× magnification.
4. Discussion
We reported previously that GTE protects against NASH in ob/ob mice by decreasing the expression of adipose lipogenic genes, improving hepatic antioxidant defenses and decreasing liver tumor necrosis factor α [18]. The present study expands on these findings by demonstrating that GTE attenuates NASH by inhibiting ROS/RNS-mediated damage that is closely related to the induction of inflammatory cell-derived enzymes. We show that GTE reduces serum ALT and decreases LPO and protein nitration, which occur concurrently with lower levels of MPO and iNOS and decreased NADPH oxidase activity. Serum ALT was correlated with liver HNE and N-Tyr, thereby supporting that GTE-mediated decreases in oxidative and nitrative modifications protect against liver injury. GTE at 0.5% effectively reduced LPO and protein nitration as well as NADPH oxidase activity. In contrast, a higher level of GTE (1%) was needed to decrease MPO and iNOS expression. Thus, ROS/RNS-mediated modifications to lipids and proteins are more responsive to GTE than iNOS and MPO expression, and the hepatoprotective activities of GTE likely extend beyond the regulation of the proinflammatory enzymes. Collectively, our findings provide novel evidence that GTE exerts anti-inflammatory activities in ob/ob mice and protects against pathogenic events contributing to liver injury associated with obesity-induced NASH.
Various intracellular sources of ROS and RNS are implicated in the “second-hit” of NASH [9]. However, our major aim was to examine whether GTE mitigates ROS/RNS-mediated events attributed to activated inflammatory cells in steatotic livers. Indeed, our findings provide new evidence that GTE protects ob/ob mice against liver injury associated with NASH by inhibiting the expression and activity of proinflammatory enzymes known to induce hepatic oxidative/nitrative damage and liver injury.
Obese mice from our study had greater hepatic LPO, which is in agreement with others [29–31] and characteristic of NASH. To our knowledge, our data provide the first evidence that greater activity of hepatic NADPH oxidase contributes to LPO and liver injury in the ob/ob model of NASH. Limited evidence in ob/ob mice suggests that NADPH oxidase activity is likely tissue specific [32,33] and possibly independent of leptin status [32]. Indeed, aorta oxidative stress in ob/ob mice was attributed partly to increased NADPH oxidase activity [33]. ob/ob mice treated with a chemical inhibitor of NADPH oxidase subunit assembly had decreased free radical generation, thereby implicating this proinflammatory enzyme as a source of ROS. In contrast, ob/ob mice had greater cardiac oxidative stress despite protein expression levels of NADPH oxidase subunits that were not significantly different lean controls [32]. Nonetheless, our observation that livers from ob/ob mice had greater hepatic NADPH oxidase activity is in agreement with others showing that obese Long-Evans Tokushima rats fed a methionine- and choline-deficient diet to induce NASH had greater expression of p47phox, a NADPH oxidase subunit [34]. In contrast, others using a knockout model for gp91phox, another subunit of NADPH oxidase, demonstrated that the absence of NADPH oxidase activity did not mitigate NASH when these mice were fed a methionine- and choline-deficient diet [35]. Regardless, our data indicate that steatotic livers of ob/ob mice have greater superoxide generation from activated inflammatory cells and complements existing knowledge that ob/ob mice have greater superoxide derived from mitochondria [31]. Following Kupffer cell activation, which is induced by bacterial products, cytokines and fatty acids [36,37], NADPH oxidase generates superoxide from oxygen. Our observations that GTE suppresses NADPH oxidase activity and that its activity correlates with HNE supports that hepatic injury is attenuated by decreasing superoxide generation. The mechanism by which GTE inhibits NADPH oxidase activity was beyond the scope of this study. However, evidence from in vitro studies [20] and a rodent model of inflammation [38] support that green tea or EGCG decreases the expression or translocation of NADPH oxidase subunits. In addition to reducing expression levels, in vitro studies indicate that EGCG and epigallocatechin have IC50 values that inhibit NADPH oxidase activity at physiologically relevant concentrations [39]. Additional work is warranted to define how GTE regulates NADPH oxidase activity in the ob/ob mouse model.
It is also likely that hepatic LPO is reduced by GTE-mediated decreases in MPO expression. The MPO–H2O2 system initiates unsaturated fatty acid oxidation [40], and greater hepatic accumulation of MPO-positive inflammatory cells has been observed in NASH patients [12]. Consistent with our histologic evidence (Fig. 1D), lower MPO expression by GTE is likely due to decreased inflammatory cell migration and infiltration [41,42]. In support, EGCG dose-dependently inhibited neutrophil migration by decreasing neutrophil chemotaxis in response to interleukin-8 [41]. Rats treated with EGCG prior to ischemia/reperfusion-induced liver injury also had lower hepatic MPO activity that was accompanied by lower LPO levels [42]. Lastly, EGCG was shown to inhibit macrophage chemotaxis as well as decrease the level of histamine [43], which activates inflammatory cells [44]. Collectively, our data support that GTE protects against LPO and liver injury by reducing inflammatory cell infiltration and MPO expression.
In addition to reducing ROS-mediated damage, our data indicate that GTE dose-dependently reduces hepatic N-Tyr and that the magnitude of protein nitration corresponds to liver injury. Greater hepatic N-Tyr in ob/ob mice was expected based on the findings of others [31] and is consistent with the two-hit mechanism of NASH [7]. GTE likely attenuated protein nitration through multiple mechanisms. Tea catechins, at least in vitro, directly scavenge superoxide and nitric oxide [14,15]. This is important because superoxide and nitric oxide spontaneously react to form peroxynitrite [45], which can then induce the nitration of protein tyrosine residues to form N-Tyr [46]. Thus, our observation that GTE decreases NADPH oxidase activity would be expected to decrease superoxide levels, and in turn, the magnitude of peroxynitrite was generated. Our finding that GTE decreases the hepatic iNOS expression would also be expected to reduce the availability of nitric oxide to react with superoxide and form peroxynitrite. Consistent with this notion, hepatic NOx concentrations were lower in obese mice fed GTE. The mechanism by which GTE decreases iNOS expression may be associated with decreases in nuclear factor κB (NFκB) activity. In lipopolysaccharide-stimulated macrophages, electrophoretic mobility shift studies indicated that EGCG inhibits NFκB binding activity and decreases iNOS activity and expression [19]. Also, unpublished observations from our group indicate that GTE decreases hepatic NFκB binding activity in rats fed a high-fat diet to induce NAFLD [47]. Thus, it is likely that GTE attenuates protein nitration by suppressing superoxide generation from NADPH oxidase and nitric oxide from iNOS, possibly through an NFκB-dependent mechanism.
GTE-mediated reductions in hepatic MPO expression would also be expected to decrease protein nitration. In addition to oxidizing lipids, MPO induces nitrative modification of proteins [48]. Using hydrogen peroxide, which forms from the reduction of superoxide during inflammatory cell activation [49], MPO catalyzes the formation of nitrogen dioxide radical, which induces tyrosine nitration [50]. In conjunction with reducing MPO, GTE may mitigate MPO-mediated damage by decreasing hydrogen peroxide availability. Indeed, we demonstrated previously that GTE increases the enzymatic activities of hepatic superoxide dismutase as well as the peroxidative enzymes catalase and glutathione peroxidase [18]. Thus, GTE increases the rate of ROS detoxification and limits the availability of reactive species for MPO-dependent reactions that would otherwise lead to oxidative/nitrative damage and hepatic injury.
Lastly, a higher level of GTE (1%) was needed to decrease the expression of proinflammatory enzymes, whereas GTE at 0.5% effectively mitigated LPO and protein nitration. These dose-dependent actions suggest that complementary mechanisms beyond its anti-inflammatory activities are likely to contribute to its hepatopro-tective effects. Indeed, we showed that GTE at 0.5% exerts antioxidant activity against liver injury in ob/ob mice by up-regulating activities of enzymatic antioxidants [18]. Also, the hypolipidemic activities of GTE [51] would be expected to decrease the flux of free fatty acids into ROS generating pathways, such as mitochondrial metabolism, that provoke oxidative damage. For example, our group [18] and others [52,53] have provided evidence that GTE decreases lipogenesis in ob/ob mice. Nonetheless, this study indicates that the maximal benefits of GTE on inflammatory parameters occur at 1%, which corresponds to ~7 cups/day of green tea in humans and is consistent with epidemiologic observations, suggesting that >10 cups/day are associated with lower levels of liver injury biomarkers [13].
No studies in humans with or at risk for NASH have identified the optimal dietary intake of GTE. This is of importance because the available data are equivocal whether GTE mitigates oxidative stress responses in humans. Indeed, some studies show that GTE reduces markers of LPO [54–56] or oxidized low-density lipoprotein [57], whereas others indicate a lack of effect of GTE on oxidative stress markers [58] or risk factors of cardiovascular disease [59]. Another area requiring attention is the potential hepatotoxic effects of GTE or its catechins. Human case reports [60,61], studies in isolated hepatocytes treated with catechins [62] and studies in mice receiving high-dose intraperitoneal injections [62,63] have suggested the possibility that green tea or its catechins may exert toxic effects. Those observations are contradicted by the findings of the present study as well as prior rodent studies by our group [17,18] and others [64], indicating that GTE is nontoxic at dietary levels up to 2%. Controlled studies in overweight men receiving green tea polyphenols (714 mg/day) for 3 weeks also indicated no adverse effects on liver or renal function or markers of cardiovascular disease risk [59]. A systematic review by the US Pharmacopeia also supports the safety of GTE when products are used and formulated appropriately [65]. Nonetheless, additional work is needed to define the safe upper limit of GTE consumption.
In conclusion, this study provides novel evidence that GTE protects against obesity-triggered NASH by suppressing the activities of proinflammatory enzymes. The findings that GTE protects against inflammatory-mediated damage associated with NASH have significant public health relevance [66]. NAFLD is largely asymptomatic [2] and steatotic livers are highly vulnerable to ROS/RNS-mediated damage that exacerbates liver-related morbidity and mortality [7]. Further studies are warranted to determine if GTE can be recommended as a dietary strategy to lower the risk of developing NASH in obese humans.
Acknowledgments
The authors thank Unilever Bestfoods (Englewood, NJ, USA) for providing the green tea extract used in this study.
Footnotes
This study was supported by grants to R.S.B. from the National Research Initiative (2007-02303) from the US Department of Agriculture Cooperative State Research, Education and Extension Service and the US Department of Agriculture–Hatch (CONS00802) program.
References
- 1.Ludwig J, McGill DB, Lindor KD. Review: nonalcoholic steatohepatitis. J Gastro-enterol Hepatol. 1997;12:398–403. doi: 10.1111/j.1440-1746.1997.tb00450.x. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–31. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 3.Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–55. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 4.Ayyad C, Andersen T. Long-term efficacy of dietary treatment of obesity: a systematic review of studies published between 1931 and 1999. Obes Rev. 2000;1:113–9. doi: 10.1046/j.1467-789x.2000.00019.x. [DOI] [PubMed] [Google Scholar]
- 5.Portincasa P, Grattagliano I, Palmieri VO, Palasciano G. Nonalcoholic steatohepatitis: recent advances from experimental models to clinical management. Clin Biochem. 2005;38:203–17. doi: 10.1016/j.clinbiochem.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 6.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 7.Day CP. Pathogenesis of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16:663–78. doi: 10.1053/bega.2002.0333. [DOI] [PubMed] [Google Scholar]
- 8.Fujita K, Nozaki Y, Yoneda M, Wada K, Takahashi H, Kirikoshi H, et al. Nitric oxide plays a crucial role in the development/progression of nonalcoholic steatohepatitis in the choline-deficient, l-amino acid-defined diet-fed rat model. Alcohol Clin Exp Res. 2010;34(suppl 1):S18–24. doi: 10.1111/j.1530-0277.2008.00756.x. [DOI] [PubMed] [Google Scholar]
- 9.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–52. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pessayre D, Fromenty B, Mansouri A. Mitochondrial injury in steatohepatitis. Eur J Gastroenterol Hepatol. 2004;16:1095–105. doi: 10.1097/00042737-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Carmiel-Haggai M, Cederbaum AI, Nieto N. A high-fat diet leads to the progression of non-alcoholic fatty liver disease in obese rats. FASEB J. 2005;19:136–8. doi: 10.1096/fj.04-2291fje. [DOI] [PubMed] [Google Scholar]
- 12.Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. 2009;175:1473–82. doi: 10.2353/ajpath.2009.080999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imai K, Nakachi K. Cross sectional study of effects of drinking green tea on cardiovascular and liver diseases. Bmj. 1995;310:693–6. doi: 10.1136/bmj.310.6981.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakagawa T, Yokozawa T. Direct scavenging of nitric oxide and superoxide by green tea. Food Chem Toxicol. 2002;40:1745–50. doi: 10.1016/s0278-6915(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 15.Paquay JB, Haenen GR, Stender G, Wiseman SA, Tijburg LB, Bast A. Protection against nitric oxide toxicity by tea. J Agric Food Chem. 2000;48:5768–72. doi: 10.1021/jf981316h. [DOI] [PubMed] [Google Scholar]
- 16.Scott BC, Butler J, Halliwell B, Aruoma OI. Evaluation of the antioxidant actions of ferulic acid and catechins. Free Radic Res Commun. 1993;19:241–53. doi: 10.3109/10715769309056512. [DOI] [PubMed] [Google Scholar]
- 17.Bruno RS, Dugan CE, Smyth JA, DiNatale DA, Koo SI. Green tea extract protects leptin-deficient, spontaneously obese mice from hepatic steatosis and injury. J Nutr. 2008;138:323–31. doi: 10.1093/jn/138.2.323. [DOI] [PubMed] [Google Scholar]
- 18.Park HJ, Dinatale DA, Chung MY, Park YK, Lee JY, Koo SI, et al. Green tea extract attenuates hepatic steatosis by decreasing adipose lipogenesis and enhancing hepatic antioxidant defenses in ob/ob mice. J Nutr Biochem. 2010 doi: 10.1016/j.jnutbio.2010.03.009. (E-Pub ahead of print) [DOI] [PubMed] [Google Scholar]
- 19.Lin YL, Lin JK. (−)-Epigallocatechin-3-gallate blocks the induction of nitric oxide synthase by down-regulating lipopolysaccharide-induced activity of transcription factor nuclear factor-kappaB. Mol Pharmacol. 1997;52:465–72. [PubMed] [Google Scholar]
- 20.Nishikawa H, Wakano K, Kitani S. Inhibition of NADPH oxidase subunits translocation by tea catechin EGCG in mast cell. Biochem Biophys Res Commun. 2007;362:504–9. doi: 10.1016/j.bbrc.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Diehl AM. Lessons from animal models of NASH. Hepatol Res. 2005;33:138–44. doi: 10.1016/j.hepres.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 22.Schattenberg JM, Galle PR. Animal models of non-alcoholic steatohepatitis: of mice and man. Dig Dis. 2010;28:247–54. doi: 10.1159/000282097. [DOI] [PubMed] [Google Scholar]
- 23.Zhao J, Chen H, Li Y. Protective effect of bicyclol on acute alcohol-induced liver injury in mice. Eur J Pharmacol. 2008;586:322–31. doi: 10.1016/j.ejphar.2008.02.059. [DOI] [PubMed] [Google Scholar]
- 24.ter Steege JC, Koster-Kamphuis L, van Straaten EA, Forget PP, Buurman WA. Nitrotyrosine in plasma of celiac disease patients as detected by a new sandwich ELISA. Free Radic Biol Med. 1998;25:953–63. doi: 10.1016/s0891-5849(98)00184-1. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Han W, Villar VA, Keever LB, Lu Q, Hopfer U, et al. D1-like receptors regulate NADPH oxidase activity and subunit expression in lipid raft microdomains of renal proximal tubule cells. Hypertension. 2009;53:1054–61. doi: 10.1161/HYPERTENSIONAHA.108.120642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umemoto S, Tanaka M, Kawahara S, Kubo M, Umeji K, Hashimoto R, et al. Calcium antagonist reduces oxidative stress by upregulating Cu/Zn superoxide dismutase in stroke-prone spontaneously hypertensive rats. Hypertens Res. 2004;27:877–85. doi: 10.1291/hypres.27.877. [DOI] [PubMed] [Google Scholar]
- 27.Giustarini D, Rossi R, Milzani A, Dalle-Donne I. Nitrite and nitrate measurement by Griess reagent in human plasma: evaluation of interferences and standardization. Methods Enzymol. 2008;440:361–80. doi: 10.1016/S0076-6879(07)00823-3. [DOI] [PubMed] [Google Scholar]
- 28.Esposito E, Iacono A, Bianco G, Autore G, Cuzzocrea S, Vajro P, et al. Probiotics reduce the inflammatory response induced by a high-fat diet in the liver of young rats. J Nutr. 2009;139:905–11. doi: 10.3945/jn.108.101808. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Ruiz I, Rodriguez-Juan C, Diaz-Sanjuan T, del Hoyo P, Colina F, Munoz-Yague T, et al. Uric acid and anti-TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology. 2006;44:581–91. doi: 10.1002/hep.21313. [DOI] [PubMed] [Google Scholar]
- 30.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci U S A. 1997;94:2557–62. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laurent A, Nicco C, Tran Van Nhieu J, Borderie D, Chereau C, Conti F, et al. Pivotal role of superoxide anion and beneficial effect of antioxidant molecules in murine steatohepatitis. Hepatology. 2004;39:1277–85. doi: 10.1002/hep.20177. [DOI] [PubMed] [Google Scholar]
- 32.Saraiva RM, Minhas KM, Zheng M, Pitz E, Treuer A, Gonzalez D, et al. Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric Oxide. 2007;16:331–8. doi: 10.1016/j.niox.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sonta T, Inoguchi T, Tsubouchi H, Sekiguchi N, Kobayashi K, Matsumoto S, et al. Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med. 2004;37:115–23. doi: 10.1016/j.freeradbiomed.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Kurita S, Takamura T, Ota T, Matsuzawa-Nagata N, Kita Y, Uno M, et al. Olmesartan ameliorates a dietary rat model of non-alcoholic steatohepatitis through its pleiotropic effects. Eur J Pharmacol. 2008;588:316–24. doi: 10.1016/j.ejphar.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 35.dela Pena A, Leclercq IA, Williams J, Farrell GC. NADPH oxidase is not an essential mediator of oxidative stress or liver injury in murine MCD diet-induced steatohepatitis. J Hepatol. 2007;46:304–13. doi: 10.1016/j.jhep.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 36.Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol. 2009;51:212–23. doi: 10.1016/j.jhep.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289–99. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 38.Papparella I, Ceolotto G, Montemurro D, Antonello M, Garbisa S, Rossi G, et al. Green tea attenuates angiotensin II-induced cardiac hypertrophy in rats by modulating reactive oxygen species production and the Src/epidermal growth factor receptor/Akt signaling pathway. J Nutr. 2008;138:1596–601. doi: 10.1093/jn/138.9.1596. [DOI] [PubMed] [Google Scholar]
- 39.Steffen Y, Gruber C, Schewe T, Sies H. Mono-O-methylated flavanols and other flavonoids as inhibitors of endothelial NADPH oxidase. Arch Biochem Biophys. 2008;469:209–19. doi: 10.1016/j.abb.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Pryor WA, Lightsey JW. Mechanisms of nitrogen dioxide reactions: initiation of lipid peroxidation and the production of nitrous acid. Science. 1981;214:435–7. doi: 10.1126/science.214.4519.435. [DOI] [PubMed] [Google Scholar]
- 41.Dona M, Dell’Aica I, Calabrese F, Benelli R, Morini M, Albini A, et al. Neutrophil restraint by green tea: inhibition of inflammation, associated angiogenesis, and pulmonary fibrosis. J Immunol. 2003;170:4335–41. doi: 10.4049/jimmunol.170.8.4335. [DOI] [PubMed] [Google Scholar]
- 42.Giakoustidis DE, Giakoustidis AE, Iliadis S, Koliakou K, Antoniadis N, Kontos N, et al. Attenuation of liver ischemia/reperfusion induced apoptosis by epigallocatechin-3-gallate via down-regulation of NF-kappaB and c-Jun expression. J Surg Res. 159:720–8. doi: 10.1016/j.jss.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 43.Mochizuki M, Hasegawa N. (−)-Epigallocatechin-3-gallate reduces experimental colon injury in rats by regulating macrophage and mast cell. Phytother Res. 24(Suppl 1):S120–2. doi: 10.1002/ptr.2862. [DOI] [PubMed] [Google Scholar]
- 44.Xie H, He SH. Roles of histamine and its receptors in allergic and inflammatory bowel diseases. World J Gastroenterol. 2005;11:2851–7. doi: 10.3748/wjg.v11.i19.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–4. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfeiffer S, Lass A, Schmidt K, Mayer B. Protein tyrosine nitration in mouse peritoneal macrophages activated in vitro and in vivo: evidence against an essential role of peroxynitrite. FASEB J. 2001;15:2355–64. doi: 10.1096/fj.01-0295com. [DOI] [PubMed] [Google Scholar]
- 47.Park HJ, Chung M-Y, Koo SI, Bruno RS. Green tea extract attenuates adipose inflammation by restoring glutathione redox status in high fat diet-induced nonalcoholic fatty liver disease. FASEB J. 2010;24:722.15. [Google Scholar]
- 48.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 49.Forman HJ, Torres M. Redox signaling in macrophages. Mol Aspects Med. 2001;22:189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- 50.Podrez EA, Schmitt D, Hoff HF, Hazen SL. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J Clin Invest. 1999;103:1547–60. doi: 10.1172/JCI5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park HJ, Bruno RS. Hepatoprotective activities of green tea in nonalcoholic fatty liver disease. Agro Food Industry Hi-Tech. 2010;21:37–40. [Google Scholar]
- 52.Kim HJ, Jeon SM, Lee MK, Jung UJ, Shin SK, Choi MS. Antilipogenic effect of green tea extract in C57BL/6J-Lep ob/ob mice. Phytother Res. 2009;23:467–71. doi: 10.1002/ptr.2647. [DOI] [PubMed] [Google Scholar]
- 53.Fiorini RN, Donovan JL, Rodwell D, Evans Z, Cheng G, May HD, et al. Short-term administration of (−)-epigallocatechin gallate reduces hepatic steatosis and protects against warm hepatic ischemia/reperfusion injury in steatotic mice. Liver Transpl. 2005;11:298–308. doi: 10.1002/lt.20348. [DOI] [PubMed] [Google Scholar]
- 54.Freese R, Basu S, Hietanen E, Nair J, Nakachi K, Bartsch H, et al. Green tea extract decreases plasma malondialdehyde concentration but does not affect other indicators of oxidative stress, nitric oxide production, or hemostatic factors during a high-linoleic acid diet in healthy females. Eur J Nutr. 1999;38:149–57. doi: 10.1007/s003940050056. [DOI] [PubMed] [Google Scholar]
- 55.Nakagawa K, Ninomiya M, Okubo T, Aoi N, Juneja LR, Kim M, et al. Tea catechin supplementation increases antioxidant capacity and prevents phospholipid hydroperoxidation in plasma of humans. J Agric Food Chem. 1999;47:3967–73. doi: 10.1021/jf981195l. [DOI] [PubMed] [Google Scholar]
- 56.Agarwal A, Prasad R, Jain A. Effect of green tea extract (catechins) in reducing oxidative stress seen in patients of pulmonary tuberculosis on DOTS Cat I regimen. Phytomedicine. 2010;17:23–7. doi: 10.1016/j.phymed.2009.10.019. [DOI] [PubMed] [Google Scholar]
- 57.Inami S, Takano M, Yamamoto M, Murakami D, Tajika K, Yodogawa K, et al. Tea catechin consumption reduces circulating oxidized low-density lipoprotein. Int Heart J. 2007;48:725–32. doi: 10.1536/ihj.48.725. [DOI] [PubMed] [Google Scholar]
- 58.Princen HM, van Duyvenvoorde W, Buytenhek R, Blonk C, Tijburg LB, Langius JA, et al. No effect of consumption of green and black tea on plasma lipid and antioxidant levels and on LDL oxidation in smokers. Arterioscler Thromb Vasc Biol. 1998;18:833–41. doi: 10.1161/01.atv.18.5.833. [DOI] [PubMed] [Google Scholar]
- 59.Frank J, George TW, Lodge JK, Rodriguez-Mateos AM, Spencer JP, Minihane AM, et al. Daily consumption of an aqueous green tea extract supplement does not impair liver function or alter cardiovascular disease risk biomarkers in healthy men. J Nutr. 2009;139:58–62. doi: 10.3945/jn.108.096412. [DOI] [PubMed] [Google Scholar]
- 60.Federico A, Tiso A, Loguercio C. A case of hepatotoxicity caused by green tea. Free Radic Biol Med. 2007;43:474. doi: 10.1016/j.freeradbiomed.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 61.Jimenez-Saenz M, del Martinez-Sanchez MC. Acute hepatitis associated with the use of green tea infusions. J Hepatol. 2006;44:616–7. doi: 10.1016/j.jhep.2005.11.041. [DOI] [PubMed] [Google Scholar]
- 62.Galati G, Lin A, Sultan AM, O’Brien PJ. Cellular and in vivo hepatotoxicity caused by green tea phenolic acids and catechins. Free Radic Biol Med. 2006;40:570–80. doi: 10.1016/j.freeradbiomed.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 63.Lambert JD, Kennett MJ, Sang S, Reuhl KR, Ju J, Yang CS. Hepatotoxicity of high oral dose (−)-epigallocatechin-3-gallate in mice. Food Chem Toxicol. 2010;48:409–16. doi: 10.1016/j.fct.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takami S, Imai T, Hasumura M, Cho YM, Onose J, Hirose M. Evaluation of toxicity of green tea catechins with 90-day dietary administration to F344 rats. Food Chem Toxicol. 2008;46:2224–9. doi: 10.1016/j.fct.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 65.Sarma DN, Barrett ML, Chavez ML, Gardiner P, Ko R, Mahady GB, et al. Safety of green tea extracts: a systematic review by the US Pharmacopeia. Drug Saf. 2008;31:469–84. doi: 10.2165/00002018-200831060-00003. [DOI] [PubMed] [Google Scholar]
- 66.Burke A, Lucey MR. Non-alcoholic fatty liver disease, non-alcoholic steatohepatitis and orthotopic liver transplantation. Am J Transplant. 2004;4:686–93. doi: 10.1111/j.1600-6143.2004.00432.x. [DOI] [PubMed] [Google Scholar]