

Abstract
Integration of clinical evaluations and whole-genome sequence data from eight individuals in a recent study demonstrates that genetic and clinical information can be combined and applied to preventive medicine. Statistical and graphical tools were developed to assess and visualize the genetic risk of common chronic conditions and to show the changes in disease risk that result from monitoring clinical symptoms over time. This approach provides a direction to consider in the adoption of genetic information in health care, but, like all provocative scientific articles, it raises as many questions as it answers.
Please see related Research: http://genomemedicine.com/content/5/6/58
The debate
There is much debate surrounding the utility of genetic information in clinical practice and general health care. Although useful in the treatment of certain cancers [1] and in the identification of pathogenic mutations in idiopathic conditions [2], there is no consensus as to whether or how to use genetic information in individual health surveillance and routine preventive medicine. A recent article by Patel et al. [3] in Genome Medicine provides a general framework for integrating genetic information into a wellness and health maintenance program. In doing so it exposes four issues at the heart of this debate: first, concern that standard clinical assessments will be replaced by genetic information; second, lack of methods to assess risk; third, the lack of good visualization tools for graphically depicting disease risk; and fourth, insufficient knowledge on how to deal with variants that are not directly related to disease risk.
A new framework
The authors [3] enrolled eight patients in the 'Center for Health Discovery and Well Being (CHDWB)' program at Emory University, USA. This program focuses on integrative approaches for health evaluation and maintenance and is an appropriate place to vet the use of genetic information in clinical practice. The patients were chosen from 500 participants in the CHDWB program on the basis of their clinical profiles. Four patients were at risk of metabolic disease according to traditional measures (such as high body mass index (BMI), body fat percentage, high-density lipoprotein levels, and triglyceride levels) and depression scores that put them in an upper disease risk category relative to the total pool of CHDWB participants; the other four had profiles that put them in a lower disease risk category. The genomes of the eight individuals were sequenced using Illumina technology (about 36X coverage) and the genetic susceptibility score for a variety of conditions was computed for each individual using a simple likelihood-based risk (LR) model. Clinical measures such as blood pressure, triglyceride levels, basophil counts and the Beck depression inventory were assessed at the time of enrolment in the CHDWB program, 6 and 12 months after enrolment, and periodically after that.
At the heart of the debate
Four issues plague the adoption of genetic information in routine clinical care, and Patel et al. [3] sought to tackle these. First, they had to overcome the misguided conception - often unknowingly perpetuated in the literature [4] - that genetic information will replace standard clinical assessments and practice. In fact, genetic information will complement the use of many standard clinical measures because of the nature of the information provided. Genetic information provides a static set of predictive biomarkers for disease susceptibility, whereas most clinical assays and instruments detect indicators that vary as a disease progresses. Information on temporally invariant genetic risks complements temporally variable clinical measures, as it helps to show whether a disease for which an individual has an inherent susceptibility has started to manifest itself or to subside. As an illustration, a study by Lyssenko et al. [5] found that genetic markers performed best when predicting long-term development of diabetes, whereas clinical laboratory assays (such as insulin and glucose levels) performed best when predicting its short-term development. This is consistent with the idea that widely used contemporary clinical assays probe the pathophysiological manifestations of a disease process that appear just before, during, or long after the onset of the disease. This is unlike risk assessments based on variants in inherited DNA sequence, which are fixed at birth.
For the eight people evaluated by Patel et al. [3], the genetic risk profiles were 'concordant' (or consistent) with the clinical assay results in many instances and 'discordant' in others. For example, four individuals at low genetic risk for obesity also had low weight and BMI levels at the time of clinical evaluation; however, one individual with a very low genetic risk of hypertension did have high blood pressure and two individuals with an elevated genetic risk of diabetes had normal glucose and insulin levels at the time of their evaluations. The discordance between high genetic risk and the healthy clinical evaluation measures in some individuals suggests that these individuals were defying their genetic susceptibility to a disease, perhaps through disease-mitigating behaviors, pharmacologic interventions, or an unknown protective effect. The success of the CHDWB program will be demonstrated if it is shown to cause an even greater reduction in patients with manifestation of clinical disease than this. For genetic medicine as a whole, the brightest future would be if clinicians can steer patients to health despite elevations in their genetic risks (Figure 1).
Figure 1.
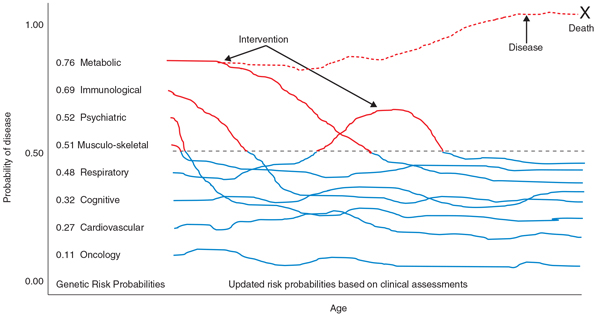
Schematic of the links between genetic risk and longitudinal clinical assessments for disease surveillance and health maintenance. In this scenario, the probabilities of disease based on genetic variants are calculated at a point in time for as many different conditions as possible, preferably at birth to facilitate lifetime health maintenance. Disease risks deemed high (such as with a probability over 0.5, denoted by the black dotted line) are noted and immediately focused on. Clinical measures are then obtained periodically to monitor disease and inform the calculation of a posterior probability of a disease manifesting itself given an individual's genetic predisposition (depicted as lines emanating from each disease category). Risks above the threshold for intervention are noted (red parts of lines). The graph also depicts a hypothetical setting (dashed red line) in which risk for metabolic disease is high, clinical measures indicate increased risk and an intervention is not undertaken, leading to disease and ultimately death from that disease.
Second, the authors [3] had to develop and implement a method to compute genetic risks, risks based on clinical examination, and integrated risks. The LR method they champion is appropriate but is ultimately dependent on summary information regarding variants associated with diseases arising from case/control-based genome-wide association studies, and not from prospective longitudinal studies [6,7]. In the authors' defense, there are precious few longitudinal studies with which one could develop or vet genetic risk prediction models, although some are emerging [8]. The Bayesian updating method also used by Patel et al. in this article [3], in which genetic risk is modified according to the current clinical profile, is also appropriate, but again should be evaluated in prospective longitudinal studies. The reasons for this are two-fold. First, appropriate weighting of clinically defined risk (based on clinician assessment and clinical laboratory results) relative to genetic risk is not trivial to determine; that is, it is not clear whether an observable clinical manifestation of a disease is worth more to a clinician in terms of immediate intervention needs than a future risk as indicated by genetic susceptibility. Second, the interpretation of the integrated Bayesian posterior probability based on a recent clinical evaluation would be problematic without an appropriate reference set of probabilities; that is, without probabilities that have been calibrated or shown to be consistent with actual data, we cannot know what a reduction in risk probability based on clinical profile means in terms of ultimate prognosis and future risk. Again, without appropriate longitudinal data, it is hard to pursue such a calibration exercise, but it would make sense to test this in existing datasets, however flawed, for example in the Framingham Heart Study data. An additional vexing problem in calculating risks for multiple diseases is taking into account the fact that many disease manifestations are correlated (such as obesity and diabetes or heart disease). The authors [3] admitted that their approach made some unrealistic assumptions in this respect (such as independence of the diseases), and this is an area worthy of further research.
The third important issue that the authors [3] had to consider is the best way to present and visualize the results of their disease risk assessments based on LR and Bayesian calculations for physicians and patients. Conveying these results must be intuitive and quick to interpret and must reflect not only multiple disease risks, but how genetic risks for these multiple diseases are modified by ongoing changes in clinical measures and evaluations. To address this the authors proposed three different graphical displays ('risk-o-grams' [6], 'gridiron plots', and 'radar plots'). Each display seeks to condense risks for multiple diseases by clustering them into broad categories such as 'immunologic' or 'metabolic', providing graphics that give a sense of how genetic risk is modified by changes in clinical measures. Although intuitive from a biomedical perspective and visually appealing, the merit of these graphical representations now requires feedback, if not overt, objective comparison among them as well as others, from clinicians, health practitioners and patients.
Finally, the authors [3] made decisions about how to deal with genetic variant information that did not directly inform their disease risk calculations. They did not focus on rare variants associated with monogenic or other diseases, novel variants likely to be damaging or possibly deleterious, or pharmacogenetically meaningful variants. In addition, they did not consider variants of unknown significance, although this is an important area for the future. In this context, highly controversial 'incidental findings' (the existence of deleterious, disease-causing variants not germane to a particular condition of focus) may not be as problematic in a setting, such as general clinical preventive medicine, that involves the comprehensive assessment of genetically mediated disease risk and health prognosis for an individual patient, as it would be in another setting, such as a search for specific disease-causing variants like BRCA1 variants [9].
Future implications
Other issues, such as costs, issues of privacy, the role of ancestry in mediating genetic risk, and, importantly, whether or not enough is known about the genetic basis of a particular disease to confidently categorize individuals as at risk, are only indirectly discussed by Patel et al. [3]. However, the approaches the authors describe are as good a set of starting points as any, and they have done a great service to the translational research community by exposing as many issues as they did. In addition, it is clear that genetic assay costs will continue to drop, greater emphasis will be placed on health information technology, and more insight into the global functional consequences of genomic variants will be obtained, paving the way for similar reports. In this light, we hope that the naysayers of the use of genetic information in general clinical practice will do with their overly critical attitude exactly what individuals genetically susceptible to the common cold hope to do when their first cold symptoms arise - just get over it.
Abbreviations
BMI: body mass index; CHDWB: Center for Health Discovery and Well Being; LR: likelihood-based risk.
Competing interests
NJS is a founder of, and consultant for, Cypher Genomics, Inc. and on the Board of Directors for MD Revolution.
References
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl WA, Markello TC, Toro C, Fajardo KF, Sincan M, Gill F, Carlson-Donohoe H, Gropman A, Pierson TM, Golas G, Wolfe L, Groden C, Godfrey R, Nehrebecky M, Wahl C, Landis DM, Yang S, Madeo A, Mullikin JC, Boerkoel CF, Tifft CJ, Adams D. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14:51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CJ, Sividas A, Tabassum R, Preeprem T, Zhao J, Arafat D, Chen R, Morgan AA, Martin GS, Brigham KL, Butte AJ, Gibson G. Whole genome sequencing in support of wellness and health maintenance. Genome Med. 2013;5:58. doi: 10.1186/gm462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts NJ, Vogelstein JT, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. The predictive capacity of personal genome sequencing. Sci Transl Med. 2012;4:133ra158. doi: 10.1126/scitranslmed.3003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med. 2008;359:2220–2232. doi: 10.1056/NEJMoa0801869. [DOI] [PubMed] [Google Scholar]
- Ashley EA, Butte AJ, Wheeler MT, Chen R, Klein TE, Dewey FE, Dudley JT, Ormond KE, Pavlovic A, Morgan AA, Pushkarev D, Neff NF, Hudgins L, Gong L, Hodges LM, Berlin DS, Thorn CF, Sangkuhl K, Hebert JM, Woon M, Sagreiya H, Whaley R, Knowles JW, Chou MF, Thakuria JV, Rosenbaum AM, Zaranek AW, Church GM, Greely HT, Quake SR. et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:525–1535. doi: 10.1016/S0140-6736(10)60210-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AA, Chen R, Butte AJ. Likelihood ratios for genome medicine. Genome Med. 2010;2:30. doi: 10.1186/gm151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer T, Sun C, Magnussen CG, Raitakari OT, Schork NJ, Venn A, Burns TL, Juonala M, Steinberger J, Sinaiko AR, Prineas RJ, Davis PH, Woo JG, Morrison JA, Daniels SR, Chen W, Srinivasan SR, Viikari JS, Berenson GS. Cohort Profile: the international childhood cardiovascular cohort (i3C) consortium. Int J Epidemiol. 2013;42:86–96. doi: 10.1093/ije/dys004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennette CS, Trinidad SB, Fullerton SM, Patrick D, Amendola L, Burke W, Hisama FM, Jarvik GP, Regier DA, Veenstra DL. Return of incidental findings in genomic medicine: measuring what patients value-development of an instrument to measure preferences for information from next-generation testing (IMPRINT). Genet Med. 2013. doi: 10.1038/gim.2013.63. [DOI] [PMC free article] [PubMed]