

Abstract
Ongoing investigation has provided new insights into the pathobiology of myocardial ischemic injury. These include an improved understanding of the roles of the major modes of cell injury and death, including oncosis, apoptosis, and unregulated autophagy, as well as the central role of the mitochondria in the progression of myocardial ischemic injury, reperfusion injury, and myocardial conditioning. This understanding is providing insights for developing new pathophysiologic, pharmacologic, and cell-based therapies, alone or in combination with percutaneous coronary interventions, for better preservation of myocardium and reduction of morbidity and mortality rates from ischemic heart disease.
Key words: Apoptosis; autophagy; cardioprotection; inflammation mediators; ischemic preconditioning, myocardial; myocardial ischemia/physiopathology; myocardial reperfusion injury/physiopathology; myocardial stunning/physiopathology; myocytes, cardiac/pathology; mitochondria; myocardial reperfusion injury; myocardium/cytology/ultrastructure; myocytes, cardiac/pathology; necrosis/prevention & control; oncosis; regeneration/physiology; reperfusion injury; signal transduction; stem cell transplantation
The dynamic alterations in cells, tissues, and organs involving molecular, biochemical, structural, and functional changes constitute collectively the pathobiology of disease. The first purpose of this review is to relate fundamental knowledge of the pathobiology of acute coronary syndromes (ACS) to clinical manifestations and the clinical course of these conditions. The second purpose is to link this knowledge to opportunities for therapeutic interventions to favorably alter the clinical course and outcomes of ACS.
Basic Pathobiology of Acute Coronary Syndrome
The basic pathobiology of ACS has been well established by extensive experimental studies and confirmation in patients. Coronary atherosclerosis is the underlying substrate for nearly all cases of ischemic heart disease.1,2 Episodes of ACS are triggered by acute alterations in coronary atheromatous plaques, particularly in thin-cap fibroatheromas, considered to be vulnerable plaques.3–6 These alterations consist of endothelial dysfunction, platelet aggregation, and spasm leading to plaque erosion, rupture, hemorrhage, and thrombosis.
After the onset of coronary occlusion, ischemic myocardium quickly loses its contractile function. Electrical instability can lead to ventricular fibrillation and sudden cardiac death. Progression from reversible to irreversible ischemic injury first develops after 15 minutes in the most profoundly ischemic subendocardium of the left ventricle and then extends in a wavefront pattern from subendocardium to subepicardium throughout the ischemic bed-at-risk.7,8 This process is associated with a transmural current-of-injury pattern on the electrocardiogram (ECG) characterized by ST-segment elevation and T-wave inversion, recognized clinically as an evolving ST-segment-elevation myocardial infarction (STEMI). In the presence of substantial collateral blood flow, the severe ischemia can be limited to the inner half of the left ventricular wall in association with prolonged ST-segment depression and T-wave flattening on ECG, recognized clinically as a non-STEMI. The evolution of acute myocardial infarction (AMI) takes place rapidly, and the full extent of myocardial necrosis is in most cases complete by 3 hours after the onset of coronary occlusion.9–11
Reperfusion profoundly alters the outcome of an evolving AMI.12–17 If instituted in a timely manner, a potential transmural AMI can be prevented: the extent of necrosis is greatly reduced and is limited to the subendocardium. However, some injured cardiomyocytes at the edge of the wavefront become irreversibly injured during reperfusion, producing a component of lethal reperfusion injury. After reperfusion, the salvaged myocardium exhibits impaired contractile function—a form of nonlethal reperfusion injury referred to as myocardial stunning. The earlier the reperfusion, the less total necrosis occurs—including the ischemia-induced component and the reperfusion-induced component—and the earlier the recovery of contractile function from the transient stunning as well. Conversely, reperfusion can be rendered less effective by the microvascular damage and obstruction that develop during the ischemic phase; this is known as the no-reflow phenomenon. The resultant lesion has the gross appearance of a hemorrhagic subendocardial infarct.11
The rapid time course of AMI and the temporal limitation on the maximal effectiveness of reperfusion constitute the pathobiological basis for the contemporary clinical strategies that emphasize rapid intervention within 1 to 2 hours after onset of symptoms.16 Increasingly effective reperfusion has been achieved through the sequential introduction of thrombolytic therapy, percutaneous transluminal angioplasty, and coronary stenting, this last referred to as percutaneous coronary intervention (PCI). The morbidity and death from AMI result from mechanical and electrical complications,9,18 but these have been substantially reduced as a result of PCI and other components of contemporary care.10 The PCI interventions, however, are palliative and subject to acute and chronic complications of clinical significance, because of untoward effects of antithrombotic therapy and pathologic changes in the perturbed coronary artery.19,20
In addition to reperfusion, the other phenomenon with a major effect on a myocardial ischemic event is myocardial conditioning.12–17 Myocardial preconditioning refers to the effect of a few brief periods of coronary occlusion and reperfusion in slowing the progression of subsequent myocardial necrosis during the first 90 to 120 minutes of prolonged coronary occlusion. Within the first 90 minutes of the sustained coronary occlusion, preconditioned myocardium develops significantly less myocardial necrosis than does non-preconditioned myocardium. When the interval between the brief coronary occlusions and the subsequent prolonged coronary occlusion is delayed for more than 30 to 60 minutes, the effect of classical preconditioning is lost. However, 24 hours later, a second window of preconditioning (SWOP) becomes manifest.
In addition, postconditioning—which is gradual or intermittent reperfusion after a coronary occlusion, rather than immediate, complete, and permanent reperfusion—has been found to have a salutary effect on myocardial ischemia-reperfusion injury.
Both preconditioning at a distance and postconditioning at a distance, produced by episodes of transient ischemia induced in skeletal muscle or other noncardiac tissues, have been identified as having salutary effects on the evolution of myocardial ischemic injury. The mechanisms responsible for these phenomena are discussed below.
Modes of Myocardial Cell Injury and Death
Necrosis literally and most accurately describes the state of cells and tissues that have passed through reversible to irreversible injury, with the subsequent onset of degenerative changes. Contemporary cell-injury research has identified several modes of cell injury and death that can lead to necrosis. The two of major importance in myocardial ischemic injury and infarction are oncosis and apoptosis.15,16,21,22 Oncosis is characterized by cellular and organellar swelling due to progressive membrane dysfunction and damage in the setting of ischemic or toxic injury, coupled with rapid energy depletion reflected by rapid loss of adenosine triphosphate (ATP). Apoptosis is characterized by cellular and organellar shrinkage and subsequent fragmentation due to activation of a caspase enzyme cascade via extrinsic or intrinsic pathways in the setting of various physiologic or pathologic stimuli and at least partial preservation of ATP. Although oncosis and apoptosis have been termed accidental and programmed cell death, respectively, both types of cell death are programmed in the sense that both can be triggered by the activation of cell surface receptors and both involve activation of distinctive gene profiles.21,22 Although the term necrosis has frequently been applied to the process of oncosis, that term is not accurate without qualification. Other recently suggested alternatives for oncosis are oncotic necrosis and programmed necrosis.22
Cardiomyocytes have been shown to have the molecular wherewithal to undergo either oncosis or apoptosis.21,22 Studies of experimental myocardial infarction have been performed using caspase inhibitors and mutant mouse models with knockout genes or increased expression of components of the apoptosis pathways. Test animals have shown partial reduction of infarct size, but not complete protection.23 These results indicate that the progression of myocardial ischemia involves both oncosis and apoptosis in approximately equal degree.15,16 The rate and magnitude of ATP depletion is probably a central factor in the progression of ischemic injury via oncotic or apoptotic pathways.21–25 Because ischemic cardiomyocytes are subject to simultaneous activation of both modes of cell death, myocardial ischemic injury can be considered to exhibit a hybrid pattern of cell injury and death.15,16
Autophagy is another mode of cell modulation that involves the segregation of cellular components, including proteins and mitochondria, into autophagic vacuoles, the merger of these autophagic vacuoles with lysosomes to form autophagolysosomes, and the subsequent degradation of the constituents in the vacuoles.15 Regulated autophagy is a mechanism of cell survival after stress, whereas unregulated autophagy leads to another distinctive form of programmed cell death.26,27 Autophagy has been shown to be capable of modulating cardiomyocyte injury and acute myocardial ischemic injury. Autophagy has also been found to have a role in cardiomyocyte survival in the setting of hibernating myocardium (chronic ischemic myocardium associated with decreased myocardial function).28
Central Role of the Mitochondria in Myocardial Ischemic Injury
Cardiomyocytes are structurally and functionally organized as follows: central nucleus (some cells are binucleate) that contains chromosomes with DNA; perinuclear lipofuscin granules (formed by regulated autophagy); lysosomes; Golgi apparatus connected to rough endoplasmic reticulum with ribosomes for protein synthesis; myofibrils arranged in sarcomeres for contraction; plasma membrane (sarcolemma) with invaginations (the T tubules) at the level of each sarcomere, and adjacent elements of smooth endoplasmic reticulum arranged in diads and triads with a central role in excitation-contraction coupling and calcium homeostasis; filaments of the cytoskeleton connecting the sarcolemma to the cell interior; and numerous mitochondria specialized for energy production (Fig. 1). Adjacent cardiomyocytes are connected to each other end-to-end by a specialized component of the sarcolemma (a plasma membrane) and by the intercalated discs, and side-to-side by desmosomes. The cardiomyocytes are supplied with oxygen and nutrients by the microvasculature: there is a one-to-one number of cardiomyocytes and capillaries. The interstitium also contains various nonmyocytic cells. Endothelium and other nonmyocytic cells constitute 75% of the number of cells in the myocardium, whereas the large, specialized cardiomyocytes constitute 75% of the volume of the myocardium.15

Fig. 1 Ultrastructure of the normal mammalian myocardium. The typical cardiomyocyte has a central nucleus (N), compact cytoplasm, a sarcolemma (plasma membrane) with invaginations called transverse tubules (TT), myofibrils arranged into sarcomeres, a few lipid droplets (LD), glycogen granules, and numerous mitochondria (M) containing many densely packed invaginations of the inner membranes, the cristae. In proximity to the cardiomyocytes are capillaries (C). Electron micrograph, ×11,000.
Cardiomyocytes are specialized for continuous contractile activity and, as a result, have a very high energy requirement. This energy is supplied by the large number of mitochondria that are organelles specialized to conduct oxidative phosphorylation. The high energy capacity of the mitochondria of cardiomyocytes is reflected by their distinctive ultrastructure, which is characterized by large numbers of tightly paced cristae formed from invaginations of the inner mitochondrial membrane. Oxidative phosphorylation and ATP generation are crucially dependent upon maintenance of the electrical potential difference (ΔΨm) across the inner mitochondrial membrane.16,17
Oncosis and apoptosis result from activation of distinct but overlapping subcellular pathways involving cell membrane death receptors, endoplasmic reticulum, and mitochondria. However, multiple lines of evidence have converged to show that the mitochondria have a central role in the pathogenesis of cell injury (Figs. 2 and 3).24,29–32 In stressed cells, deleterious and salutary effects acting on mitochondria are mediated by death channels and salvage pathways, respectively.29,30 The mitochondrial death channels include the mitochondrial permeability transition pore (mPTP) and the mitochondrial apoptosis channel (mAC). The mPTP is a voltage-dependent channel that is regulated by calcium and oxidative stress. Three different proteins influence the function of the mPTP. These are the voltage-dependent anion channel (VDAC), the adenine nucleotide translocator (ANT), and cyclophilin D (CypD). The VDAC is located in the outer membrane, the ANT in the inner membrane, and CypD on the matrix side of the inner membrane. Together, these proteins span the 2 mitochondrial membranes and provide a path from the mitochondrial matrix to the cytoplasm. The second channel, the mAC, is responsible for mediating many forms of apoptosis. The activity of the mAC is highly regulated by the Bcl-2 family of proteins.33
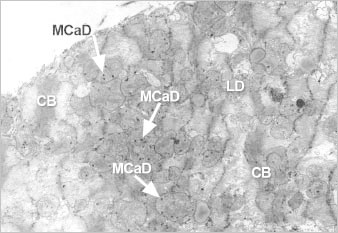
Fig. 2 Ultrastructure of ischemic cardiomyocyte with features of contraction-band formation and calcium overload as seen in the peripheral zone of an evolving canine myocardial infarct. Note the foci of myofibrils condensed into bands (CB), the lipid droplets (LD), and granular mitochondrial calcium phosphate deposits (MCaD). Electron micrograph, ×6,500. Reproduced with permission.14

Fig. 3 Ultrastructural detail of ischemic cardiomyocyte with lipid deposits (LD) and swollen mitochondria containing very electron-dense, annular-granular calcium phosphate deposits (MCaD).
As adenosine triphosphate (ATP) is reduced, mitochondrial oxidative capacity is decreased, leading to an accumulation of reesterified fatty acids as lipid droplets. As the sarcolemmal function becomes impaired, an increase in calcium influx occurs. With reperfusion, further calcium influx is coupled with an oxidative burst, generating toxic oxygen-based radicals. The excess calcium triggers hypercontraction of the myofibrils, which manifests as contraction bands. The overload of calcium ions and toxic oxygen-based radicals leads to opening of the mitochondrial permeability transition pore (mPTP), loss of membrane potential, swelling, and collapse of ATP generation. Electron micrograph, ×26,000. Reproduced with permission.14
Apoptosis can be initiated both by an extrinsic pathway that involves the activation of certain membrane receptors with death domains and by intrinsic pathways that involve the endoplasmic reticulum and the mitochondria. There are levels of interaction between these pathways. A central event in many forms of apoptosis is mitochondrial outer-membrane permeabilization (MOMP), which is produced by activation of the mAC by certain stimuli.29–33 Opening of the MOMP leads to release of cytochrome c and other molecules that join with cytoplasmic components, including procaspase 9, apoptotic protease-activating factor-1 (Apaf-1), and dATP, to form the apoptosome. The apoptosome then triggers the activation of caspase 3 and other effector caspases in the cytoplasm, leading to downstream effects on the nucleus. The mitochondrial pathway (rather than the extrinsic pathway) appears to be dominant in the apoptosis component of myocardial ischemic injury. This was confirmed in a recent experimental study in which the extent of myocardial ischemia and reperfusion damage was reduced in transgenic mice overexpressing the anti-apoptotic Bcl-2 but not in transgenic mice overexpressing a truncated form of the surface death receptor.34 A central event in oncosis is early opening of the mPTP in the inner mitochondrial membrane, which occurs without cytochrome c release. The opening of the mPTP results in the immediate loss of the electrical potential difference across the inner membrane, with resultant cessation of ATP synthesis, influx of solute, and mitochondrial swelling. Among other mechanisms, a modest increase in the expression of uncoupling protein 2 (UCP-2) can cause a rapid decline in mitochondrial membrane potential and ATP level, resulting in oncosis.35 The interrelationships between apoptosis and oncosis has been confirmed by models that involve initial apoptosis followed by oncosis and, conversely, oncosis followed by apoptosis.24,31–33 Regarding the mitochondria, the oncotic trigger (loss of the mPTP) can occur in close temporal relationship to cytochrome c release via the mAC.
In ischemic cardiomyocytes, lack of oxygen causes the electron transport in mitochondria to back up, thereby priming various components of the electron transport chain to generate oxygen free radicals when oxygen returns. Indeed, upon reperfusion there occurs, in the form of an oxidative burst, a massive diversion of electrons from the electron transport system to the generation of oxygen radicals. Simultaneously, a large influx of calcium occurs. A prime target of the excess oxygen radicals and calcium is the mPTP, which results in opening of the mPTP and collapsing of mitochondrial function.17 This is a central event in ischemic reperfusion injury.
Conditioning is a complex phenomenon that begins with the activation of various G-protein coupled receptors by various autocoids, including adenosine, bradykinin, and opioids, which are released during the brief periods of ischemia and reperfusion.36–38 Activation of these receptors initiates a complex signaling cascade, including multiple kinases, that leads to the opening of potassium channels in the mitochondrial membrane and maintenance of the mPTP and the electrical potential of the inner mitochondrial membrane. Preservation of mitochondrial function and ATP production is the primary mechanism for the protective effect of conditioning. The SWOP, or late preconditioning, is due to the activation of genes that produce molecules that participate in the subcellular pathways of conditioning.
The protective effect of conditioning involves, among other things, the activation of a reperfusion injury salvage kinase (RISK) pathway in the mitochondria.16,17,39 One component of this pathway is the action of phosphatidylinositol-3 kinase (PI-3K) on Akt (protein kinase B) and on the mammalian target of rapamycin (mTOR). The other component involves mitogen-associated protein kinase (MAPK) and p42/p44 extracellular signal-related kinase (ERK). The 2 arms of this pathway converge on p70s6 kinase to activate glycogen synthase kinase β, which acts to prevent opening of the mPTP. An isoform of the ATP-sensitive potassium channel (K+ ATP) also regulates mPTP opening.
Pathophysiologic and Pharmacologic Considerations
The cumulative effects of episodes of ACS and the subsequent extent of myocardial infarction have major significance for prognosis. Acutely, extensive myocardial infarction is associated with acute heart failure, cardiogenic shock, and malignant arrhythmias.18 Chronically, the cumulative effect of ischemic damage leads to chronic heart failure.15 Chronic heart failure is characterized not only by the loss of myocardium but by progressive pathologic remodeling in the viable myocardium. Myocardial remodeling involves complex changes, including hypertrophy, hyperfunction, and dysfunction of cardiomyocytes; death of cardiomyocytes by oncosis, apoptosis, and autophagy; attempts at replacement of cardiomyocytes by activation of stem cell mechanisms; connective-tissue proliferation; microcirculatory changes; and altered ventricular geometry.12–16 Coronary artery stenting and coronary artery bypass grafting, unless introduced early, probably will not be effective in stabilizing or reversing the process that leads to fixed structural dilation of the heart and manifests itself clinically as ischemic cardiomyopathy.15
Progress in understanding the basic pathobiology of ischemic heart disease has led to many years of research aimed at developing pharmacologic approaches to limiting myocardial ischemic damage. Unfortunately, initially promising experimental work in this area has too often been followed by negative or equivocal clinical trials. Although myocardial ischemia-reperfusion injury is clearly mediated by calcium overload, toxic oxygen-based molecules, inflammatory mediators, and neutrophils, agents aimed against these components of ischemic injury have not been consistently effective in clinical trials.16 A number of reasons for this situation have been brought to light.16,17,40 The major advances in salvage of ischemic myocardium have come from the introduction of thrombolytic therapy and PCI, rather than from pharmacologic regimens. Nevertheless, increased understanding of the pathobiology of myocardial ischemic injury and the central role of the mitochondria has led to renewed interest and promise in novel therapeutic approaches, used alone or in combination with PCI, for the treatment of patients with ACS. These treatments involve pathophysiologic and pharmacologic approaches as well as cell-based therapies (Table I).15,16,29–33,41–51
TABLE I. New Cardioprotective Strategies for Patients with ACS

Ischemic preconditioning has to be instituted before the onset of sustained myocardial ischemia, making its application impractical for a patient who presents with an evolving AMI. However, ischemic preconditioning has been effectively applied in settings wherein the clinical event can be planned, including PCI and cardiac surgery.42–47 Postconditioning has been applied with success in the catheterization laboratory during PCI. Some small clinical trials have reported beneficial effects, including, in one study, a 36% reduction in infarct size as measured by creatine kinase release.46 Remote ischemic conditioning appears to involve the release of humoral factors or the activation of neural signals from the transiently ischemic organ, which initiates the protective conditioning pathways in the heart.17,47 One small trial has reported improved myocardial salvage when ischemic remote conditioning was applied in the field en route to the hospital. Larger clinical trials to test the efficacy of this approach are now under way. Another pathophysiologic intervention, mild hypothermia, is receiving renewed interest.47 There, the broad mechanism of action is slowing the metabolism, which also slows alterations of the signaling pathways.
The promising results of conditioning protocols has renewed interest in finding pharmacologic approaches to activating the cellular pathways responsible for the protective effect of conditioning, with one goal being the reduction or prevention of lethal reperfusion injury.17 The prototype for this pharmacologic approach is cyclosporine. Cyclosporine is known to inhibit the formation and opening of the mPTP by binding to CypD. In a proof-of-concept clinical trial involving 58 patients, cyclosporine administered as a 2.5-mg/kg intravenous bolus at the time of PCI was found to reduce the size of myocardial infarction compared with placebo. Infarct size was reduced by 40% as measured by creatine kinase release. Evaluation by MRI also showed less myocardial damage.41,42 A large multicenter trial of 1,000 patients is now under way. Other pharmacologic agents are being designed and tested for their ability to activate the RISK pathway in the mitochondria and to improve mitochondrial energy metabolism.17,39,47,48 Other pharmacologic agents under recent or active investigation include adenosine, atrial naturetic peptide, β-adrenergic–blocking drugs, nitrates, phosphodiesterase-5 inhibitors, and supersaturated oxygen therapy and antioxidant peptides targeted at mitochondria.47–50 There clearly is a resurgence of activity in the area of pharmacologic cardioprotection.51
In experimental models, caspase inhibitors and other suppressors of apoptosis have been shown to reduce myocardial infarct size.43 However, approaches to retard apoptosis during evolving myocardial ischemic injury in patients have not yet been demonstrated. Molecules termed necrostatins have been found to have inhibitory effects that are thought to be mediated by activity against receptors involved in the hybrid form of combined apoptosis and oncosis, designated as necroptosis.43 Exploration of the conditioning effects on nonmyocytic components of the myocardium, including endothelial cells, fibroblasts, and extracellular matrix, also is under consideration.44
Cell-Based Therapeutic Approaches
Intense interest has developed in the use of stem cells (primitive replicating cells) as a completely different approach for the reduction of myocardial damage and pathologic remodeling and for the regeneration of normal myocardium.15,52–56 A wide variety of stem cells and protocols has been explored in experimental models and in clinical trials. Stem cell populations are defined by specific cell surface markers. The stem cells include harvested endogenous cardiac stem cells, as well as stem cell populations enriched from bone marrow. Induced pluripotent stem cells prepared from adult cells also are being studied. Stem cell therapy is intended to augment the activity of resident cardiac stem cells, which contribute to cardiac plasticity and provide for some level of cardiac-cell turnover but cannot prevent the manifestations of myocardial aging and heart failure.53
The proposed mechanisms of benefit in regard to stem cells focus on the restoration of myocardial function and the regeneration of cardiomyocytes and vessels, either directly via administered stem cells or indirectly via the paracrine effects on myocardial constituents, including endogenous cardiac stem cells.15 Although some experimental studies have evaluated the effects of autologous stem cells, many of these studies have used allogeneic or xenogeneic stem cells, usually with some immunosuppressive regimen, in various species or in immunodeficient mice.15,52,54,57,58 Evidence of cardiomyocyte regeneration has been variable in experimental models.15,52,54,57,58 The results of clinical trials have also been variable, with some showing improvement in values such as ejection fraction and others reporting insignificant effects.15,52,54–56
Initial clinical trials have involved autologous stem cell administration. Given the complexities involved in autologous stem cell programs, increasing interest is developing in the use of allogeneic stem cell preparations, based on the concept that various stem cell preparations are immunoprivileged and immunologically inert. There is reason for concern, however, that nonautologous stem cells can become immunogenic when exposed to a perturbed environment such as an evolving myocardial infarct, with potential consequences of greater inflammation and destruction of the stem cells.57–59 The evidence for long-term retention and differentiation of stem cells, even autologous stem cells, into new cardiomyocytes and vessels is inconsistent.15,52,59 Investigators in the National Institutes of Health-sponsored Cardiovascular Cell Therapy Research Network have recognized that variability in the response to cell-based therapy determines the fate of transplanted stem cells and the relationship of cellular transplantation to changes in cardiac function.56 An alternative view is that cell-based therapy might prove to be a logistically complex system in which specialized cells are used for the delivery and induction of mediator and effector molecules. These molecules then exert effects of variable duration on the myocardium, even though there is inconsequential retention or differentiation of the transplanted precursor and stem cells. Much further investigation is needed to determine the proper role of cell-based therapy in the treatment of patients with ischemic heart disease.
Conclusion
Prevention of atherosclerotic disease on the basis of understanding the risk factors clearly is the most cost-effective and preferred approach.60 Nevertheless, ischemic heart disease due to established coronary atherosclerosis remains a major clinical problem. Considerable progress has been made in the treatment of patients with ACS through the application of electrophysiologic monitoring and PCI.10 However, given the complexities of limiting myocardial damage, morbidity, and death from ACS, additional and potentially more effective treatment options for patients with ischemic heart disease are still needed. In this context, increased understanding of the pathobiology of myocardial ischemic injury is leading to new thinking regarding approaches to limit the extent of myocardial damage and remodeling and to ensure salvage of the optimal amount of viable, functional myocardium. The goals remain to decrease the morbidity and mortality rates from ischemic heart disease and to reduce the number of patients with end-stage heart disease that requires costly care, including chronic-heart-failure therapy, ventricular-assist-device support, implantable-cardiac-defibrillator support, and cardiac transplantation.61,62
Footnotes
Address for reprints: L. Maximilian Buja, MD, Department of Pathology and Laboratory Medicine, The University of Texas Medical School at Houston, 6431 Fannin St., MSB 2.276, Houston, TX 77030
E-mail: l.maximilian.buja@uth.tmc.edu
References
- 1.Gimbrone MA Jr, Garcia-Cardena G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol 2013;22(1):9–15. [DOI] [PMC free article] [PubMed]
- 2.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 2012;32(9):2045–51. [DOI] [PMC free article] [PubMed]
- 3.Buja LM, Willerson JT. The role of coronary artery lesions in ischemic heart disease: insights from recent clinicopathologic, coronary arteriographic, and experimental studies. Hum Pathol 1987;18(5):451–61. [DOI] [PubMed]
- 4.Libby P. Molecular and cellular mechanisms of the thrombotic complications of atherosclerosis. J Lipid Res 2009;50 Suppl:S352–7. [DOI] [PMC free article] [PubMed]
- 5.Fishbein MC. The vulnerable and unstable atherosclerotic plaque. Cardiovasc Pathol 2010;19(1):6–11. [DOI] [PubMed]
- 6.Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists' view. Eur Heart J 2013;34(10):719–28. [DOI] [PubMed]
- 7.Reimer KA, Ideker RE. Myocardial ischemia and infarction: anatomic and biochemical substrates for ischemic cell death and ventricular arrhythmias. Hum Pathol 1987;18(5):462–75. [DOI] [PubMed]
- 8.Reimer KA, Jennings RB. Myocardial ischemia, hypoxia, and infarction. In: Fozzard HA, Haber E, Jennings RB. The heart and cardiovascular system: scientific foundations. 2nd ed. New York: Raven Press; 1991. p. 1875–973.
- 9.Burke AP, Virmani R. Pathophysiology of acute myocardial infarction. Med Clin North Am 2007;91(4):553–72. [DOI] [PubMed]
- 10.Thiene G, Basso C. Myocardial infarction: a paradigm of success in modern medicine. Cardiovasc Pathol 2010;19(1):1–5. [DOI] [PubMed]
- 11.Basso C, Rizzo S, Thiene G. The metamorphosis of myocardial infarction following coronary recanalization. Cardiovasc Pathol 2010;19(1):22–8. [DOI] [PubMed]
- 12.Buja LM. Modulation of the myocardial response to ischemia. Lab Invest 1998;78(11):1345–73. [PubMed]
- 13.Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol 2005;14(4):170–5. [DOI] [PubMed]
- 14.Willerson JT, Buja LM. Myocardial reperfusion: biology, benefits, and consequences. Dialog Cardiovasc Med 2006; 11(4):267–78.
- 15.Buja LM, Vela D. Cardiomyocyte death and renewal in the normal and diseased heart. Cardiovasc Pathol 2008;17(6): 349–74. [DOI] [PubMed]
- 16.Buja LM, Weerasinghe P. Unresolved issues in myocardial reperfusion injury. Cardiovasc Pathol 2010;19(1):29–35. [DOI] [PubMed]
- 17.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007;357(11):1121–35. [DOI] [PubMed]
- 18.Lavie CJ, Gersh BJ. Mechanical and electrical complications of acute myocardial infarction. Mayo Clin Proc 1990;65(5): 709–30. [DOI] [PubMed]
- 19.Buja LM. Vascular responses to percutaneous coronary intervention with bare-metal stents and drug-eluting stents: a perspective based on insights from pathological and clinical studies. J Am Coll Cardiol 2011;57(11):1323–6. [DOI] [PubMed]
- 20.Otsuka F, Finn AV, Yazdani SK, Nakano M, Kolodgie FD, Virmani R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat Rev Cardiol 2012;9 (8):439–53. [DOI] [PubMed]
- 21.Weerasinghe P, Hallock S, Brown RE, Loose DS, Buja LM. A model for cardiomyocyte cell death: insights into mechanisms of oncosis. Exp Mol Pathol 2013;94(1):289–300. [DOI] [PubMed]
- 22.Weerasinghe P, Buja LM. Oncosis: an important non-apoptotic mode of cell death. Exp Mol Pathol 2012;93(3):302–8. [DOI] [PubMed]
- 23.Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest 2005;115(3):565–71. [DOI] [PMC free article] [PubMed]
- 24.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 2010;72:19–44. [DOI] [PubMed]
- 25.Gottlieb RA. Cell death pathways in acute ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther 2011;16(3–4):233–8. [DOI] [PMC free article] [PubMed]
- 26.Gottlieb RA, Mentzer RM Jr. Autophagy: an affair of the heart. Heart Fail Rev 2012 Nov 28. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- 27.Hickson-Bick DL, Jones C, Buja LM. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J Mol Cell Cardiol 2008;44(2):411–8. [DOI] [PubMed]
- 28.Yan L, Sadoshima J, Vatner DE, Vatner SF. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy 2009;5(5):709–12. [DOI] [PubMed]
- 29.Webster KA. Programmed death as a therapeutic target to reduce myocardial infarction. Trends Pharmacol Sci 2007;28 (9):492–9. [DOI] [PubMed]
- 30.Webster KA. Mitochondrial death channels. Am Sci 2009;97 (5):384–91. [DOI] [PMC free article] [PubMed]
- 31.Kung G, Konstantinidis K, Kitsis RN. Programmed necrosis, not apoptosis, in the heart. Circ Res 2011;108(8):1017–36. [DOI] [PubMed]
- 32.Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol 2012;32(7):1552–62. [DOI] [PMC free article] [PubMed]
- 33.Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A 2012;109(17):6566–71. [DOI] [PMC free article] [PubMed]
- 34.Kristen AV, Ackermann K, Buss S, Lehmann L, Schnabel PA, Haunstetter A, et al. Inhibition of apoptosis by the intrinsic but not the extrinsic apoptotic pathway in myocardial ischemia-reperfusion. Cardiovasc Pathol 2013 Feb 11. Epub ahead of print. [DOI] [PubMed]
- 35.Mills EM, Xu D, Fergusson MM, Combs CA, Xu Y, Finkel T. Regulation of cellular oncosis by uncoupling protein 2. J Biol Chem 2002;277(30):27385–92. [DOI] [PubMed]
- 36.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev 2003;83(4):1113–51. [DOI] [PubMed]
- 37.Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemic preconditioning. Heart Fail Rev 2007;12(3–4):181–8. [DOI] [PubMed]
- 38.Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther 2010;24(3):225–34. [DOI] [PMC free article] [PubMed]
- 39.Smith CC, Yellon DM. Adipocytokines, cardiovascular pathophysiology and myocardial protection. Pharmacol Ther 2011;129(2):206–19. [DOI] [PubMed]
- 40.Miura T, Miki T. Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol 2008;103(6):501–13. [DOI] [PubMed]
- 41.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 2008;359(5):473–81. [DOI] [PubMed]
- 42.Morel O, Perret T, Delarche N, Labeque JN, Jouve B, Elbaz M, et al. Pharmacological approaches to reperfusion therapy. Cardiovasc Res 2012;94(2):246–52. [DOI] [PubMed]
- 43.Oerlemans MI, Koudstaal S, Chamuleau SA, de Kleijn DP, Doevendans PA, Sluijter JP. Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int J Cardiol 2013;165(3):410–22. [DOI] [PubMed]
- 44.Bell RM, Yellon DM. Conditioning the whole heart–not just the cardiomyocyte. J Mol Cell Cardiol 2012;53(1):24–32. [DOI] [PubMed]
- 45.Hausenloy DJ, Boston-Griffiths E, Yellon DM. Cardioprotection during cardiac surgery. Cardiovasc Res 2012;94(2):253–65. [DOI] [PMC free article] [PubMed]
- 46.Sharma V, Bell RM, Yellon DM. Targeting reperfusion injury in acute myocardial infarction: a review of reperfusion injury pharmacotherapy. Expert Opin Pharmacother 2012; 13(8):1153–75. [DOI] [PubMed]
- 47.Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol 2012;59(11): 969–78. [DOI] [PubMed]
- 48.Whittington HJ, Hall AR, McLaughlin CP, Hausenloy DJ, Yellon DM, Mocanu MM. Chronic metformin associated cardioprotection against infarction: not just a glucose lowering phenomenon. Cardiovasc Drugs Ther 2013;27(1):5–16. [DOI] [PubMed]
- 49.Rocha M, Hernandez-Mijares A, Garcia-Malpartida K, Banuls C, Bellod L, Victor VM. Mitochondria-targeted antioxidant peptides. Curr Pharm Des 2010;16(28):3124–31. [DOI] [PubMed]
- 50.Szeto HH, Schiller PW. Novel therapies targeting inner mitochondrial membrane–from discovery to clinical development. Pharm Res 2011;28(11):2669–79. [DOI] [PubMed]
- 51.Bell R, Beeuwkes R, Botker HE, Davidson S, Downey J, Garcia-Dorado D, et al. Trials, tribulations and speculation! Report from the 7th Biennial Hatter Cardiovascular Institute Workshop. Basic Res Cardiol 2012;107(6):300. [DOI] [PubMed]
- 52.Buja LM, Vela D. Current status of the role of stem cells in myocardial biology and repair. Cardiovasc Pathol 2011;20(5): 297–301. [DOI] [PubMed]
- 53.Kajstura J, Rota M, Cappetta D, Ogorek B, Arranto C, Bai Y, et al. Cardiomyogenesis in the aging and failing human heart. Circulation 2012;126(15):1869–81. [DOI] [PMC free article] [PubMed] [Retracted]
- 54.Karantalis V, Balkan W, Schulman IH, Hatzistergos KE, Hare JM. Cell-based therapy for prevention and reversal of myocardial remodeling. Am J Physiol Heart Circ Physiol 2012;303(3):H256–70. [DOI] [PMC free article] [PubMed]
- 55.Ptaszek LM, Mansour M, Ruskin JN, Chien KR. Towards regenerative therapy for cardiac disease. Lancet 2012;379 (9819):933–42. [DOI] [PubMed]
- 56.Rodriguez-Porcel M, Kronenberg MW, Henry TD, Traverse JH, Pepine CJ, Ellis SG, et al. Cell tracking and the development of cell-based therapies: a view from the Cardiovascular Cell Therapy Research Network. JACC Cardiovasc Imaging 2012;5(5):559–65. [DOI] [PMC free article] [PubMed]
- 57.Vela DC, Silva GV, Assad JA, Sousa AL, Coulter S, Fernandes MR, et al. Histopathological study of healing after allogenic mesenchymal stem cell delivery in myocardial infarction in dogs. J Histochem Cytochem 2009;57(2):167–76. [DOI] [PMC free article] [PubMed]
- 58.Gahremanpour A, Vela D, Zheng Y, Silva GV, Fodor W, Cardoso CO, et al. Xenotransplantation of human unrestricted somatic stem cells in a pig model of acute myocardial infarction. Xenotransplantation 2013;20(2):110–22. [DOI] [PubMed]
- 59.Buja LM, Vela D. Immunologic and inflammatory reactions to exogenous stem cells: implications for experimental studies and clinical trials for myocardial repair. J Am Coll Cardiol 2010;56(21):1693–700. [DOI] [PubMed]
- 60.Frohlich J, Al-Sarraf A. Cardiovascular risk and atherosclerosis prevention. Cardiovasc Pathol 2013;22(1):16–8. [DOI] [PubMed]
- 61.Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol 2011(1);8:30–41. [DOI] [PMC free article] [PubMed]
- 62.Liu SS, Monti J, Kargbo HM, Athar MW, Parakh K. Frontiers of therapy for patients with heart failure. Am J Med 2013 (1);126:6–12. [DOI] [PubMed]