

Abstract
Previously, DFNB89, a locus associated with autosomal-recessive nonsyndromic hearing impairment (ARNSHI), was mapped to chromosomal region 16q21–q23.2 in three unrelated, consanguineous Pakistani families. Through whole-exome sequencing of a hearing-impaired individual from each family, missense mutations were identified at highly conserved residues of lysyl-tRNA synthetase (KARS): the c.1129G>A (p.Asp377Asn) variant was found in one family, and the c.517T>C (p.Tyr173His) variant was found in the other two families. Both variants were predicted to be damaging by multiple bioinformatics tools. The two variants both segregated with the nonsyndromic-hearing-impairment phenotype within the three families, and neither mutation was identified in ethnically matched controls or within variant databases. Individuals homozygous for KARS mutations had symmetric, severe hearing impairment across all frequencies but did not show evidence of auditory or limb neuropathy. It has been demonstrated that KARS is expressed in hair cells of zebrafish, chickens, and mice. Moreover, KARS has strong localization to the spiral ligament region of the cochlea, as well as to Deiters’ cells, the sulcus epithelium, the basilar membrane, and the surface of the spiral limbus. It is hypothesized that KARS variants affect aminoacylation in inner-ear cells by interfering with binding activity to tRNA or p38 and with tetramer formation. The identification of rare KARS variants in ARNSHI-affected families defines a gene that is associated with ARNSHI.
Main Text
Hearing impairment (HI) affects nearly 300 million people of all ages globally and increases in prevalence per decade of life.1 Children and adults with bilateral, moderate-to-profound HI have a poorer quality of life, which encompasses not only problems in physical function but also socioemotional, mental, and cognitive difficulties.2,3 In particular, children with congenital HI must be identified and habilitated within the first 6 months of life so that delays in the acquisition of speech, language, and reading skills can be prevented.4
Among children with congenital sensorineural HI, more than 80% do not display syndromic features and ∼60% have a family history of HI or a confirmed genetic etiology.5 Because of the complex cellular organization of the inner ear, hundreds of genes and proteins are predicted to influence auditory mechanisms. To date, for nonsyndromic HI (NSHI), about 170 loci have been localized and mutations in ∼75 genes have been identified in humans (Hereditary Hearing Loss Homepage). Of the gene variants that have been implicated in NSHI, almost 60% are autosomal recessive (AR) in inheritance, and 95% of the genes that harbor mutations that cause ARNSHI were initially mapped and identified in consanguineous families. The knowledge that has been gained from functional, expression, and localization studies after the identification of genes with mutations that cause NSHI has immensely expanded our understanding of inner-ear physiology.
Previously, an ARNSHI-associated locus, DFNB89, was mapped to chromosomal region 16q21–q23.2 in two unrelated, consanguineous Pakistani families.6 The two families, 4338 and 4406 (Figures 1A and 1B), had maximum multipoint LOD scores of 6.0 and 3.7, respectively. The homozygosity regions that overlap in the two families led to the identification of a 16.1 Mb locus (chr16: 63.6–79.7 Mb) that includes 180 genes. Additionally, a third consanguineous Pakistani family, 4284 (Figure 1C), was identified, and showed suggestive linkage to the DFNB89 region with a maximum multipoint parametric LOD score of 1.93. For family 4284, linkage analysis was performed for ∼6,000 SNP markers that were genotyped across the genome with the Illumina Linkage Panel IVb.
Figure 1.
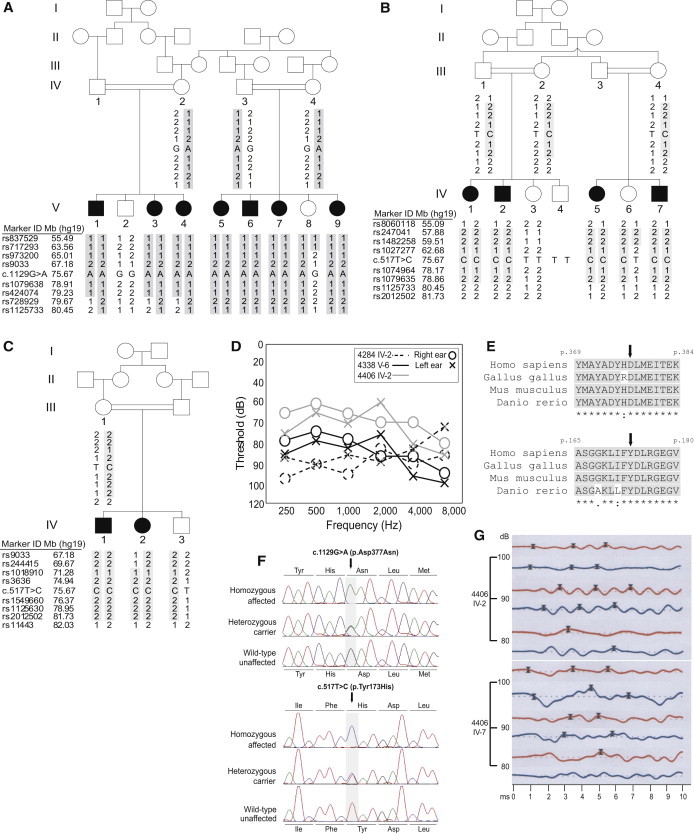
Pedigree Drawings, Clinical Data, and Mutation Information for Families Affected by KARS Variants
(A–C) Family 4338 (A), affected by the c.1129G>A (p.Asp377Asn) variant, and families 4406 (B) and 4284 (C), affected by the c.517T>C (p.Tyr173His) variant segregating with hearing impairment. Each family has a unique haplotype within the DFNB89 locus.
(D) Air-conduction audiograms for individuals V-6 of family 4338 (black solid), IV-2 of family 4406 (gray), and IV-2 of family 4284 (black dotted). Circles are for the right ear, and crosses are for the left ear. All three individuals have symmetric, moderate-to-profound impairment across all frequencies.
(E) Multiple-sequence alignment showing evolutionary conservation of amino acid residues Asp377 and Tyr173 (black arrows) in humans, chickens, mice, and zebrafish.
(F) Chromatograms comparing an HI individual who is homozygous for each KARS variant, a heterozygous carrier, and an individual with normal hearing.
(G) Auditory brainstem response (ABR) tracings at 80–100 dB from two individuals from family 4406. Red lines are for the right ear, and blue lines are for the left ear. Crosses mark the latency peaks at waves I, III, and V. ABR waveforms were well formed in both individuals but showed unilateral delay in interpeak latencies I-III or I-V and a difference > 0.2 ms in the interaural latencies of wave V. For individual IV-2, the delay in interpeak I-III or I-V was seen only at 100 dB, but not at 90 dB. Additionally, individual IV-7 had an absent compliance peak from tympanometry on the left ear, suggesting otitis media or middle-ear effusion at the time of testing, which could explain the latency delays on the left ear compared to the right ear. Otoacoustic emissions (OAEs) were bilaterally absent in both individuals, ruling out auditory neuropathy.
Consanguineous families 4284, 4338, and 4406 from Pakistan are affected by ARNSHI, which was found to segregate with unique haplotypes within the DFNB89 locus (Figures 1A–1C). From the medical history, no other risk factors were identified as a possible cause of HI. For all HI individuals, there were no reports of vertigo or other neurologic complaints. Additionally, from physical examination, no other syndromic, vestibular, neurologic, or systemic abnormalities were detected. One HI individual from each family, namely V-6 from family 4338, IV-2 from family 4406, and IV-2 from family 4284 (Figures 1A–1C), was tested for air-conduction audiometry (Figure 1D). Bilateral, symmetric severe-to-profound hearing impairment was demonstrated in HI individuals from families 4284 and 4338, whereas individual IV-2 from family 4406 had symmetric moderate-to-severe hearing impairment across all frequencies tested (Figure 1D).
Before the commencement of the study, approval was obtained from the institutional review boards of Quaid-i-Azam University and Baylor College of Medicine and the affiliated hospitals. Informed consent was obtained from all family members who participated in the study. DNA samples of HI individuals V-7 from family 4338, IV-5 from family 4406, and IV-1 from family 4284 (Figures 1A–1C) were submitted for whole-exome sequencing at the University of Washington Center for Mendelian Genomics. For families 4338 and 4406, sequence capture was performed in solution with the Roche NimbleGen SeqCap EZ Human Exome Library v.2.0 (∼36.6 Mb of target sequence), whereas for family 4284, the NimbleGen Big Exome 2011 Library (EZ Exome v.3.0 with ∼64 Mb target sequence) was used. Sequencing was performed with an Illumina HiSeq, and average read depths were 121× for sample V-7 (family 4338), 99 × for sample IV-5 (family 4406), and 53 × for sample IV-1 (family 4284). Fastq files were aligned to the human reference sequence (UCSC Genome Browser hg19) with the Burrows-Wheeler Aligner7 for the generation of demultiplexed BAM files. Realignment of indel regions, recalibration of base qualities, and variant detection and calling were performed with various components of the Genome Analysis Toolkit8 for the production of VCF files. Variant sites that were of low quality and more likely to be false positives were flagged. Annotation was performed with SeattleSeq 137.
For all variants that were homozygous for the minor allele and that were identified within the DFNB89 interval through exome sequencing, potentially functional variants, including nonsense, missense, splice-site, and indel variants, were screened for quality and against public variant databases dbSNP, 1000 Genomes, and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server. Within the DFNB89 interval, five rare missense variants within four genes, namely COG4 (MIM 606976), ZFHX3 (MIM 104155), KARS (MIM 601421), and CNTNAP4 (MIM 610518), were homozygous for the nonreference allele and were not observed in variant databases (Table 1). Of these five missense variants, only variants within KARS were observed in all three families affected by DFNB89-associated ARNSHI (Table 1). Additionally, for individual IV-1 from family 4284, only one homozygous KARS missense variant, c.517T>C (p.Tyr173His) (isoform 1 [RefSeq accession number NM_001130089.1]), was found within the DFNB89 locus and not in variant databases and also passed quality measures. This same variant was also homozygous in individual IV-5 from family 4406. On the other hand, individual V-7 from family 4338 was homozygous for KARS variant c.1129G>A (p.Asp377Asn).
Table 1.
Bioinformatic Evaluation of Rare Missense Variants Observed from Exome Sequence Data to Be within the DFNB89 Region at 16q21–q23.2
|
Sample ID |
|||||
|---|---|---|---|---|---|
| 4406 IV-5 | 4406 IV-5 | 4406 IV-5 and 4284 IV-1 | 4338 V-7 | 4338 V-7 | |
| hg19 coordinate | 70,543,852 | 72,830,796 | 75,670,401 | 75,665,624 | 76,587,774 |
| Gene | COG4 | ZFHX3 | KARS | KARS | CNTNAP4 |
| RefSeq accession number | NM_015386.2 | NM_006885.3 | NM_001130089.1 | NM_001130089.1 | NM_033401.3 |
| Nucleotide mutation | c.811A>G | c.5785C>T | c.517T>C | c.1129G>A | c.3703G>A |
| PhyloPa | 2.37 | 1.18 | 4.89 | 6.04 | 2.24 |
| GERPb | 5.61 | 2.48 | 5.96 | 6.03 | 3.47 |
| Amino acid substitution | p.Ile271Val | p.Pro1929Ser | p.Tyr173His | p.Asp377Asn | p.Ala1235Thr |
| PolyPhen-2c | benign | benign | probably damaging | probably damaging | benign |
| SIFT | tolerated | tolerated | damaging | damaging | tolerated |
| MutationTaster | disease causing | polymorphism | disease causing | disease causing | disease causing |
| Mutation Assessor | neutral | neutral | functional, high | functional, high | not available |
| LRT | deleterious | neutral | deleterious | deleterious | deleterious |
All variants listed are absent from publically available databases. The following abbreviation is used: LRT, likelihood ratio test.
PhyloP scores indicate nucleotide conservation under a null hypothesis of neutral evolution.
GERP provides position-specific estimates of evolutionary constraint.
Based on HumVar data set for PolyPhen-2.
For missense variants that were of good quality and not observed in variant databases, five bioinformatics tools were used to evaluate potential functionality: PolyPhen-2,9 SIFT,10 MutationTaster,11 MutationAssessor,12 and the likelihood ratio test (LRT).13 Only the two variants within KARS—c.1129G>A (p.Asp377Asn) and c.517T>C (p.Tyr173His)—were deemed damaging by all five bioinformatics tools used for prediction of functionality of missense variants (Table 1).
To estimate the evolutionary conservation of the nucleotides and the amino acid residues at which the variants occur, we derived GERP14 and phyloP15 (with the phyloP46wayall option) scores from the UCSC Genome Browser. Nucleotides c.1129G and c.517T within KARS are highly conserved according to both PhyloP and GERP scores (Table 1). Protein sequences from nonhuman species were derived from the UniProtKB database with blastp and were aligned with ClustalW2.16 Multiple-sequence alignment of human KARS with 165 nonhuman KARS and similar proteins from primates to fungi provided further strong evidence of evolutionary conservation at the amino acid residues where the two variants occur (Figure 1E and Figure S1, available online). The tyrosine residue at position 173 is highly conserved except in three insect species, whereas the aspartic acid residue at position 377 is identical in all aligned sequences (Figure S1).
We sequenced variants predicted to be damaging in all family members and 325 ethnically matched control individuals to check for segregation with HI. We designed primers by using Primer3 software17 to amplify exons 5 and 9 of KARS. PCR-amplified DNA products were purified with ExoSAP-IT (Affymetrix-USB) and sequenced with the BigDye Terminator v.3.1 Cycle Sequencing Kit and an Applied Biosystems 3730 DNA Analyzer (Life Technologies). DNA sequences were analyzed with Sequencher software v.4.9 (Gene Codes). DNA samples from all available family members were sequenced for KARS variant c.1129G>A (p.Asp377Asn) in family 4338 and c.517T>C (p.Tyr173His) in both families 4284 and 4406 (Figures 1A–1C and 1F). In all three families, the KARS variants segregated with HI status. Neither KARS variant was identified in 650 ethnically matched control chromosomes.
Previously, an individual with both recessive intermediate Charcot-Marie-Tooth disease B (MIM 613641) and bilateral acoustic neuroma was described in the literature to have compound-heterozygous mutations in KARS.18 Additional testing was performed for the evaluation of both conditions within the families affected by DFNB89-associated ARNSHI. For HI individuals IV-2 and IV-7 from family 4406 (Figure 1B), tests for tympanometry, otoacoustic emissions (OAEs), and auditory brainstem response (ABR) were performed (Figure 1G). In both HI individuals, OAEs were absent and well-formed ABR waveforms were present (Figure 1G), thus ruling out auditory neuropathy and supporting the occurrence of cochlear pathology, particularly of the outer hair cells. Although the ABR waveforms in both individuals showed unilateral delay in interpeak latencies I-III or I-V and a difference > 0.2 ms in the interaural latencies of wave V (Figure 1G and Table S1), the delay in interpeak I-III or I-V for individual IV-2 was seen only at 100 dB, but not at 90 dB. Additionally, individual IV-7 had an absent compliance peak during tympanometry of the left ear, which is suggestive of otitis media or middle-ear effusion at the time of testing. To further exclude Charcot-Marie-Tooth disease (CMT), we tested HI individual V-6 from family 4338 (Figure 1A) for nerve conduction velocity of the median, tibial, common peroneal, and sural nerves and for electromyography of the abductor pollicis brevis, first dorsal interosseous, extensor hallucis longus, and tibialis anterior muscles. All motor and sensory action potentials were bilaterally normal; thus, in this individual, CMT is unlikely.
To predict the possible effect of KARS variants on protein structure, we used InterProScan19 to search for protein signatures within KARS and used SWISS-MODEL20 for protein modeling. The KARS c.517T>C (p.Tyr173His) variant was identified in two families (4284 and 4406) affected by DFNB89-associated ARNSHI. When modeled on the basis of the known structure of human KARS (Protein Data Bank ID 3BJU),21 position 173 occurs within β strand 2, and the change from the aromatic side chain of tyrosine to the basic side chain of histidine results in the loss of an H-bond with the asparagine residue at position 201 within α-helix 3 (Figures S2A and S2B). MutationTaster predicted the p.Tyr173His variant to cause the loss of the β strand. The residue at position 173 also forms two H-bonds with valine at position 184 within β strand 3; however, despite the amino acid change, these two bonds, which contribute to the maintenance of the antiparallel sheet, remain unchanged (Figures S2A and S2B).
The tyrosine residue at position 173 is predicted to occur within the oligomer-binding (OB)-fold motif of the nucleic-acid- or anticodon-binding domain. The OB-fold motif is composed of a highly conserved five-stranded β-barrel that is further organized into a β-meander made up of β strands 1–3 and a β-hairpin made up of β strands 4 and 5; the β-meander and β-hairpin are connected by a less conserved α-helix (i.e., α-helix 3 in KARS), which caps the entire β-barrel.22 Of the β-barrel components in KARS, strands β-1 and β-2 are least resistant to denaturation,23 which might imply the importance of the stability of these strands to the domain structure. The tyrosine residue at position 173 lies in the middle of the β-2 strand, and the loss of the H-bond with an asparagine residue at α-helix 3 probably affects the conformation of the β-barrel within the OB-fold motif. Moreover, the tyrosine residue at position 173 is adjacent to a phenylalanine residue that aligns with the active binding site of the anticodon-binding domain of aspartyl-tRNA synthetase (DARS).22 The p.Tyr173His variant is therefore hypothesized to interfere with tRNA binding and consequently the catalytic activity of KARS.
For family 4338, the KARS c.1129G>A (p.Asp377Asn) variant was found to segregate with HI. For position 377 within α-helix 9 of KARS, when the acidic side chain of the aspartic acid residue is changed to the amide side chain of asparagine, the H-bonds with glutamic acid at position 380 and isoleucine at 381 are maintained (Figures S2C and S2D). On the other hand, one out of three H-bonds with the aspartic acid residue at position 374, particularly the bond between the amine group at 374 and the carboxyl side chain at 377, is lost (Figures S2C and S2D). Also, the amino acid change introduces an H-bond with the lysine residue at position 126 of another KARS monomer within the tetrameric structure (Figures S2C and S2D). The amide side chain of asparagine has a different rotation angle than does the carboxyl side chain of aspartic acid. MutationTaster predicted the p.Asp377Asn variant to cause the loss of the α-helical structure. It can be seen that the H-bond-formation changes due to p.Asp377Asn interfere with the formation of the α-helix and cause the subsequent loss of the H-bond between the C=O group of tyrosine at position 375 and the NH group of leucine at 378 (Figures S2C and S2D).
In addition to having a role in charging lysyl-tRNA molecules, KARS plays a role in the stabilization of the multisynthetase complex (MSC) because of the high affinity of MSC scaffold proteins (e.g., p38) to KARS.24 The aspartic acid residue at position 377 within α-helix 9 of the aminoacylation domain is three residues upstream of the KARS tetramer interface that not only creates binding surfaces for KARS dimers to interact but also binds p38 scaffolding protein within the MSC.21 Loss of α-helix 9 due to the c.1129G>A (p.Asp377Asn) variant that was identified in family 4338 is predicted to affect the configuration of the tetramer interface. Additionally, the fact that the p.Asp377Asn variant causes the gain of an H-bond with another KARS monomer might affect dimer or tetramer formation. KARS is least active in aminoacylation as a tetramer that is unbound to p38, whereas the p38-bound KARS dimer and tetramer are both more than twice as active as the unbound tetramer.25 The p.Asp377Asn variant might also affect KARS activity by changing the equilibrium of the bound and unbound dimer-tetramer forms of KARS.
To further elucidate the role of KARS in hearing, we tested the occurrence of KARS cDNA in hair cells in chickens, zebrafish, and mice. All animals were kept with the approval of the Case Western Reserve University Institutional Animal Care and Use Committee. Sensory patches were dissected from white leghorn chickens that were less than 3 weeks old and incubated for 20 min in dissociation chicken saline solution (154 mM NaCl, 6 mM KCl, 0.1 mM CaCl2, 8 mM glucose, and 5 mM HEPES [pH = 7.4]) with Type XXVI endopeptidase (Sigma-Aldrich) at 0.1 mg/ml. After trituration, 200 hair cells were isolated by picking with a glass micropipette.26 RNA was isolated from these cells, and this was followed by synthesis and amplification of cDNA (Ovation RNA-Seq System, NuGen Technologies), which was subjected to massively parallel sequencing with an Illumina HiScan for the production of single-end reads. Quality control was performed on the raw sequencing data with FASTQC software, and TopHat27 was used for aligning the reads to the galGal3 reference genome. Cufflinks28 was then used on these alignment files for the generation of a transcriptome assembly. There were 103,045,330 total mapped reads. Expression values for each transcript were calculated as read counts (1,012 reads aligned to KARS and 1,286 reads aligned to protocadherin 15 or PCDH15), and the read alignments to galGal4 were viewed with the Integrated Genome Viewer29 through the Galaxy platform.30–32 KARS is expressed in chicken hair cells (Figure 2), as determined by massively parallel sequencing of the hair cell transcriptome. The reads per kilobase per million mapped reads was 4.61 for KARS (RefSeq NM_001030583) and 1.84 for PCDH15 (RefSeq NM_001044654), a gene which is known to be expressed specifically in hair cells.33
Figure 2.
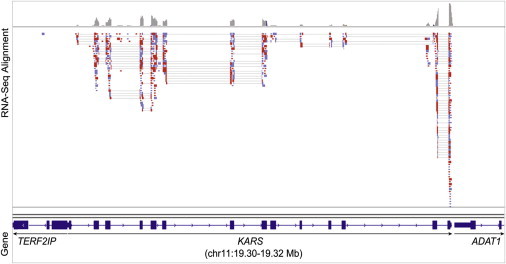
Expression of KARS in Chicken Hair Cells
Expression of KARS in purified chicken hair cells was detected by RNA-seq. The alignment of reads was visualized with the Integrative Genomics Viewer; blue read color represents forward orientation relative to the reference genome, and red color represents reverse reads.
For RT-PCR experiments, zebrafish cDNA was produced from purified adult hair cells, adult maculae, and whole embryos. Adult hair cell and macula cDNA were produced as previously described.26 To make cDNA from the whole fish, we isolated RNA from approximately 20 embryos that were 7 days postfertilization (RNeasy Mini Kit, QIAGEN) and used it to synthesize randomly primed cDNA (SuperScript III Reverse Transcriptase, Life Technologies). We followed the same procedure to produce cDNA from maculae of three adult mice of the FVB/NJ strain. PCR amplifications were performed (Ex Taq DNA Polymerase, Takara Bio) with interexonic primers designed to recognize different exons of Kars of either the zebrafish or the mouse. Each primer set (Table S2) was used with PCR parameters designed to amplify a segment of the Kars cDNA, but not the genomic locus. Kars expression was detected in zebrafish hair cells and in maculae of both zebrafish and mice (Figures 3A and 3B).
Figure 3.
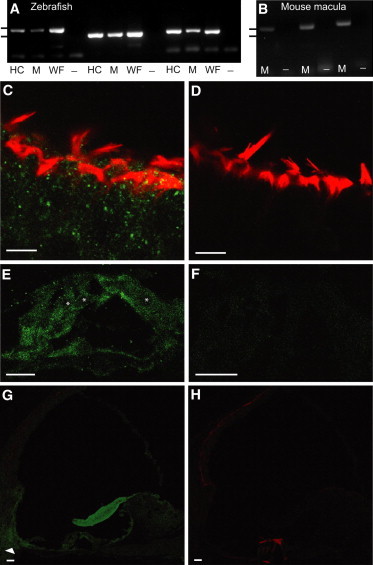
Expression and Localization of Kars in Zebrafish and Mice
RT-PCR analyses of zebrafish (A) and mouse (B) Kars mRNAs with the use of three separate primer sets for each species (Table S2). Tick marks in (A) and (B) correspond to molecular weights of 500 bp (upper) and 400 bp (lower). Confocal images of whole-mount mouse vestibular tissue (C and D) and cochlear cryosections (E–H) are displayed. Scale bars represent 1 μm in (A) and (B) and 10 μm in (C)–(H).
(A) Agarose gel confirms the expected amplicon size with the use of primer set 1, directed toward kars cDNA with zebrafish hair cell (HC), macula (M), and whole fish (WF) cDNA templates (lanes 1, 2, and 3, respectively). No product was observed without the template (–) (lane 4). The same pattern of template cDNAs was repeated for primer sets 2 (lanes 5–8) and 3 (lanes 9–12).
(B) Agarose gel confirms the expected Kars amplicon size with the use of primer set 1 and mouse macula cDNA (lane 1), but no product was observed without the template (lane 2). The same pattern of template cDNA was used for primer sets 2 (lanes 3–4) and 3 (lanes 5–6).
(C) Mouse vestibular tissue labeled with KARS polyclonal antibody (green) and phalloidin (red) reveals the broad distribution of KARS in hair cells and supporting cells.
(D) Control mouse vestibular tissue labeled with only secondary antibody and phalloidin.
(E) Mouse organ of Corti section labeled with KARS antibody and phalloidin demonstrates KARS localization to inner hair cells, outer hair cells, and supporting cells. Hair cell nuclei are denoted by asterisks.
(F) Control organ of Corti tissue labeled exclusively with secondary antibody.
(G) A cochlear section reveals KARS localization to an area that contains otic fibrocytes (arrowhead). Localization to this area was observed in 41% (n = 21) of sections.
(H) Control, cochlear section labeled with only secondary antibody and phalloidin.
For immunolabeling experiments, 6- to 12-month-old FVB/NJ mice were used. The vestibular tissue was extracted and fixed in 4% paraformaldehyde (PFA) in PBS for 1 hr. After three washes with PBS, the tissue was permeabilized with 0.05% Triton X-100 in PBS for 2 hr and then blocked with 1% BSA in 0.05% Triton X-100 for 2 hr. The tissue was then incubated overnight at 4°C with primary antibody (rabbit polyclonal to Lysyl tRNA synthetase ab31532, Abcam) at a 1:300 dilution in blocking solution. This antibody has been effectively used for immunolabeling fixed cultured cells.34 After a 30 min wash in PBS, the tissue was incubated with secondary antibody (Alexa Fluor 488 goat anti-rabbit IgG, Life Technologies) at a 1:200 dilution, and actin filaments were labeled with phalloidin (Alexa Fluor 568 phalloidin, Life Technologies) at a 1:50 dilution. After a final 30 min wash in PBS, the tissue was mounted on a slide with mounting medium (Vectashield, Vector Laboratories). For immunolabeling of the cochlea, the internal ears of mice were removed, fixed, and sectioned as previously described.26 Sections were postfixed with 4% PFA for 15 min and then washed three times with PBS. We performed antigen retrieval by incubating sections for 5 min with 1% sodium dodecyl sulfate and washing them three times with PBS. Sections were blocked with 5% goat serum and 0.1% Triton X-100 in PBS for 1 hr. Specimens were sequentially incubated with primary and secondary antibodies diluted in 5% goat serum and 0.1% Triton X-100. Prior to imaging, sections were set in Vectashield with a coverslip. All immunolabeled images were captured with a laser-scanning confocal microscope (Leica Microsystems) and a 40× oil-immersion objective.
Hair cells of the vestibular system exhibited labeling with KARS antibody (Figures 3C and 3D). Additionally, within the mouse organ of Corti, KARS was localized to the inner and outer hair cells, Deiters’ cells, and the basilar membrane (Figure 3E). The tectorial membrane showed a strong affinity for KARS antibody, and KARS labeling was strongest within the spiral ligament, particularly in the area containing type II and Type IV fibrocytes (Figure 3G). KARS was also strongly localized to the outer and inner sulcus cells and spiral limbus epithelium. On the other hand, KARS showed weak localization to the stria vascularis, Reissner’s membrane, and cochlear nerve endings (Figure 3G).
In this report, three ARNSHI-affected families that segregate different haplotypes overlapping within the DFNB89 interval6 were found to each have a missense mutation within KARS, which encodes a ubiquitously expressed protein, lysyl-tRNA synthetase (UniGene 131351 – Hs.3100). Many genes that are involved in the etiology of ARNSHI also encode ubiquitously expressed proteins, e.g., ESRRB (DFNB35),35 HGF (DFNB39),36 ILDR1 (DFNB42),37 and MSRB3 (DFNB74)38 to name a few.
Fourteen human phenotypes, all of which manifest as neurologic defects, are due to mutations in amino-acyl tRNA synthetase (ARS)-encoding genes, nine of which are mitochondrial. Of these phenotypes, five include sensorineural hearing impairment, namely Perrault syndrome (from mutations in mitochondrial leucyl-tRNA synthetase 2 [LARS2 [MIM 604544]39 and histidyl-tRNA synthetase 2 [HARS2 [MIM 600783]),40 Usher syndrome type IIIB (MIM 614504) (caused by a homozygous variant in cytoplasmic histidyl-tRNA synthetase [HARS (MIM 142810)]),41 CMT type 2N (MIM 613287) (due to a variant in dominant alanyl-tRNA synthetase [AARS (MIM 601065)]),42 and pontocerebellar hypoplasia type 6 (caused by compound-heterozygous mutations in arginyl-tRNA synthetase 2 [RARS2 (MIM 611524)]).43 Functional studies for the missense mutations in these ARS-encoding genes showed adequate localization but decreased aminoacylation activity.40–42 Alternatively, ARS variants that have normal enzymatic and localizing capabilities can also have decreased aminoacylation activity as a result of misfolding and failure to adopt a globule-like state.44
For KARS, studies using bacteria and cultured mammalian cell lines have demonstrated that KARS mutations result in a temperature-regulated decrease in KARS activity or cell apoptosis.45,46 An individual with CMT was found to have compound-heterozygous KARS variants18 c.398T>A (p.Leu133His) and a duplication of TT nucleotides, which results in a frameshift change and leads to a null allele via nonsense-mediated decay. It was previously shown that these two KARS variants significantly reduce the enzyme activity of the protein.18 Additional testing in HI individuals from the two families affected by DFNB89-associated ARNSHI ruled out auditory neuropathy and CMT, thus identifying NSHI as a phenotype associated with KARS mutations.
KARS mRNA was detected in isolated hair cells of chickens and zebrafish, and additional mouse studies demonstrated strong localization of KARS to otic fibrocytes and the hair cells and supporting cells of the cochlea. The presence of this protein within inner-ear structures that are involved in mechanotransduction provides further evidence of a role for KARS in hearing. Because of the localization pattern of KARS within the cochlea, perturbations in aminoacylation might affect many of the cellular processes of the different specialized cells of the cochlea and therefore result in hearing impairment. The identification of rare KARS variants in ARNSHI-affected families defines a gene that is associated with NSHI.
Acknowledgments
We are very thankful to the families who participated in the study. This study was funded by grants DC003594, DC011651, and DC009437 from the National Institute on Deafness and Other Communication Disorders of the National Institutes of Health (NIH), grant HG006493 from the National Human Genome Research Institute of the NIH, and the Higher Education Commission of Pakistan. Genotyping of the families was performed at the Center for Inherited Disease Research, which is funded through the NIH to The Johns Hopkins University under contract number N01-HG-065403.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/
Cufflinks, http://cufflinks.cbcb.umd.edu/
FASTQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Galaxy, https://main.g2.bx.psu.edu/
Genome Analysis Toolkit, http://www.broadinstitute.org/gatk/
Hereditary Hearing Loss Homepage, http://hereditaryhearingloss.org
Integrative Genomics Viewer, www.broadinstitute.org/igv/
InterProScan, http://www.ebi.ac.uk/Tools/pfa/iprscan/
Likelihood Ratio Test, http://www.genetics.wustl.edu/jflab/
MutationAssessor, http://mutationassessor.org
MutationTaster, http://www.mutationtaster.org
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer3, http://primer3.wi.mit.edu/
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
SIFT, http://sift.jcvi.org/
SWISS-MODEL, http://swissmodel.expasy.org/
TopHat, http://tophat.cbcb.umd.edu/
UCSC Genome Browser, http://genome.ucsc.edu/
UniGene, http://www.ncbi.nlm.nih.gov/unigene/
UniProt, http://www.uniprot.org/
References
- 1.Lin F.R., Niparko J.K., Ferrucci L. Hearing loss prevalence in the United States. Arch. Intern. Med. 2011;171:1851–1852. doi: 10.1001/archinternmed.2011.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fellinger J., Holzinger D., Sattel H., Laucht M. Mental health and quality of life in deaf pupils. Eur. Child Adolesc. Psychiatry. 2008;17:414–423. doi: 10.1007/s00787-008-0683-y. [DOI] [PubMed] [Google Scholar]
- 3.Chia E.M., Wang J.J., Rochtchina E., Cumming R.R., Newall P., Mitchell P. Hearing impairment and health-related quality of life: the Blue Mountains Hearing Study. Ear Hear. 2007;28:187–195. doi: 10.1097/AUD.0b013e31803126b6. [DOI] [PubMed] [Google Scholar]
- 4.Yoshinaga-Itano C., Sedey A.L., Coulter D.K., Mehl A.L. Language of early- and later-identified children with hearing loss. Pediatrics. 1998;102:1161–1171. doi: 10.1542/peds.102.5.1161. [DOI] [PubMed] [Google Scholar]
- 5.Yaeger D., McCallum J., Lewis K., Soslow L., Shah U., Potsic W., Stolle C., Krantz I.D. Outcomes of clinical examination and genetic testing of 500 individuals with hearing loss evaluated through a genetics of hearing loss clinic. Am. J. Med. Genet. A. 2006;140:827–836. doi: 10.1002/ajmg.a.31179. [DOI] [PubMed] [Google Scholar]
- 6.Basit S., Lee K., Habib R., Chen L., Umm-e-Kalsoom, Santos-Cortez R.L., Azeem Z., Andrade P., Ansar M., Ahmad W., Leal S.M. DFNB89, a novel autosomal recessive nonsyndromic hearing impairment locus on chromosome 16q21-q23.2. Hum. Genet. 2011;129:379–385. doi: 10.1007/s00439-010-0934-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 12.Reva B., Antipin Y., Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chun S., Fay J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 17.Rozen S., Skaletsky H.J. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S., Misener S., editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press; New Jersey: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 18.McLaughlin H.M., Sakaguchi R., Liu C., Igarashi T., Pehlivan D., Chu K., Iyer R., Cruz P., Cherukuri P.F., Hansen N.F., NISC Comparative Sequencing Program Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet. 2010;87:560–566. doi: 10.1016/j.ajhg.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zdobnov E.M., Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 20.Arnold K., Bordoli L., Kopp J., Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 21.Guo M., Ignatov M., Musier-Forsyth K., Schimmel P., Yang X.L. Crystal structure of tetrameric form of human lysyl-tRNA synthetase: Implications for multisynthetase complex formation. Proc. Natl. Acad. Sci. USA. 2008;105:2331–2336. doi: 10.1073/pnas.0712072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murzin A.G. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 1993;12:861–867. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandrescu A.T., Jaravine V.A., Dames S.A., Lamour F.P. NMR hydrogen exchange of the OB-fold protein LysN as a function of denaturant: the most conserved elements of structure are the most stable to unfolding. J. Mol. Biol. 1999;289:1041–1054. doi: 10.1006/jmbi.1999.2813. [DOI] [PubMed] [Google Scholar]
- 24.Robinson J.C., Kerjan P., Mirande M. Macromolecular assemblage of aminoacyl-tRNA synthetases: quantitative analysis of protein-protein interactions and mechanism of complex assembly. J. Mol. Biol. 2000;304:983–994. doi: 10.1006/jmbi.2000.4242. [DOI] [PubMed] [Google Scholar]
- 25.Fang P., Zhang H.M., Shapiro R., Marshall A.G., Schimmel P., Yang X.L., Guo M. Structural context for mobilization of a human tRNA synthetase from its cytoplasmic complex. Proc. Natl. Acad. Sci. USA. 2011;108:8239–8244. doi: 10.1073/pnas.1100224108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDermott B.M., Jr., Baucom J.M., Hudspeth A.J. Analysis and functional evaluation of the hair-cell transcriptome. Proc. Natl. Acad. Sci. USA. 2007;104:11820–11825. doi: 10.1073/pnas.0704476104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorvaldsdóttir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goecks J., Nekrutenko A., Taylor J., Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blankenberg D., Von Kuster G., Coraor N., Ananda G., Lazarus R., Mangan M., Nekrutenko A., Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010;Chapter 19:19.10.1–19.10.21. doi: 10.1002/0471142727.mb1910s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giardine B., Riemer C., Hardison R.C., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alagramam K.N., Murcia C.L., Kwon H.Y., Pawlowski K.S., Wright C.G., Woychik R.P. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat. Genet. 2001;27:99–102. doi: 10.1038/83837. [DOI] [PubMed] [Google Scholar]
- 34.David A., Netzer N., Strader M.B., Das S.R., Chen C.Y., Gibbs J., Pierre P., Bennink J.R., Yewdell J.W. RNA binding targets aminoacyl-tRNA synthetases to translating ribosomes. J. Biol. Chem. 2011;286:20688–20700. doi: 10.1074/jbc.M110.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collin R.W., Kalay E., Tariq M., Peters T., van der Zwaag B., Venselaar H., Oostrik J., Lee K., Ahmed Z.M., Caylan R. Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am. J. Hum. Genet. 2008;82:125–138. doi: 10.1016/j.ajhg.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schultz J.M., Khan S.N., Ahmed Z.M., Riazuddin S., Waryah A.M., Chhatre D., Starost M.F., Ploplis B., Buckley S., Velásquez D. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am. J. Hum. Genet. 2009;85:25–39. doi: 10.1016/j.ajhg.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borck G., Ur Rehman A., Lee K., Pogoda H.M., Kakar N., von Ameln S., Grillet N., Hildebrand M.S., Ahmed Z.M., Nürnberg G. Loss-of-function mutations of ILDR1 cause autosomal-recessive hearing impairment DFNB42. Am. J. Hum. Genet. 2011;88:127–137. doi: 10.1016/j.ajhg.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmed Z.M., Yousaf R., Lee B.C., Khan S.N., Lee S., Lee K., Husnain T., Rehman A.U., Bonneux S., Ansar M. Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am. J. Hum. Genet. 2011;88:19–29. doi: 10.1016/j.ajhg.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierce S.B., Gersak K., Michaelson-Cohen R., Walsh T., Lee M.K., Malach D., Klevit R.E., King M.C., Levy-Lahad E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 2013;92:614–620. doi: 10.1016/j.ajhg.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puffenberger E.G., Jinks R.N., Sougnez C., Cibulskis K., Willert R.A., Achilly N.P., Cassidy R.P., Fiorentini C.J., Heiken K.F., Lawrence J.J. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McLaughlin H.M., Sakaguchi R., Giblin W., Wilson T.E., Biesecker L., Lupski J.R., Talbot K., Vance J.M., Züchner S., Lee Y.C., NISC Comparative Sequencing Program A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N) Hum. Mutat. 2012;33:244–253. doi: 10.1002/humu.21635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glamuzina E., Brown R., Hogarth K., Saunders D., Russell-Eggitt I., Pitt M., de Sousa C., Rahman S., Brown G., Grunewald S. Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2. J. Inherit. Metab. Dis. 2012;35:459–467. doi: 10.1007/s10545-011-9413-6. [DOI] [PubMed] [Google Scholar]
- 44.Banerjee R., Reynolds N.M., Yadavalli S.S., Rice C., Roy H., Banerjee P., Alexander R.W., Ibba M. Mitochondrial aminoacyl-tRNA synthetase single-nucleotide polymorphisms that lead to defects in refolding but not aminoacylation. J. Mol. Biol. 2011;410:280–293. doi: 10.1016/j.jmb.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Racine F.M., Steinberg W. Defects of two temperature-sensitive lysyl-transfer ribonucleic acid synthetase mutants of Bacillus subtilis. J. Bacteriol. 1974;120:372–383. doi: 10.1128/jb.120.1.372-383.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukushima K., Motomura S., Kuraoka A., Nakano H., Nishimoto T. A single point mutation of hamster aminoacyl-tRNA synthetase causes apoptosis by deprivation of cognate amino acid residue. Genes Cells. 1996;1:1087–1099. doi: 10.1046/j.1365-2443.1996.d01-225.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.