

Abstract
α-Tropomyosin (Tm) carrying hypertrophic cardiomyopathy mutation D175N or E180G was expressed in Escherichia coli. We have assembled dimers of two polypeptide chains in vitro that carry one (αα*) or two (α*α*) copies of the mutation. We found that the presence of the mutation has little effect on dimer assembly, thereby predicting that individuals heterozygous for the Tm mutations are likely to express both αα* and α*α* Tm. Depending on the expression level, the heterodimer may be the predominant form in individuals carrying the mutation. Thus, it is important to define differences in the properties of Tm molecules carrying one or two copies of the mutation. We examined the Tm homo- and heterodimer properties: actin affinity, thermal stability, calcium regulation of myosin subfragment 1 binding, and calcium regulation of myofibril force. We report that the properties of the heterodimer may be similar to those of the wild-type homodimer (actin affinity, thermal stability, D175N αα*), similar to those of the mutant homodimer (calcium sensitivity, D175N αα*), intermediate between the two (actin affinity, E180G αα*), or different from both (thermal stability, E180G αα*). Thus, the properties of the homodimer are not a completely reliable guide to the properties of the heterodimer.
Tropomyosin is an α-helical, coiled-coil protein that, with troponin, is involved in the calcium-dependent regulation of actomyosin activity.1 Each Tm dimer lies along the surface of an actin filament and associates in a head-to-tail manner into a linear polymer in which the C-terminal section of a Tm dimer associates with the N-terminus of the next Tm dimer. The overlap between adjacent Tms is relatively short, 11 amino acids,2 compared to the overall length of the molecule (284 amino acids per monomer). In the off or B-state, each Tm covers the myosin binding site on seven actin monomers. Calcium binding to troponin releases TnI from its binding site on actin, and the Tm relaxes back to its preferred position on the actin surface (C-state) where most of the myosin binding site is now exposed. Strong binding of myosin to actin induces the Tm to move even farther from the B-state to the M-state.3,4 The movement of Tm over seven actins and the associated communication with the adjacent Tms can result in significant cooperativity in the binding of calcium to a single Tn or the binding of a single myosin to actin.5
The human cardiac muscle tissue contains α- and βTm isoforms expressed as a mixture of αα homodimers and αβ heterodimers. To date, 11 missense mutations in αTm have been associated with hypertrophic cardiomyopathy (HCM).6 HCM is a pathophysiologic condition associated with an impaired myocardial function resulting from enlargement or hypertrophy of myocytes and consequent remodeling of the heart. This can in some cases lead to sudden cardiac death.7 HCM is primarily a genetic disorder inherited in an autosomal dominant manner with an estimated incidence of one in 500 individuals. A plethora of disease-causing mutations has been identified in the sarcomeric proteins of the heart,8 including the protein in which we are interested, α-tropomyosin (Tm).
Missense mutations in Tm associated with HCM have so far only been reported in the αTm isoform. Therefore αα, αα*, and α*α* as well as αβ and α*β Tm dimers (where an asterisk represents a point mutation) are expected in affected individuals with a heterozygous background. Two of these mutations, D175N and E180G, have been intensively studied9−19 to characterize the biochemical and biophysical properties of the protein and subsequently to understand the consequences of these mutations for cardiac function. All of these in vitro studies of Tm carrying cardiomyopathy mutations to date were conducted exclusively with homodimers because of the lack of a reliable method for the assembly and purification of Tm heterodimers. The reported results are not always consistent with those found from in vivo models of the disease (human slow muscle fibres,20 transgenic mouse models,21,22 and adenovirus-mediated infected cardiomyocytes23). In these in vivo models, Tm heterodimers may be the predominant forms of Tm.
The aim of this study is then the identification of the biophysical, biochemical, and functional consequences of Tm HCM mutations in homo- or heterodimeric form, based on the recently described method for the formation in vitro and the subsequent isolation of Tm αβ heterodimers.24
We report here the optimization of this method for the assembly and purification of human cardiac αTm heterodimers carrying HCM mutation D175N or E180G in one of the two α chains. We go on to investigate the in vitro biochemical and biophysical properties of these mutants at the molecular level. Our results demonstrate that both D175N- and E180G-Tm readily form αα* heterodimers and these assemble on actin with small changes in the apparent affinity for actin. We also measure the thermal stability of the heterodimers compared to that of the wild type (WT) and the homodimers carrying the mutation in both chains. In vitro, the calcium activation of actin filaments assembled with mutant Tms showed small changes to a lower calcium level required for activation (0.08–0.14 pCa unit). The largest change in calcium sensitivity was seen with E180G homodimers, and this result was confirmed using rabbit skeletal myofibrils in which the TmTn complex was extracted and replaced with human cardiac TmTn with or without the mutation in Tm.
All of the mutations produced relatively small effects, but noticeably, the effect of the heterodimer was not always intermediate between those of the WT and mutant homodimers. The study of homodimers is not therefore necessarily a good predictor of the properties of the heterodimer.
Materials and Methods
Protein Preparations
Native rabbit skeletal actin and myosin S1 were prepared by the methods of Spudich and Watt25 and Margossian and Lowey,26 respectively. F-Actin used in the kinetic experiments was labeled at Cys374 with pyrene iodoacetamide27 and stabilized by overnight incubation at 10 μM with a 1:1 ratio of phalloidin.
The rat α isoforms of Tm (WT-Tm,
D175N-Tm, and E180G-Tm) were expressed in Escherichia coli and had the Ala-Ser N-terminal extension shown to mimic the N-terminal
acetylation of the native αTm.28,29 WT-Tm and
its HCM mutants were cloned as described previously.15 Note that the rat αTm protein sequence differs from
the human sequence at a single site, R220 in human versus K220 in
rat (UniProt entries P09493 and P04692 for human and rat forms, respectively).
The formation and isolation of defined Tm heterodimers required the
production of WT-Tm with a His6 affinity tag followed by
the Factor Xa protease recognition site and Ala-Ser N-terminal extension.
His-Tm was cloned as described in ref (24) with the following N-terminal sequence.
All the generated Tm clones were transformed into BL-21 E. coli and expressed and harvested as previously described.30 All nontagged forms of Tm with the Ala-Ser N-terminal extension were purified by ion exchange chromatography using HiTrap Q HP columns (GE Healthcare). The His-Tm (αα wild type) purification was conducted via a Ni-NTA column matrix (QIAGEN Ltd.). Both purification methods are described in detail by Kalyva et al.24
Recombinant human cardiac Tn subunits hcTnC (UniProt entry P63316), hcTnI (UniProt entry P19429), and hcTnT (UniProt entry P45379-6) were overexpressed in BL-21 E. coli and reconstituted and purified as described by Al-Sarayreh.31
Thin filaments used in the kinetic binding assays were assembled by incubating F-actin, cTn, and Tm in a 2.5:1:1 ratio for 1 h. The calcium sensitivity of thin filaments was tested by the stopped flow. The experimental procedure is described in detail in Transient Kinetics.
Formation and Purification of WT-D175N- and WT-E180G-Tm Heterodimers
The formation of defined αα* Tm heterodimers was performed as previously described by Kalyva et al.24 αα* Tm heterodimers were assembled by mixing WT His-Tm with a nontagged homodimer of the Tm carrying a point HCM mutation (α*α* Tm) in a ratio 1:4. The high ratio of untagged to tagged Tm resulted in a low level of doubly tagged dimers simplifying the purification. The sample was heated to 58 °C in the presence of DTT to generate a mixture of Tm monomers and then cooled to 37 °C for 1 h, and the monomers were allowed to reanneal into dimers. Tm carrying zero, one, or two His tags was separated using a TALON Superflow metal affinity resin column (Clontech) connected to an FPLC system (ÄKTA). Samples were collected and analyzed on a 10% sodium dodecyl sulfate (SDS) gel (see Figure 1). For further details, see Results.
Figure 1.

Protein content of eluted fractions from the affinity purification of α His-tagged WT-D175N-Tm heterodimers. Proteins were eluted by linear gradient from 0 to 250 mM imidazole: lane 1, molecular mass markers; lane 2, mixture of His-Tm dimers and monomers; lane 3, D175N-Tm dimers; lanes 4–12, His-tagged WT-D175N-Tm heterodimers; lanes 13 and 14, mixture of both His-tagged WT-D175N–Tm heterodimers and His-Tm homodimers. Samples were run on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel under nonreducing conditions.
Thermal Unfolding of Recombinant Tropomyosins Using Circular Dichroism
CD spectra and unfolding isotherms were collected on a Jasco 715 spectropolarimeter (software, spectra management version 1.51.00) in a stoppered 1 mm cuvette (Starna Scientific Ltd.) as described by Kalyva et al.24 All measurements were performed in 20 mM KPi (pH 7.0), 0.5 M KCl, and 5 mM MgCl2. Thermal unfolding was recorded at a fixed wavelength of 222 nm over a temperature range from 5 to 65 °C using a Peltier device (Jasco PTC 423S/15) and a temperature increasing at a rate of 1 °C/min. The final concentration of all Tm samples was 7 μM. The reversibility of the unfolding–refolding process was assessed by reheating the Tm sample directly after it had been cooled from the previous temperature scan. The temperature scans were repeated three times without and three to four times with 1 mM DTT added.
The CD data from thermal unfolding experiments were analyzed using MicroCal Origin version 8.6. The isotherms were smoothed (Savitzky–Golay method, 50-point window), differentiated, and then fit to multiple Gaussian peaks.
Cosedimentation and Densitometry
Cosedimentation assays were performed as previously described by Coulton et al.30 F-Actin at 7 μM was mixed with increasing concentrations of Tm (0.2–2.4 μM) at 20 °C in 20 mM MOPS (pH 7.0), 100 mM KCl, and 5 mM MgCl2 to a final volume of 100 μL. The samples were incubated for 1 h. The Tm bound to F-actin was pelleted by centrifugation at 370000g for 20 min (Beckman TL100A). Equivalent samples of the pellet and supernatant were then assessed via 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). Densitometry analysis was conducted by using an Epson Perfection V750 Pro scanner and ScionImage (Scion Corp., Frederick, MD). The free Tm concentration was plotted against the fractional actin saturation (θ), and the Hill equation [θ = [Tm]h/(K50%h + [Tm]h)] was used for determination of the Hill coefficient (h) and the midpoint of the fitted curve (K50%) (Table 1).
Table 1. Affinities of Tm for Actina.
| Tm dimer | K50% (μM) | h |
|---|---|---|
| WT-Tm | 0.20 ± 0.02 | 2.5 ± 0.8 |
| WT-D175N-Tm | 0.23 ± 0.05b | 2.5 ± 0.5 |
| D175N-Tm | 0.52 ± 0.15c | 2.8 ± 0.1 |
| WT-E180G-Tm | 0.27 ± 0.03d | 2.1 ± 0.5 |
| E180G-Tm | 0.42 ± 0.12d,e | 2.2 ± 0.4 |
The actin binding affinity (K50%) and the Hill coefficient (h), both derived for the fit of the Hill equation to data of panels B and C of Figure 5. The data represent an average of at least four measurements (n ≥ 4) with given standard deviations.
Different from that of the D175N heterodimer (p ≤ 0.01).
Different from that of the WT (p ≤ 0.01).
Different from that of the WT (p ≤ 0.05).
Different from that of the E180G heterodimer (p ≤ 0.05).
Transient Kinetics
The thin filaments were reconstituted by mixing 5 μM phalloidin-stablilized pyrene actin (ppA) with 2 μM Tm and 2 μM hcTn in 20 mM MOPS (pH 7.0), 100 mM KCl, 5 mM MgCl2, and 1 mM DTT. All the proteins were incubated for 1 h at 4 °C to ensure the full saturation of the actin filament and then diluted as required. All concentrations refer to those after mixing in a 1:1 ratio in the stopped flow.
The change in the pyrene actin fluorescence was monitored by the stopped-flow fluorimeter (SF-61DX2 spectrophotometer, HiTech Scientific) at 20 °C. Pyrene was excited at 365 nm, and the fluorescence emission was then detected through a KV 399 nm cutoff filter. All the measured data were analyzed with the Kinetassist software provided with the instrument. Each transient was the average of at least five shots.
Initially, the quality of the thin filament proteins was assayed by mixing 0.25 μM S1 in the absence of calcium with a 10-fold excess of actin (2.5 μM) and 1 μM WT-Tm and increasing concentrations of hcTn. The observed reaction was a single-exponential reaction, and the observed rate constant (kobs) decreased as the hcTn concentration increased until saturation, typically at 2.5 μM hcTn with a kobs of 2.0 s–1. The hcTn required for saturation was used in all subsequent measurements with the same batch of hcTn. Repeating the reaction with a saturating level of Tn in the presence of calcium typically increased kobs 3.5-fold to 7.0 s–1, close to the value in the absence of Tn (8.0 s–1).11,32 The ratio of the kobs value in the presence (2 mM Ca-EGTA) and absence of calcium (2 mM EGTA) was used to calculate the equilibrium constant KB, the equilibrium between the blocked and closed states of the thin filament as defined by the three-state model of thin filament regulation. The rate constant for binding of S1 to an excess of actin sites is defined as kobs = [A]k × KB/(1 + KB), where [A]k is the pseudo-first-order rate constant for binding of S1 to actin and KB/(1 + KB) is the fraction of actin sites in the on state. In the presence of Ca2+, all the sites are available and kobs = [A]k. KB was therefore calculated from the ratio of the observed rate constants in the presence and absence of calcium: kobs+Ca/kobs–Ca = (1 + KB)/KB.3
For experiments in which [S1] ≫ [actin], thin filament proteins were diluted to final concentrations of 0.25 μM actin, 0.1 μM Tm, and 0.1 μM hcTn and mixed with 2.5 μM S1.11 The experimental conditions were as described above with the addition of 2 mM EGTA and 2 mM Ca-EGTA to give defined Ca2+ concentrations from pCa 9.8 to 4.6. The volume ratios of EGTA and Ca-EGTA were calculated with WEBMAXCLITE version 1.15.
Myofibril Experiments: Preparation, Endogenous Regulatory Protein Replacement, and Force Recording
Myofibrils were prepared by homogenization of glycerinated rabbit psoas muscles, as described previously.33 All solutions were kept around 0 °C and contained a cocktail of protease inhibitors, including leupeptin (10 μM), pepstatin (5 μM), phenylmethanesulfonyl fluoride (200 μM), E64 (10 μM), NaN3 (500 μM), and DTT (0.5 mM).
Endogenous Tm and Tn were replaced in rabbit skeletal muscle myofibrils with hcTn and αTm (WT; WT-E180G-Tm and E180G-Tm) as previously described.31,34 The extent of the Tm–Tn extraction and replacement was 98% complete as assessed by 12% SDS–PAGE analysis.34
For force recording,35 a small volume of the myofibril suspension was transferred to a chamber (15 °C) filled with a relaxing solution (pCa 9.0) on an inverted microscope and mounted horizontally between two glass microtools. One tool was connected to a length control motor that could produce rapid (<1 ms) length changes. The second tool was a calibrated cantilevered force probe (2–6 nm/nN; frequency response of 2–5 kHz). Myofibrils were activated and relaxed by rapid translation of two continuous streams of relaxing and activating solutions with different pCa values (9 and 4.5) flowing from a double-barrelled glass pipet placed within 0.5–1 mm of the preparation. The maximal force (P0), the rate of force development (kACT), and the rate of force redevelopment following release–restretch protocols (kTR)36 were measured at submaximal and saturating pCa levels.
Activating and relaxing solutions, calculated as previously described,37 were at pH 7 and contained 10 mM total EGTA (Ca-EGTA:EGTA ratio set to obtain the different pCa used), 5 mM MgATP, 1 mM free Mg2+, creatine phosphate (CP, 10 mM), 10 mM MOPS, propionate, and sulfate to adjust the final solution to an ionic strength of 200 mM and monovalent cation concentration of 155 mM. The concentration of contaminant inorganic phosphate (Pi) from spontaneous breakdown of MgATP and CP was ∼500 μM.
Results
Formation and Purification of Tm Heterodimers
The method of preparing heterodimers is essentially that of Kalyva et al.,24 as outlined in Materials and Methods. Briefly, His-Tm (αα) was mixed with the Tm carrying a point HCM mutation (α*α*) in a ratio of 1:4. The mixture was heated at 58 °C for 10 min in the presence of 20 mM DTT and then cooled to 37 °C for 45 min to allow dimer formation. DTT was removed via gel filtration (Econo-Pac 10DG). The Tm dimers were catalytically cross-linked at Cys190 by incubation in 10 mM K3Fe(CN)6, 2 μM CuSO4, 2 M NaCl, 10 mM MOPS (pH 7) buffer at 35 °C overnight.38 The cross-link was formed to trap Tm dimers for purification. The sample was then dialyzed in 300 mM KCl, 20 mM KPi, 2 mM MgCl2 (pH 7) column buffer.
The mixture of dimers was purified by affinity chromatography using a 25 mL TALON Superflow metal affinity resin column. The mixture of Tm dimers applied to the column was washed with 100 mL of wash buffer. The flow-through contained only nontagged Tm. The His-tagged dimers were eluted with a linear gradient from 0 to 250 mM imidazole. The high concentration of imidazole prevents the monitoring of Tm dimer elution by the absorbance at 280 nm. The protein content of eluted fractions was therefore monitored by running a 10% SDS–PAGE gel under nonreducing conditions as shown in Figure 1. Lanes 2 and 3 show His-WT and D175N-Tm homodimers as controls, respectively. Heterodimers with only one His tag were eluted first (lanes 4–12), followed by a mixture of singly and doubly tagged Tm dimers at higher imidazole concentrations, and both migrated with apparent molecular masses close to 85 kDa. Lanes 4–14 show also a minor fraction of heterodimers that were not cross-linked (two bands of tagged and nontagged Tm monomers with similar intensity at ∼30 and 25 kDa, respectively). The elution profile of the WT-E180G-Tm heterodimer is shown in Figure S1 of the Supporting Information.
The fractions containing Tm heterodimers were pooled together, and the purity was checked via 4–12% SDS–PAGE (Figure 2). The heterodimers were concentrated by precipitation at pH 4.6 and collected by centrifugation at 4200 rpm for 12 min. Pelleted protein was dissolved in FXa protease reaction buffer [20 mM Tris-HCl, 50 mM NaCl, and 1 mM CaCl2 (pH 6.5)], and the concentration was measured. The His tag was proteolytically cleaved from heterodimers with FXa protease digestion. Figure 2 shows the isolation and purity of the WT-D175N-Tm heterodimer before and after His tag digestion. Lanes 2 and 3 show His-Tm and D175N-Tm homodimer controls under nonreducing conditions, respectively. The His-Tm dimer does not form a cross-link as readily as nontagged Tm and is shown as a mixture of monomers and dimers that migrated with apparent molecular masses of 39 and 85 kDa, respectively. Lane 4 shows the expected 1:1 ratio of tagged and nontagged Tm arising from the heterodimer under reducing conditions [β-mercaptoethanol (BME) was used to reduce disulfide bonds]. The same sample under nonreducing conditions (Figure 2, lane 5) shows a single dimer band as evidence of the pure His-tagged WT-D175N-Tm heterodimer with an apparent molecular mass of 83 kDa. The final WT-D175N-Tm heterodimer after His tag digestion (Figure 2, lane 6) showed a single band under reducing conditions as WT and D175N-Tm run together under these conditions. The purity of heterodimers was measured by gel densitometry (described in Cosedimentation and Densitometry). Two bands of the purified His-tagged WT-D175N-Tm heterodimer under reducing conditions (Figure 2, lane 4) showed a 4% difference in density, which indicates a 96% purity of heterodimers. Additionally, the intensities of the WT and D175N-Tm bands from cosedimentation assays (Figure 4A) were compared, and a 5% difference in the density between two Tm bands was measured (95% heterodimers). The ratio of α and α* monomers was checked routinely for both tagged and nontagged heterodimers. The isolation and purity of the WT-E180G-Tm heterodimer is shown in Figure S2 of the Supporting Information.
Figure 2.

Isolation and purity check of Tm heterodimers: lane 1, molecular mass markers; lane 2, mixture of His-Tm dimers and monomers; lane 3, D175N-Tm dimers; lanes 4 and 5, assembled and purified His-tagged WT-D175N-Tm heterodimer under reducing conditions and cross-linked, respectively; lane 6, WT-D175N-Tm heterodimer after removal of the His tag under reducing conditions. Samples were run on a 4–12% SDS–PAGE gel.
Figure 4.

Affinity of Tm for actin determined by cosedimentation analysis. Actin (7 μM) incubated with 0.2–2.4 μM Tm dimers at 20 °C for 1 h and then centrifuged at 370000g. (A) SDS gels (10%) were used for analysis of actin and Tm in the pellet and supernatant. (B and C) Plots of the fractional saturation of actin by Tm as a function of free Tm concentration for (B) WT-Tm (■), WT-D175N-Tm (○), and D175N-Tm (▲) and (C) WT-Tm (■), WT-E180G-Tm (○), and E180G-Tm (▲). The best fit to the Hill equation is superimposed on the data. The actin affinities (K50%) and the Hill coefficients (h) are listed in Table 2. Buffer conditions: 20 mM MOPS, 100 mM KCl, 5 mM MgCl2 (pH 7.0).
The yield of the heterodimer was 11 mg from a starting mixture of 23 mg of His-Tm and 92 mg of α*-Tm. The homodimer proteins can be recycled in further rounds of heterodimer assembly.
The molecular mass of the purified recombinant Tm proteins was determined by mass spectrometry (Bruker Daltonics micrOTOF-Q II). The measured molecular mass of the WT-Tm dimer was 65676.04 Da compared to the expected mass of 65675 Da. The masses of both heterodimers were within 0.9 Da of the expected masses. Compared to the WT dimer, the WT-E180G-Tm heterodimer had the predicted 72.1 Da decrease in mass, which was 144.3 Da for the homodimer. The D175N-Tm homodimer is predicted to have a mass 2 Da larger than that of the WT and 1 Da larger than that of the heterodimer. These masses were observed for D175N-Tm but are not very distinct between the WT and D175N dimers.
Thermal Stability Assays
Tm has the characteristic CD spectrum of a coiled-coil α-helical protein (see the inset of Figure 3A). The thermal stability of cross-linked or reduced Tm dimers was examined using CD at 222 nm. Figure 3 shows unfolding isotherms of homo- and heterodimers over the temperature range of 5–65 °C. Each sample was heated to 65 °C and then cooled a total of seven times: three times in the absence of 1 mM DTT (the thiol-cross-linked Tm dimers) and four times in the presence of 1 mM DTT. The unfolding profile measurements under nonreducing conditions were almost identical for every sample, indicating the reversibility of the unfolding reaction. The first measurement after addition of 1 mM DTT showed both a decrease in thermal stability and the oxidation of the cross-link. The three subsequent measurements were again identical.
Figure 3.

Thermal unfolding of αTm homo- and heterodimers carrying HCM mutations compared to WT-Tm. Normalized unfolding profiles of 7 μM homodimers (solid lines) and heterodimers (dashed lines) of D175N (A and B) and E180G (C and D) in the presence (B and D) or absence (A and C) of 1 mM DTT. Buffer conditions: 0.5 M KCl, 5 mM MgCl2, 20 mM potassium phosphate buffer (pH 7.0). The CD spectrum of 7 μM αD175N-Tm in the range of 195–250 nm is shown in the inset of panel A.
Figure 3A shows the unfolding profile for the third heating cycle of the cross-linked WT homodimer superimposed on the profile of the D175N homodimer and the WT-D175N-Tm heterodimer. All these almost identical profiles show a major unfolding event at ∼57 °C and a minor one at ∼37 °C. The first derivative of the curves resolves a third, smaller domain with a midpoint at ∼49 °C (see the Supporting Information).
After addition of DTT (Figure 3B) to reduce the Cys190 cross-link, the WT profile showed a reduction in the size of the event at 37 °C. The 57 °C event occurred at a lower temperature of ∼53 °C, and the intermediate event was unchanged at 49 °C. The D175N homodimer was more stable than WT by ∼1 °C, and the heterodimer was similar to the WT below 40 °C but closer to the D175N homodimer above 45 °C. The details of what is happening to the unfolding domains are shown more clearly in the first differential of the data (see the Supporting Information).
The unfolding profiles for the third heating cycle of cross-linked homo- and heterodimers of E180G-Tm are shown in Figure 3C. There is a significant loss of thermal stability at ∼30 °C that is proportional to the number of Tm mutations. The profile of the double E180G-Tm mutant exhibits its first thermal transition at ∼30 °C, that of the WT-E180G-Tm heterodimer at 35 °C, and that of the WT at 40 °C. Above 45 °C all three Tm constructs are identical. Reduction of the cross-link at Cys190 shows several changes. Compared to WT, both dimers are less stable over the range of 30–50 °C. The homodimer has a new transition at ∼42 °C, and both hetero- and homodimers are apparently more stable without the cross-link at 30 °C. Again the interpretation in terms of three folding domains is clear when looking at the differentiated data in the Supporting Information.
It is important to mention that both WT-D175N and WT-E180G-Tm heterodimers under reducing conditions will reanneal into a mixture of αα, αα*, and α*α* (the asterisk indicates a mutation) in a ratio of approximately 1:2:1. For the WT-D175N-Tm heterodimer, this has little consequence as all dimers have similar unfolding characteristics. For the WT-E180G-Tm heterodimer, this complicates the interpretation. By subtracting 25% of the expected transitions for WT and E180G homodimers (the expected proportion after reannealing), we can estimate the unfolding transitions for the heterodimer. The error on such a process is large, but it can indicate the underlying properties and is shown as a thick gray line in Figure S3F of the Supporting Information. This does not alter the positions of the three transitions but does alter their relative proportions where the area of all three transitions is reduced by 55%.
Actin Binding Assays
The affinities of WT-D175N-Tm and WT-E180G-Tm heterodimers for F-actin were measured using cosedimentation assays (see Materials and Methods). Figure 4A shows the SDS–PAGE analysis of the supernatant and pellets of centrifuged 7 μM actin with 0.2–2.4 μM WT-D175N-Tm heterodimer.
Pellets show that almost all of the actin sediments with a small fraction, probably G-actin, remaining in the supernatant. Note that the 10% gel used here under reducing conditions resolves the WT- and D175N-Tm monomers. The similarity of the density of the two bands is consistent with a pure heterodimer.
Figure 4B shows the sigmoid binding curves of WT- and D175N-Tm constructs with F-actin as expected for Tm polymerizing on the actin surface. Fitting the data to the Hill equation gives an estimate of the affinity (K50%). The apparent affinity (K50%) of the WT-D175N-Tm heterodimer for actin (0.23 μM) was tighter in comparison to that of the D175N-Tm homodimer (0.52 μM; p ≤ 0.05). K50% for WT-Tm was indistinguishable from that of the WT-D175N-Tm heterodimer, indicating that the introduction of the D175N mutation in only one Tm chain does not have any effect on binding to actin.
The binding curves of WT and E180G-Tm constructs with F-actin are shown in Figure 4C. The affinity of the E180G-Tm homodimer was significantly weaker (0.42 μM; p ≤ 0.05) that that of the WT (0.20 μM), with the heterodimer at an intermediate value (0.27 μM; p ≤ 0.05). The obtained K50% values for HCM Tm homodimers are consistent with previously published data.15 The Hill coefficients showed similar values (2.1–2.8) for all αTm measured.
Calcium Sensitivity of S1 Binding to a Thin Filament
The following stopped-flow experiments were used to evaluate the calcium-dependent occupancy of the blocked state of the thin filament.3 The thin filament was reconstituted with pyr-actin, human cardiac Tn, and αTm carrying an HCM mutation (D175N or E180G) in one or both chains of the Tm dimer. S1 (0.25 μM) was rapidly mixed with a 10-fold excess of the pyr-actin·Tm·Tn complex. Figure 5 shows the set of observed pyrene actin fluorescence transients with each Tm construct in the absence (pCa 8.9) and presence of calcium (pCa 4.6). Each transient is fit to a single exponential, and for WT-Tm, the kobs values were 7.68 and 2.21 s–1, respectively, giving a ratio of the two values of 3.5 (Table 2). These are in good agreement with previous measurements for cardiac Tn.32 The reduction in kobs at pCa 8.9 has been interpreted as being caused by a fraction of the unavailable actin sites and the ratio of rate constants kobs(−Ca2+)/kobs(+Ca2+) = KB/(1 + KB), where KB is the equilibrium constant between actin·Tm·Tn complexes in the blocked and closed states. For the WT, the ratio therefore defines KB = 0.4. The kobs and KB values for all Tm homo- and heterodimers in the presence and absence of calcium are listed in Table 3. The derived values of KB indicate that the value is independent of the mutations as shown previously for D175N- and E180G-Tm homodimers.11
Figure 5.

S1 binding to an excess of the pyr-actin·sk αTm·cTn complex in the presence and absence of calcium. Observed transients for excess actin (2.5 μM ppA, 1 μM hcTn, and 1 μM sk αTm) binding to 0.25 μM S1 with or without Ca2+. Thin filaments were reconstituted with each Tm dimer. Five transients are shown (one for each Tm) in the presence or absence of calcium. E180G-Tm (gray line) in the presence of Ca2+ showed a slight variation. The best fit single-exponential kobs values are listed in Table 3. Experimental conditions: as for Figure 4 with addition of 2 mM EGTA or 2 mM Ca-EGTA.
Table 2. Thin Filament Ca2+ Sensitivity for Thin Filaments Reconstituted with cTn and αTm Carrying HCM Mutationsa.
|
kobs (s–1) |
|||||
|---|---|---|---|---|---|
| Tm dimer | pCa50% | h | without Ca2+ | with Ca2+ | KB |
| WT-Tm | 5.89 ± 0.01 | 1.23 ± 0.03 | 2.21 ± 0.07 | 7.68 ± 0.18 | 0.40 |
| WT-D175N-Tm | 6.00 ± 0.06b | 1.20 ± 0.11 | 2.31 ± 0.01 | 8.00 ± 0.14 | 0.40 |
| D175N-Tm | 5.97 ± 0.05b | 1.24 ± 0.03 | 2.41 ± 0.06 | 8.48 ± 0.07 | 0.41 |
| WT-E180G-Tm | 5.92 ± 0.03 | 1.20 ± 0.09 | 2.31 ± 0.04 | 7.58 ± 0.14 | 0.47 |
| E180G-Tm | 6.03 ± 0.06b,c | 1.24 ± 0.12 | 2.71 ± 0.07 | 8.46 ± 0.16 | 0.44 |
The pCa50% value is the apparent calcium affinity, and h is the Hill coefficient for calcium switching of the thin filaments, both derived from the fit of the Hill equation to the data of panels A and B of Figure 6. All pCa50% and h values are averages of three measurements with two combined independent Tm and cTn samples. The kobs values are the observed exponential rate constants of S1 binding to excess thin filaments (2.5 μM ppA, 1 μM hcTn, and 1 μM αTm) in the presence and absence of calcium. kobs values were derived from observed transients shown in Figure 5. The kobs rate constants given are averages of two measurements with two independent Tm samples. KB represents the equilibrium between blocked and closed states of the thin filament. KB was calculated by using the equation kobs(−Ca2+)/kobs(+Ca2+) = KB/(1 + KB).
Different from the WT value (p ≤ 0.05).
Different from the E180G heterodimer value (p ≤ 0.05).
Table 3. Mechanical Behavior of Skeletal Myofibrils Replaced with WT-Tm and α-Tm Homo- and Heterodimers with the E180G Mutation at 15 °Ca.
| myofibril batch | sarcomere length (μm) | Po 4.5 (mN mm–2) | kACT (s–1) | kTR (s–1) | Po 5.80/Po 4.50 | Po 5.85/Po 4.50 | Po 5.90/Po 4.50 |
|---|---|---|---|---|---|---|---|
| E180G-Tm Homodimer | |||||||
| mutant Tm | 2.24 ± 0.04 (8) | 280 ± 33 (9) | 5.00 ± 0.26 (9) | 4.60 ± 0.53 (9) | 0.50 ± 0.03 (8)b | 0.10 ± 0.03 (7) | |
| control WT-Tm | 2.29 ± 0.04 (8) | 286 ± 25 (9) | 4.81 ± 0.70 (7) | 4.46 ± 0.35 (8) | 0.30 ± 0.04 (7) | 0.04 ± 0.02 (6) | |
| WT-E180G-Tm Heterodimer | |||||||
| mutant Tm | 2.41 ± 0.04 (10) | 282 ± 32 (10) | 5.82 ± 0.42 (8) | 5.82 ± 0.32 (10) | 0.14 ± 0.01 (8) | ||
| control WT-Tm | 2.42 ± 0.06 (8) | 321 ± 32 (9) | 5.73 ± 0.55 (9) | 5.72 ± 0.43 (6) | 0.13 ± 0.01 (6) | ||
All values are given as means ± the standard error of the mean and refer to different myofibril batches; the number in parentheses is the number of myofibrils. Po is the steady isometric tension, and the index refers to pCa (4.5 for maximal activation and 5.8, 5.85, and 5.9 for submaximal activation). kACT is the rate constant of the maximally activated tension rise following a stepwise pCa decrease (9.0 → 4.5) by fast solution switching. kTR is the rate constant of tension redevelopment following release and restretch of maximally activated myofibrils. Ratios of submaximal to maximal activated tension were obtained from jump experiments.
p ≤ 0.001 (Student's t test) vs the same parameter measured in the control preparations.
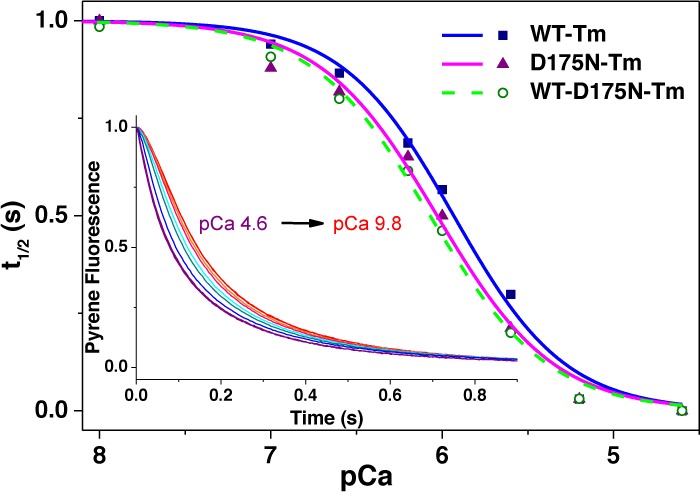
The calcium sensitivity of the thin filaments containing the various Tm dimers was measured as described previously.11,39 The experiment was conducted using a 10-fold excess of S1 binding to thin filaments because of the limited yields of αα* Tm heterodimers. In this assay, 2.5 μM S1 was rapidly mixed with preincubated 0.25 μM pyr-actin, 0.1 μM cTn, and 0.1 μM αTm over a range of calcium concentrations. The inset of Figure 6A shows the set of observed sigmoid transients at different calcium concentrations (pCa from 4.6 to 9.8) with thin filaments containing WT-D175N-Tm. Measurements for each Tm were repeated three times (two independent samples of Tm were combined with two independent samples of hcTn). Plots of the half-time of the reaction (t1/2) against pCa for WT-D175N and WT-E180G-Tm heterodimers are shown in panels A and B of Figure 6, respectively, with the best fit to the Hill equation superimposed. The panels show the data for the WT and double mutants for comparison. The midpoints (pCa50%) of the calcium-induced change in t1/2 and Hill coefficients derived from the fit to the Hill equation are listed in Table 2.
Figure 6.

Dependence of the observed transient of S1 binding to the pyr-actin·Tm·cTn complex on calcium concentration. (A) Transients observed for the preincubated pyr-actin·αTm·cTn complex (0.25 μM ppA, 0.1 μM WT-D175N-Tm, and 0.1 μM hcTn) rapidly mixed with 2.5 μM S1 at various Ca2+ concentrations. The fractional half-time (t1/2) is plotted vs pCa concentration. Data for WT-Tm (■), WT-D175N-Tm (○), and D175N-Tm (▲) are shown in panel B and data for WT-Tm (■), WT-E180G-Tm (○), and E180G-Tm (▲)in panel C. The best fit to the Hill equation is superimposed with midpoints (pCa50%) and Hill coefficients (h) listed in Table 3. (D) Comparison of measured pCa midpoints for the five Tm constructs, including the standard deviation. Each column represents an average of three measurements. Experimental conditions: with addition of 2 mM pCa buffers at various Ca2+ concentrations.
The pCa50% measurements exhibit increased calcium sensitivity of D175N- and E180G-Tm homodimers by 0.086 and 0.143 pCa unit, respectively. WT-D175N- and WT-E180G-Tm heterodimer pCa values are 0.116 and 0.03 pCa unit higher, respectively, than that of the WT. The differences in pCa50% are plotted in a bar graph (Figure 6C). The Hill coefficient did not show any significant changes in the HCM mutants in comparison to that of the WT.
Functional Assay in Myofibrils Replaced for the Endogenous Tm·Tn Complex
The following mechanical measurements in myofibrils were taken to test the functional impact of human cardiac αTm carrying an HCM mutation in one (heterodimer) or both chains (homodimer) when present in the sarcomere. For this reason, we extracted endogenous Tm and Tn and replaced them in rabbit skeletal muscle myofibrils31,34 with hcTn and αTm (WT, WT-E180G-Tm, and E180G-Tm). Only the E180G mutation has been used in these experiments, based on in vitro findings showing the largest effect in increasing the sensitivity of calcium activation of actin filaments.
As shown in Table 3, the presence of the E180G mutation in both heterodimeric and homodimeric forms did not significantly affect maximally activated force development (Po 4.5) or the kinetics of its development (kACT and kTR). Resting sarcomere lengths were in the optimal overlap range of rabbit psoas muscle, and differences in the two sets of experiments reported in Table 3 were caused by different myofibril batches. Interestingly, a clear effect was present with the E180G-Tm homodimer when tension development at submaximal calcium levels (pCa 5.8–5.9) was compared to the full activation in Ca2+ concentration-jump experiments. As shown in Figure 7 and Table 3, myofibrils replaced with the E180G-Tm heterodimer did not significantly differ from control WT-Tm, while the presence of the E180G-Tm homodimer significantly enhanced (see Table 3) tension development (i.e., thin filament activation) at low Ca2+ concentrations. The differential effect of αTm homodimers versus heterodimers on submaximal calcium-activated force seems clear even though for αTm heterodimers the comparison was made at only one submaximal pCa that was eliciting a relatively small amount of tension (Table 3). On the other hand, calcium-independent tension [pCa 9 (see Figure 7)], which signals the presence of some myosin heads that can cycle in the absence of Ca2+, was not significantly affected by the E180G mutation in the heterodimeric or homodimeric form.
Figure 7.

Force response of skeletal myofibrils replaced for the endogenous Tm·Tn complex at submaximal and maximal levels of calcium activation at 15 °C. Myofibrils replaced with the αTm heterodimer (A) and homodimer (B) with the E180G mutation (right panels) or with WT-Tm (left panels) were activated at submaximal Ca2+ concentrations (pCa 5.85 in panel A and pCa 5.8 in panel B) and then subjected to a Ca2+ concentration jump to full activation (pCa 4.5). Bars above traces correspond to the timing of the solution change. Top traces show the force; bottom traces show the release and restretch (30%) of myofibril length to measure the exponential redevelopment of force (kTR). Calibrations are indicated by the horizontal and vertical bars corresponding to 1 s and 100 mN mm–2. (A) Left: sarcomere length of 2.47 μm, resting tension of 102 mN mm–2, Po 5.85/Po 4.50 = 0.11. Right: sarcomere length of 2.45 μm, resting tension of 67 mN mm–2, Po 5.85/Po 4.50 = 0.18. (B) Left: sarcomere length of 2.52 μm, resting tension of 96 mN mm–2, Po 5.80/Po 4.50 = 0.31. Right: sarcomere length of 2.36 μm, resting tension of 87 mN mm–2, Po 5.80/Po 4.50 = 0.61.
Discussion
The results of our study show a method for the in vitro formation of defined, cardiac αTm heterodimers, using bacterially expressed αTm homodimers, and is optimized for αTm heterodimers with HCM mutations D175N and E180G. The Tm HCM mutants do not show any preference for homodimer over heterodimer formation as using a combination of 50% α*Tm with 50% His-αTm produced a mixture of dimers close to that predicted for random association with a ratio of 1:2:1 (αα:αα*:α*α*). The ratio of formed dimers shows that the introduction of the His tag into the Tm molecule does not affect dimerization. This suggests that in the heterozygous cell, if both WT and mutant proteins are expressed in similar amounts then the heterodimer will form a large fraction of the assembled dimer. Studies of the expression of Tm mutations in cultured cardiac myocytes23 or in human skeletal muscle biopsies20 show expression levels of 40–50% of the mutant Tm. Thus, assembly of a significant proportion of heterodimers is likely, but to date, this has not been investigated in cell cultures, biopsies, or transgenic animals. It ought to be possible to detect heterodimers in tissue samples if the Tm can be cross-linked before isolation and analysis. However, note that the difference between the WT and mutant may be too small to detect by SDS–PAGE or mass spectrometry. Whether such Tm dimers assemble into the thin filament or alter the calcium activation of contraction will depend on how the heterodimers interact with the other thin filament components.
Because the mutations do not affect dimer formation, we considered what other role the side chains of D175N and E180G in the homo- or heterodimer might play in the overall function. These surface charge residues are in a position to alter the stability of the Tm–Tm contacts16 or alter the interaction with actin (in position B, C, or M on the actin surface)40 or with troponin and hence alter calcium sensitivity.
Actin binding assays with D175N- and E180G-Tm homodimers showed a weaker affinity of approximately 2.5-fold in comparison to that of the WT (K50% = 0.21 μM), which agrees with previous measurements.11 The WT-D175N-Tm heterodimer had an actin affinity (0.23 μM) that was not distinguishable from that of the WT, while the value of the WT-E180G-Tm heterodimer (0.27 μM) was intermediate between those of the E180G homodimer and WT. However, the physiological significance in a muscle fiber is likely negligible because of much higher concentrations of actin and Tm. These values show that all Tm dimers bind actin with a tight apparent affinity, and under physiological conditions, all will bind to actin. If all of the protein were present in the same cell, there would be a very marginal preference for the WT over the mutant proteins.
The thermal unfolding measurements of the reduced WT and D175N-Tm homodimer show two major thermal transitions at 47.5 °C (N-terminal region) and 53.1 °C (C-terminal region) and a third at 41.2 °C. These observations agree with previously measured data for WT-αTm at high salt concentrations (0.5–1 M).24,41 A high salt concentration was used to prevent end-to-end polymerization of Tm that can confuse the melting isotherm. Similar studies of Tm thermal unfolding at a low salt concentration (100 mM KCl) and an ∼3-fold higher protein concentration using differential scanning calorimetry (DSC)15 reported the thermal transition of C-terminal region Tm at 50 °C and the N-terminal region Tm at 42.7 °C. These differences are probably caused by differences in the ionic strengths of the buffers. When the Tm is cross-linked at Cys190, the most thermally stable domains are stabilized with some loss of stability of the least stable domain.
The thermal unfolding of the D175N-Tm homodimer, both reduced and cross-linked, was indistinguishable from that of the WT as previously reported,15 and unsurprisingly, the D175N heterodimer was therefore also very similar to the WT.
The E180G-Tm homodimer shows a decreased thermal stability compared to that of the WT in agreement with the work of Kremneva et al.15 For the cross-linked homodimer, the least stable domain unfolds at ∼26 °C, well below physiological temperature, while the other two domains are similar to those of WT. When reduced, the most stable domain is similar to the WT while the domain with midrange stability is less stable by 5 °C at (42 °C). In contrast, the least stable domain is only a little less stable than the WT at 39 °C. This temperature is close to body temperature, which led Kremneva et al. to speculate that this domain unfolding could contribute to the pathology of this mutation particularly under conditions where the body temperature is elevated, i.e., during fever or intense exercise. For the heterodimer, the least stable domain is intermediate between the homodimer and WT when cross-linked and similar to the WT for the more stable domains.
It is important to note that both mutations are close to the disulfide cross-link at Cys190 and the cross-link could perturb the structure around the mutation. This is difficult to rule out for the E180G-Tm homodimer, but the observation that the D175N mutant has a stability identical to that of the WT suggests little interaction between the cross-link and the residue at position 175.
In the absence of the cross-link, the interpretation is more difficult because the protein will exist as a mixture of homodimers and heterodimers. Even so, the mixture appears to be less stable than the pure E180G homodimer at physiological temperatures with a midpoint transition close to 36 °C. The presence of the WT and E180G homodimers would tend to make this domain appear to be more stable, suggesting that the heterodimer may be significantly less stable than either WT or E180G homodimers and could be partially folded even at normal body temperature. Note, however, that Tm is stabilized by binding to actin and once bound would be resistant to unfolding. We conducted some preliminary sedimentation studies of the E180G-Tm heterodimer binding to actin at 37 °C and did not observe a significant loss of affinity.
Tropomyosin thermal stability may be directly related to the stiffness or flexibility of the Tm, and flexibility is widely recognized as an important parameter defining Tm function.42−44 Any mutation-induced change in Tm flexibility could have a significant effect on the interaction with actin, the movement of Tm between sites on actin, and the accessibility of myosin to its binding sites on actin. The D175N mutation has shown no change in stability and is therefore not predicted to alter flexibility. Recently, it has been shown that the E180G mutation increases the local flexibility of the Tm molecule (in the homodimer) even when bound to actin,17 and the same could be true for the heterodimer. If the Tm thermal instability plays a role in the pathology of HCM, it may occur through the metabolic load imposed by the loss and clearing of misfolded Tm that is not bound to actin.
Many HCM mutations, at least in thin filament proteins, are associated with an increase in calcium sensitivity; i.e., less calcium is required to activate the thin filament, and conversely, if the same amount of calcium is released because of a stimulus, more calcium has to be removed from the cell before relaxation occurs. Previous work has shown that D175N and E180G homodimers do show changes in calcium sensitivity when in a fully assembled actin filament in in vitro motility assays (IVM) or reconstituted fiber experiments,9,10,19 with E180G showing a greater shift than D175N. Our measurements of the apparent calcium affinity for calcium switching of the actin·Tm·Tn filament for homodimers (Table 2) confirm this and show ΔpCa values of 0.08 for D175N and 0.14 for E180G. These are similar to the values we reported using the same method (0.09 and 0.13, respectively)11 or estimated using IVM by Bing et al. (0.082 and 0.115, respectively). Larger values, with lower precision, were reported with IVM by Wang et al. (pCa50% values of 0.2 for D175N and 0.64 for E180G19) and by Bai et al using reconstituted cardiac preparations (pCa50% values of 0.03 for D175N and 0.64 for E180G).9
Interestingly, important differences exist upon comparison of homodimers with heterodimers: in the case of the heterodimers, the WT-E180G-Tm heterodimer had only a small ΔpCa (0.03) compared to that of the WT, much smaller than that of the homodimer (0.14), while the D175N heterodimer had a larger ΔpCa (0.11) that was not significantly different from that of the homodimer (0.09). Thus, our data are compatible with both homo- and heterodimers showing the positive shift in calcium sensitivity expected for these HCM mutations and with the fact that the value of the E180G heterodimer surprisingly is very small and not significant.
The pCa shifts are small, and we tested if the same change in sensitivity could be seen in the sarcomere via replacement of the Tm·Tn complex in rabbit psoas myofibrils with human cardiac Tm·Tn with either WT-Tm or the E180G homo- or heterodimer. The results (Figure 7 and Table 3) do show an increased calcium sensitivity for the homodimer compared to that of the WT but not for the heterodimer. This is compatible with the solution data using pure actin·Tm·Tn filaments.
This work is significant for two reasons. First, we establish that heterodimers with only one mutation in the αTm can be formed easily in vitro and therefore can be expected to form in vivo. Second, we establish that the properties of the heterodimers are not simply means of the properties of the two homodimers. Some properties are intermediate like the affinity of E180G-Tm for actin, but others like the thermal stability of E180G-Tm and the calcium sensitivity of D175N-Tm heterodimers are more similar to that of the mutant homodimer than to that of the WT homodimer. Tm αβ heterodimers showed intermediate thermal stability, and the affinity for actin was weaker than that of homodimers.24 This observation is important also upon comparison of in vitro results, obtained exclusively with homodimers of Tm carrying cardiomyopathy mutations, which results in transgenic (TG) animals or human biopsies and clinical data. In these last cases, the ratios of WT and mutant homodimers or heterodimers are probably highly variable, and this would produce significant differences in the extent of the modification of calcium sensitivity. Our results provide insight into the molecular mechanism of phenotype modification in the presence of cardiomyopathy mutations of Tm.
It should be noted that the approach taken here to examine the behavior of distinct heterodimers of Tm is distinct from the approach taken by others44,45 who have assembled a mixture of homodimers (in various ratios) onto actin. If heterodimers are formed in the cell, then the actin filaments may contain a combination of both types of homodimer and the heterodimer on a single filament. This is more complex than the situation described here and is more difficult to assess. Different types of tags, probably fluorescent tags, would be required to assess the Tm composition of individual actin filaments.
Acknowledgments
We thank Samantha Lynn for the preparation of actin and myosin S1.
Glossary
Abbreviations
- HCM
hypertrophic cardiomyopathy
- Tm
tropomyosin
- αα*
heterodimer of αTm in which one chain carries the mutation
- hcTn
human cardiac troponin
- DTT
dithiothreitol
- ppA
pyrene-labeled, phalloidin-stabilized actin
- CD
circular dichroism
- S1
myosin subfragment 1
- His-Tm
wild-type α-tropomyosin homodimer with a histidine tag
- WT-D175N-Tm
α-tropomyosin heterodimer with the D175N mutation
- WT-E180G-Tm
α-tropomyosin heterodimer with the E180G mutation
- IVM
in vitro motility.
Supporting Information Available
Elution profile from WT-E180G-Tm heterodimer purification and its purity and detailed results from thermal stability assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Molecular Cardiology Laboratory, Faculty of Medicine, University of Crete, Heraklion, Crete, Greece.
The authors declare no competing financial interest.
M.J. was supported by a University of Kent studentship, A.K. by a BHF studentship, the M.G. group by a Wellcome Trust program grant (085309), and the University of Florence group by the 7th Framework Program of the European Union (“BIG-HEART”, Grant 241577) and the Italian Ministry of University and Research (PRIN 2008).
Supplementary Material
References
- Gordon A. M.; Regnier M.; Homsher E. (2001) Skeletal and cardiac muscle contractile activation: Tropomyosin “rocks and rolls”. News Physiol. Sci. 16, 49–55. [PubMed] [Google Scholar]
- Greenfield N. J.; Huang Y. J.; Swapna G. V.; Bhattacharya A.; Rapp B.; Singh A.; Montelione G. T.; Hitchcock-DeGregori S. E. (2006) Solution NMR structure of the junction between tropomyosin molecules: Implications for actin binding and regulation. J. Mol. Biol. 364, 80–96. [DOI] [PubMed] [Google Scholar]
- McKillop D. F.; Geeves M. A. (1993) Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 65, 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman W.; Craig R. (2008) Tropomyosin and the steric mechanism of muscle regulation. Adv. Exp. Med. Biol. 644, 95–109. [DOI] [PubMed] [Google Scholar]
- Maytum R.; Lehrer S. S.; Geeves M. A. (1999) Cooperativity and switching within the three-state model of muscle regulation. Biochemistry 38, 1102–1110. [DOI] [PubMed] [Google Scholar]
- Genomics of Cardiovascular Development and Remodeling. NHLBI Program for Genomic Applications, Harvard Medical School. (http://www.cardiogenomics.org) (accessed October 2012).
- Seidman J. G.; Seidman C. (2001) The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 104, 557–567. [DOI] [PubMed] [Google Scholar]
- Marston S. B. (2011) How do mutations in contractile proteins cause the primary familial cardiomyopathies?. Journal of Cardiovascular Translational Research 4, 245–255. [DOI] [PubMed] [Google Scholar]
- Bai F.; Weis A.; Takeda A. K.; Chase P. B.; Kawai M. (2011) Enhanced active cross-bridges during diastole: Molecular pathogenesis of tropomyosin’s HCM mutations. Biophys. J. 100, 1014–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing W.; Knott A.; Redwood C.; Esposito G.; Purcell I.; Watkins H.; Marston S. (2000) Effect of hypertrophic cardiomyopathy mutations in human cardiac muscle α-tropomyosin (Asp175Asn and Glu180Gly) on the regulatory properties of human cardiac troponin determined by in vitro motility assay. J. Mol. Cell. Cardiol. 32, 1489–1498. [DOI] [PubMed] [Google Scholar]
- Boussouf S. E.; Maytum R.; Jaquet K.; Geeves M. A. (2007) Role of tropomyosin isoforms in the calcium sensitivity of striated muscle thin filaments. J. Muscle Res. Cell Motil. 28, 49–58. [DOI] [PubMed] [Google Scholar]
- Chang A. N.; Harada K.; Ackerman M. J.; Potter J. D. (2005) Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in α-tropomyosin. J. Biol. Chem. 280, 34343–34349. [DOI] [PubMed] [Google Scholar]
- Golitsina N.; An Y.; Greenfield N. J.; Thierfelder L.; Iizuka K.; Seidman J. G.; Seidman C. E.; Lehrer S. S.; Hitchcock-DeGregori S. E. (1997) Effects of two familial hypertrophic cardiomyopathy-causing mutations on α-tropomyosin structure and function. Biochemistry 36, 4637–4642. [DOI] [PubMed] [Google Scholar]
- Hilario E.; da Silva S. L.; Ramos C. H.; Bertolini M. C. (2004) Effects of cardiomyopathic mutations on the biochemical and biophysical properties of the human α-tropomyosin. Eur. J. Biochem. 271, 4132–4140. [DOI] [PubMed] [Google Scholar]
- Kremneva E.; Boussouf S.; Nikolaeva O.; Maytum R.; Geeves M. A.; Levitsky D. I. (2004) Effects of two familial hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys. J. 87, 3922–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. E.; Suphamungmee W.; Janco M.; Geeves M. A.; Marston S. B.; Fischer S.; Lehman W. (2012) The flexibility of two tropomyosin mutants, D175N and E180G, that cause hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 424, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly S.; Lehrer S. S. (2012) Long-range effects of familial hypertrophic cardiomyopathy mutations E180G and D175N on the properties of tropomyosin. Biochemistry 51, 6413–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur M. C.; Chase P. B.; Chalovich J. M. (2011) Several cardiomyopathy causing mutations on tropomyosin either destabilize the active state of actomyosin or alter the binding properties of tropomyosin. Biochem. Biophys. Res. Commun. 406, 74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Brunet N. M.; Grubich J. R.; Bienkiewicz E. A.; Asbury T. M.; Compton L. A.; Mihajlovic G.; Miller V. F.; Chase P. B. (2011) Facilitated cross-bridge interactions with thin filaments by familial hypertrophic cardiomyopathy mutations in α-tropomyosin. J. Biomed. Biotechnol. 2011, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottinelli R.; Coviello D. A.; Redwood C. S.; Pellegrino M. A.; Maron B. J.; Spirito P.; Watkins H.; Reggiani C. (1998) A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circ. Res. 82, 106–115. [DOI] [PubMed] [Google Scholar]
- Muthuchamy M.; Pieples K.; Rethinasamy P.; Hoit B.; Grupp I. L.; Boivin G. P.; Wolska B.; Evans C.; Solaro R. J.; Wieczorek D. F. (1999) Mouse model of a familial hypertrophic cardiomyopathy mutation in α-tropomyosin manifests cardiac dysfunction. Circ. Res. 85, 47–56. [DOI] [PubMed] [Google Scholar]
- Prabhakar R.; Boivin G. P.; Grupp I. L.; Hoit B.; Arteaga G.; Solaro J. R.; Wieczorek D. F. (2001) A familial hypertrophic cardiomyopathy α-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J. Mol. Cell. Cardiol. 33, 1815–1828. [DOI] [PubMed] [Google Scholar]
- Michele D. E.; Albayya F. P.; Metzger J. M. (1999) Direct, convergent hypersensitivity of calcium-activated force generation produced by hypertrophic cardiomyopathy mutant α-tropomyosins in adult cardiac myocytes. Nat. Med. 5, 1413–1417. [DOI] [PubMed] [Google Scholar]
- Kalyva A.; Schmidtmann A.; Geeves M. A. (2012) In vitro formation and characterization of the skeletal muscle α·β tropomyosin heterodimers. Biochemistry 51, 6388–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich J. A.; Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871. [PubMed] [Google Scholar]
- Margossian S. S.; Lowey S. (1982) Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 85(Part B), 55–71. [DOI] [PubMed] [Google Scholar]
- Criddle A. H.; Geeves M. A.; Jeffries T. (1985) The use of actin labelled with N-(1-pyrenyl)iodoacetamide to study the interaction of actin with myosin subfragments and troponin/tropomyosin. Biochem. J. 232, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maytum R.; Geeves M. A.; Konrad M. (2000) Actomyosin regulatory properties of yeast tropomyosin are dependent upon N-terminal modification. Biochemistry 39, 11913–11920. [DOI] [PubMed] [Google Scholar]
- Monteiro P. B.; Lataro R. C.; Ferro J. A.; Reinach Fde C. (1994) Functional α-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 269, 10461–10466. [PubMed] [Google Scholar]
- Coulton A. T.; Koka K.; Lehrer S. S.; Geeves M. A. (2008) Role of the head-to-tail overlap region in smooth and skeletal muscle β-tropomyosin. Biochemistry 47, 388–397. [DOI] [PubMed] [Google Scholar]
- Al-Sarayreh S. (2011) Effect of hypertrophic and dilated cardiomyopathies associated mutations in troponin I on cardiac thin filament dynamics. Ph.D. Dissertation, University of Leicester, Leicester, U.K. [Google Scholar]
- Maytum R.; Westerdorf B.; Jaquet K.; Geeves M. A. (2003) Differential regulation of the actomyosin interaction by skeletal and cardiac troponin isoforms. J. Biol. Chem. 278, 6696–6701. [DOI] [PubMed] [Google Scholar]
- Tesi C.; Colomo F.; Nencini S.; Piroddi N.; Poggesi C. (1999) Modulation by substrate concentration of maximal shortening velocity and isometric force in single myofibrils from frog and rabbit fast skeletal muscle. J. Physiol. 516(Part 3), 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scellini B.; Piroddi N.; Poggesi C.; Tesi C. (2010) Extraction and replacement of the tropomyosin-troponin complex in isolated myofibrils. Adv. Exp. Med. Biol. 682, 163–174. [DOI] [PubMed] [Google Scholar]
- de Tombe P. P.; Belus A.; Piroddi N.; Scellini B.; Walker J. S.; Martin A. F.; Tesi C.; Poggesi C. (2007) Myofilament calcium sensitivity does not affect cross-bridge activation-relaxation kinetics. Am. J. Physiol. 292, R1129–R1136. [DOI] [PubMed] [Google Scholar]
- Brenner B. (1988) Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: Implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 85, 3265–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesi C.; Piroddi N.; Colomo F.; Poggesi C. (2002) Relaxation kinetics following sudden Ca2+ reduction in single myofibrils from skeletal muscle. Biophys. J. 83, 2142–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancso A.; Graceffa P. (1991) Smooth muscle tropomyosin coiled-coil dimers. Subunit composition, assembly, and end-to-end interaction. J. Biol. Chem. 266, 5891–5897. [PubMed] [Google Scholar]
- Head J. G.; Ritchie M. D.; Geeves M. A. (1995) Characterization of the equilibrium between blocked and closed states of muscle thin filaments. Eur. J. Biochem. 227, 694–699. [DOI] [PubMed] [Google Scholar]
- Lehman W.; Hatch V.; Korman V.; Rosol M.; Thomas L.; Maytum R.; Geeves M. A.; Van Eyk J. E.; Tobacman L. S.; Craig R. (2000) Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol. 302, 593–606. [DOI] [PubMed] [Google Scholar]
- Hayley M.; Chevaldina T.; Heeley D. H. (2011) Cold adaptation of tropomyosin. Biochemistry 50, 6559–6566. [DOI] [PubMed] [Google Scholar]
- Geeves M.; Griffiths H.; Mijailovich S.; Smith D. (2011) Cooperative [Ca2+]-dependent regulation of the rate of myosin binding to actin: Solution data and the tropomyosin chain model. Biophys. J. 100, 2679–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes K. C.; Lehman W. (2008) Gestalt-binding of tropomyosin to actin filaments. J. Muscle Res. Cell Motil. 29, 213–219. [DOI] [PubMed] [Google Scholar]
- Lakdawala N. K.; Dellefave L.; Redwood C. S.; Sparks E.; Cirino A. L.; Depalma S.; Colan S. D.; Funke B.; Zimmerman R. S.; Robinson P.; Watkins H.; Seidman C. E.; Seidman J. G.; McNally E. M.; Ho C. Y. (2010) Familial dilated cardiomyopathy caused by an α-tropomyosin mutation: The distinctive natural history of sarcomeric dilated cardiomyopathy. J. Am. Coll. Cardiol. 55, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza M.; Marston S.; Willott R.; Ashley C.; Mogensen J.; McKenna W.; Robinson P.; Redwood C.; Watkins H. (2005) Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J. Biol. Chem. 280, 28498–28506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.