

Summary
Mitochondria are strategically and dynamically positioned in the cell to spatially coordinate ATP production with energy needs and to allow the local exchange of material with other organelles. Interactions of mitochondria with the sarco-endoplasmic reticulum (SR/ER) have been receiving much attention owing to emerging evidence on the role these sites have in cell signaling, dynamics and biosynthetic pathways. One of the most important physiological and pathophysiological paradigms for SR/ER–mitochondria interactions is in cardiac and skeletal muscle. The contractile activity of these tissues has to be matched by mitochondrial ATP generation that is achieved, at least in part, by propagation of Ca2+ signals from SR to mitochondria. However, the muscle has a highly ordered structure, providing only limited opportunity for mitochondrial dynamics and interorganellar interactions. This Commentary focuses on the latest advances in the structure, function and disease relevance of the communication between SR/ER and mitochondria in muscle. In particular, we discuss the recent demonstration of SR/ER–mitochondria tethers that are formed by multiple proteins, and local Ca2+ transfer between SR/ER and mitochondria.
Key words: Mitochondria, Sarcoplasmic reticulum, Mitochondria-associated membranes, MAM, Ryanodine receptor, Uniporter, Ca2+ signalling, reactive oxygen species, ROS
Introduction
Isolated mitochondria are competent to produce energy on their own and are not particularly responsive to changes in their environment that mimic intracellular signals. These observations supported the early view that mitochondria, the putative descendents of free-living proteobacteria, maintained autonomy inside the cells and became powerplants that are regulated individually by substrate availability (Kurland and Andersson, 2000). However, in the cells, mitochondria are commonly visualized at specific locations and in physical interaction with other structures (Ardail et al., 1993; Rizzuto et al., 1998; Csordás et al., 2010) and dynamically change their distribution in response to various stimuli (McBride et al., 2006). Furthermore, recording mitochondrial function inside the cells provided evidence that mitochondria not only sense and respond to cellular signals, but they also, in many instances, are locally controlled by their strategic localization or transfer signals through local interactions (Rizzuto et al., 1998; Csordás et al., 2010).
A few aspects of the dynamic encounters of mitochondria with cellular structures are illustrated in Fig. 1, including mitochondria forming an interface with the sarco-endoplasmic reticulum (SR/ER), which allows the local transport of material and effective signal transduction between the two organelles (Fig. 1A). The emerging information on the structure and specific functions of SR/ER–mitochondria interactions in muscle cells is the main focus of this Commentary. In addition, there are areas of high mitochondrial density in which soluble molecules released from a mitochondrion into the cytoplasm can locally affect the function of neighboring mitochondria (Fig. 1B). This mechanism allows the release of Ca2+ or reactive oxygen species (ROS) from a subset of mitochondria to trigger Ca2+ or ROS release from the adjacent mitochondria (Ichas et al., 1997; Zorov et al., 2000; Pacher and Hajnóczky, 2001; Aon et al., 2003). The initial signal can thus be propagated throughout the mitochondrial population in a regenerative manner. Pro-apoptotic Bcl-2 family proteins also employ propagation mechanisms in the mitochondrial population to execute cell death (Lartigue et al., 2008; Garcia-Perez et al., 2012). In response to apoptotic stimuli, mitochondria release activators of the cytoplasmic components of the apoptotic cascade (e.g. cytochrome c), as well as factors that act on neighboring mitochondria to trigger additional release events (Pacher and Hajnóczky, 2001). This lateral signaling between mitochondria can promote propagation of a full strength apoptotic signal inside a cell – a distance that can be over several hundred micrometers – within a few minutes. Therefore, this mechanism might be particularly important for coordinated execution of apoptosis in large cells abundant in mitochondria, such as myocytes of cardiac muscle and fibers of skeletal muscle.
Fig. 1.

The ‘social life’ of mitochondria. (A) Illustration of the interactions of SR/ER with mitochondria. Fluorescent proteins targeted to ER (ER–GFP) and mitochondria (MitoRFP) were expressed in RLB-2H3 cells and viewed using confocal microscopy followed by three-dimensional reconstruction. The physical tethering between ER and mitochondria (blue rectangle in scheme) facilitates local transfer of Ca2+ (green arrow) and membrane constituents. (B) Intermitochondrial coupling mediated by soluble molecules. Imaging of the mitochondrial membrane potential loss in H9c2 myotubes as reported by a potentiometric probe (TMRE) illustrates the spatial organization of mitochondrial apoptosis as a regenerative wave. The propagation of membrane permeabilization among individual mitochondria is mediated by factors that are released from the first responding mitochondria during permeabilization and promote permeabilization of the neighboring mitochondria. (C) Intermitochondrial content exchange mediated by fusion. Time course of a typical mitochondrial transient fusion is shown by photoactivatable fluorescent protein technology. The donor mitochondrion containing the photoactivated Kindling protein (MitoKP) aligns with the acceptor (MitoGFP), followed by content mixing. Within seconds the pair has reseparated at the apparent site of fusion and moved apart. (D) Mitochondrial transport along microtubules. In the image mitochondria (MitoDSRed) are aligned with microtubules (TubulinGFP). The images shown in A and C are adapted with permission from Spät et al., 2008 with permission from Elsevier, and Liu et al., 2009, respectively. Images shown in B and D were acquired as described in Pacher and Hajnóczky, 2001 and Yi et al., 2004.
Mitochondria can also be in physical contact with each other and undergoing membrane fusion (Fig. 1C). Mitochondrial fusion supports the uniform distribution of the soluble matrix and intermembrane space (IMS) constituents, as well as of components of the outer and inner mitochondrial membranes (OMM and IMM, respectively) in the mitochondrial population (Chan et al., 2006; Twig et al., 2008; Liu et al., 2009). The mitochondrial fusion–fission cycle is also a means for segregating damaged mitochondrial components (Twig et al., 2008). On the basis of the functional impairments observed upon deletion of OMM fusion proteins, mitochondrial fusion has been proposed to be essential for both skeletal (Chen et al., 2010) and cardiac (Chen et al., 2011) muscle development and function.
The positioning of mitochondria for the above-described interactions is supported by mitochondrial anchorage to cytoskeletal structures (Fig. 1D). In many mammalian cell types, mitochondria are aligned with and move along microtubules, but can also interact or travel along microfilaments or intermediate filaments (Milner et al., 2000; Yi et al., 2004; Anesti and Scorrano, 2006). Microtubule-assisted mitochondrial movements are required for their deposition throughout neuronal processes (Saxton and Hollenbeck, 2012) and for both mitochondrial fusion and fission (Liu et al., 2009). Because of the strict structural arrangements of cardiomyocytes and muscle fibers, the existence and relevance of mitochondrial movements in muscle is less clear. Cytoskeleton-dependent positioning of mitochondria is also necessary for heterotypic organellar interactions (Fig. 1). The different interactions of mitochondria described above can impact on each other (e.g. ER–mitochondrial associations form sites for mitochondrial fission) (Friedman et al., 2011). Collectively, these findings illustrate that mitochondria can receive various local inputs from many cell constituents and produce signals that can locally and globally affect a range of cellular components. In this Commentary, we will focus on the bidirectional coupling of mitochondria with the SR/ER, analyzing first the structural, then the functional interaction between these organelles, and finally, the possible role of its dysregulation in muscle-specific pathologies.
Structural interactions between SR/ER and mitochondria in different cell types
A close association between ER and mitochondria was initially described in electron microscopy images of liver tissue (Morré et al., 1971; Shore and Tata, 1977). A stable interorganellar interaction was indicated when tissue fractionation studies revealed an ER membrane contamination in isolated mitochondria, referred to as mitochondria-associated membranes (MAMs) (Vance, 1990). A close proximity between ER and mitochondria in live cells was first observed by co-expression of two differently colored fluorescent proteins targeted to these organelles and the three-dimensional reconstruction of high-resolution fluorescent microscopy images (Ardail et al., 1993; Rizzuto et al., 1998). The actual physical links, known as tethers, were first visualized by electron tomography, and found to be made of protein(s) as they could be disrupted by limited proteolysis (Csordás et al., 2006).
A number of molecular entities have been described to support the physical interaction between the ER and mitochondria. These include protein complexes, such as that formed between the molecular chaperone Grp75 (also known as HSPA9) and the inositol trisphosphate (InsP3) receptor Ca2+ channel (IP3R) in the ER, the voltage-dependent anion channel 1 (VDAC1) located in the OMM (Szabadkai et al., 2006), or the complex between mitofusin 2 (MFN2) located in the ER with either mitofusin 1 (MFN1) or MFN2 in the OMM (de Brito and Scorrano, 2008), as well as the ERMES complex that has been described in yeast (Kornmann et al., 2009). Besides IP3R and VDAC1, other Ca2+-binding proteins localize to the MAM, including AMF-R (Wang et al., 2000), Miro1 (Fransson et al., 2003; Kornmann et al., 2011) and the ryanodine receptor Ca2+ channel (RyR2) in the cardiac muscle (Chen et al., 2012). Additionally, many signaling proteins have been described to reside at the MAM (Fujimoto and Hayashi, 2011; Lynes and Simmen, 2011).
The interactions of mitochondria are subject to a dynamic environment. In the peripheral cytoplasm, SR/ER and mitochondria commonly appear as discrete structures that form SR/ER–mitochondrial complexes and move in a coordinated manner, maintaining an unaltered interface (Spät et al., 2008; Friedman et al., 2010). However, in the perinuclear zone of cells, the extensive SR/ER and mitochondria usually form continuous networks with only limited long-range movements. Here, mitochondria often appear to slide along the SR/ER (David Weaver and G.H., unpublished observations). This movement would force the interface-forming structures to continually reorganize or to move laterally in the membrane(s). Control of the ER–mitochondrial movements takes place both at the level of the tracks, for example, acetylation of microtubules favors the ER–mitochondrial sliding (Friedman et al., 2010), and at motor complexes, which can be inhibited by cytoplasmic [Ca2+] elevations (Yi et al., 2004; Brough et al., 2005). The interface also shows tightening during ER stress conditions (Csordás et al., 2006) and disruption during high [Ca2+] exposure (Goetz et al., 2007). Thus, SR/ER–mitochondrial coupling is modulated by both cell dynamics and signaling.
Cardiomyocytes and skeletal muscle fibers present a complex and unique architecture (Fig. 2A; for details see Box 1). SR/ER–mitochondria associations have been visualized in vivo using synthetic interorganellar linkers in cardiac-muscle-derived cell lines (Fig. 2B) (Csordás et al., 2010), rat cardiomyocytes and skeletal muscle fibers (G.C., V.E. and G.H., unpublished data).
Fig. 2.

SR–mitochondrial physical coupling in the muscle. (A) Close proximity of SR and mitochondria in a Flexor Digitorum Brevis (FDB) SM fiber is illustrated by labeling with an SR-specific dye, BODIPY–Ryanodine (left) and the potentiometric dye TMRE (middle). The overlay image on the right shows that SR (green) and mitochondria (red) run in parallel, both in longitudinal and transversal orientation. The yellow areas indicate the close proximity between SR and mitochondria. (B) Visualization of the sites of close SR/ER–mitochondrial associations in a live cardiac-muscle-derived cell (H9c2) cell by drug-inducible synthetic interorganellar linkers. The scheme on the left illustrates the rapamycin (Rapa)-inducible bridge-forming modules. Specifically, the OMM- and SR/ER-targeting sequences were coupled with the two components of the FKBP (FK506 binding protein 12)-FRB (FRB domain of mTOR) heterodimerization system, respectively. Addition of rapamycin causes heterodimerization between adjacent FKBP and FRB domains to rapidly connect the SR/ER- and OMM-targeted anchors. Induction of the bridge formation is initially confined to the areas where the SR/ER and OMM were naturally close. On the right, confocal images show the broad SR/ER and mitochondrial distribution of the respective fluorophores before and enrichment (colocalization of the CFP and RFP tags) at the sites of the SR/ER–mitochondrial interface after 3 minutes of 100 nM rapamycin treatment (bottom row). Note the rapamycin-induced colocalization of CFP and mRFP appears in white on the overlay images. For technical details see Csordás et al., 2010. The scheme is adapted with permission from Csordás et al., 2010 with permission from Elsevier. (C) TEM image of a membrane complex formed by TT and jSR (the diad) in close association with a mitochondrion (M) in cardiac muscle. In the magnified schematics of the jSR-mitochondrial interface (right), two forms of the tethering mechanisms are depicted. ‘Professional’ tethers refer to those proteins that are known to provide only structural support for the interface. MFN2 is a candidate to support SR-to-mitochondria juxtaposition. Signaling complexes provide both structural support and communication between SR/ER and mitochondria, and include the σ1 receptor, mitostatin, RAB32, phosphofurin acidic cluster sorting protein 2 (PACS2), ryanodine receptor (RyR) and voltage-dependent anion channels (VDACs). TT, transversal tubule; jSR, junctional SR; OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane.
Box 1. Structural organization of the muscle.
The structure of cardiomyocytes and skeletal muscle fibers is determined by the sarcomeres that contain the contractile myofibrils and are interrupted by invaginations of the sarcolemma, known as the transversal tubules (TTs) that are juxtaposed to a specialized region of the SR, the junctional SR (jSR) (see figure). The voltage-dependent L-type Ca2+ channels (Cav1.2, also known as CACNA) are concentrated in the TT, whereas the RyRs are primarily present on the regions of the jSR, facing the TT. The jSR stores large amounts of Ca2+ that is mostly bound to calsequestrin (CSQ, also known as CASQ). When an action potential propagates along the sarcolemma and the TT, Cav1.2s are stimulated and spread the activation to RyRs, which release Ca2+ from the SR to trigger contraction, a phenomenon known as excitation–contraction (EC) coupling. Both Cav1.2 and RyR have tissue-specific subunits that form the so-called Ca2+ release units (CRUs). Heart expresses Cav1.2α1c (CACNA1C) and RyR2, whereas skeletal muscle expresses Cav1.2α1s (CACNA1S) and RyR1 (Dulhunty, 2006). The coupling between the Cav1.2 and RyR is also tissue specific. In the heart, Cav1.2α1c to RyR2 coupling depends on Ca2+ entry, whereas in the skeletal muscle, Cav1.2α1s and RyR1 are physically coupled and RyR1 activation relies on the conformational change encountered by Cav1.2 upon voltage sensing (see Fig. 3B). The jSR and TT form diads (consisting of one jSR and one TT) in the heart, and triads (two jSRs and one TT) in skeletal muscle (see figure) (Franzini-Armstrong et al., 1998). In addition to the jSR, other regions of the SR, such as the corbular SR and the network SR (nSR) of cardiomyocytes also contain RyR2 (Franzini-Armstrong et al., 2005; Lukyanenko et al., 2007).
Mitochondria occupy ∼40% of the cell volume in cardiomyocytes and ∼20% in skeletal muscle fibers (Isaeva et al., 2005) and are mostly located among the myofibrils. In ventricular cardiomyocytes, intermyofibrillar mitochondria fill the space parallel with the myofibrils, whereas in skeletal muscle fibers, mitochondria are localized close to the CRUs (see Fig. 2A). Neonatal cardiomyocytes or skeletal myotubes, as well as adult atrial myocytes have a less developed architecture where every cellular organelle enjoys a higher level of freedom.
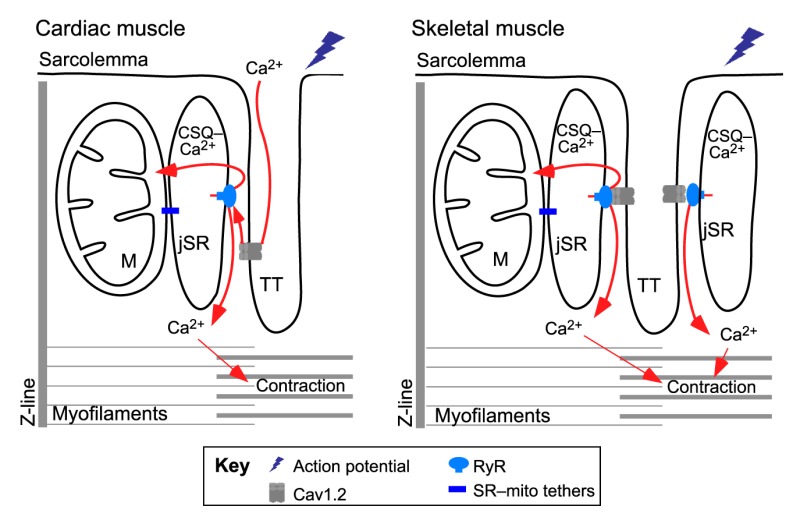
The structural relationship between SR and mitochondria in cardiac and skeletal muscle
In transmission electron microscopy (TEM) of cardiac muscle, 90% of the Ca2+ release units (CRUs) are close to mitochondria, with an average distance of 37 nm between the RyR ‘foot’ on the junctional SR (jSR) and the mitochondrial surface (Sharma et al., 2000). Most RyR2 molecules are located as part of the CRU in the gap between SR and transversal tubules (TTs) (Fig. 3A). A small subgroup of RyR2s can be localized close to perinuclear mitochondria (Lukyanenko et al., 2007; Salnikov et al., 2009). In the areas of jSR–mitochondria associations, the highly folded IMM always has at least one contact point with the OMM. Furthermore, the contact points are enriched in the VDAC channels that mediate Ca2+ transfer across the OMM (García-Pérez et al., 2011). Thus, the arrangements of the jSR–OMM–IMM might provide a ‘highway’ for Ca2+ delivery from the SR to the mitochondrial matrix.
Fig. 3.

Specific functions preferentially localized at the SR–mitochondrial interface. (A) A range of cellular activities, including several biosynthetic and signaling pathways and events involving membrane dynamics take place at the ER/SR–mitochondrial interface. (B) Local Ca2+ transport between RyR and mitochondria (red arrows). Upon action potential stimulation, the voltage dependent Ca2+ channel Cav1.2 activates RyR. In the heart, the Cav1.2-specific subunit α1c permeates Ca2+ from the extracellular medium to activate the RyR2. In skeletal muscle, Cav1.2 α1s interacts physically with RyR1, and the activation of Cav1.2 α1s directly promotes RyR1-mediated release of Ca2+ from the jSR to the cytoplasm. A high-Ca2+ microdomain is generated at the mouth of the RyRs, and it is sensed by the mitochondrial Ca2+ uniporter. Mitochondrial Ca2+ uptake activates matrix Ca2+-dependent dehydrogenases (CSMDH) and ATP synthesis. (C) Local interactions between ROS producers and ROS-sensitive proteins at the SR–mitochondria interface. The mitochondria electron transport chain generates superoxide anion (· O2−) at the level of complex I and III (ETC, electron transport chain), which is converted into H2O2 by superoxide dismutase 2 (SOD2) in the mitochondrial matrix, or SOD1 in the cytosol or intermembrane space. H2O2 serves as substrate for glutathione peroxidase (GPX) that mediates the conversion of glutathione (GSH) to glutathione disulfide (GSSG; oxidized glutathione). GSSG targets proteins that contain reactive cysteines, such as RyR or the SR calcium transport ATPase (SERCA). SR- and TT-localized NADPH oxidases (NOXs) also contribute to · O2− generation. For further details see Csordás and Hajnóczky, 2009.
Tethers have been observed in the muscle between mitochondria and jSR or network SR (nSR), and less frequently TTs (Boncompagni et al., 2009; Hayashi et al., 2009). The role of MFN2 in tethering is currently debated since in MFN2-ablated heart, both unaltered SR–mitochondrial associations (Papanicolaou et al., 2011) and structural derangements (Chen et al., 2012) have been reported. The reason for the discrepancy between these works might be due to the different parameters that have been evaluated or the different approaches used for the establishment of the knockouts. Interestingly, biochemical analyses have demonstrated that full-length MFN2 is predominantly present in the OMM, whereas a lower-molecular-mass protein that is recognized by antibodies against the MFN2 N-terminus is found in the fractions that contain MAM (García-Pérez et al., 2008; García-Pérez et al., 2011). Another study provided evidence for an RyR2- and VDAC2-containing protein complex in cardiac muscle (Min et al., 2012). Thus, MFN2-related proteins and VDAC2 probably have a role in stabilizing SR–mitochondrial interactions in cardiac muscle.
Transmission electron microscopy (TEM) of skeletal muscle has shown that the distance between the RyR foot and the OMM is 130 nm. Opposite to the RyR-containing SR surface, there are tethers between the jSR and OMM (Fig. 3A), with an average length of 10 nm. Tethers have also been visualized between the nSR and OMM. Notably, the distribution of mitochondria in mouse skeletal muscle changes during development, from a longitudinal arrangement towards a triad-adjacent location running in parallel to the z-lines. In parallel, the frequency of tethers increases during the first four postnatal months (Boncompagni et al., 2009). Furthermore, there are extensive associations between SR and mitochondria in mammalian muscle, whereas lower vertebrates display a less-extensive interface (Franzini-Armstrong and Boncompagni, 2011). TEM studies have, thus far, only been complemented by the demonstration of co-purification of SR with mitochondria (Mitchell et al., 1983) and with live-cell co-localization studies of SR-mitochondrial interactions (Fig. 2A).
In summary, the existence of SR–mitochondria tethers has been demonstrated in both cardiac and skeletal muscle. However, the molecular entities that constitute the tethers remain elusive. With regard to a possible role of MFN2, a question remains as to whether the knockout phenotype (Chen et al., 2011) is indeed caused by impaired SR–mitochondria tethering or the lack of other functions of this protein. The impact SR–mitochondria coupling has on the entire mitochondrial population depends on the fusion state of the mitochondria, as connectivity among mitochondria helps to spread signals generated by the SR/ER–mitochondria coupling to other mitochondria. Functional consequences of knockout of MFN1 or MFN2, including mitochondrial fragmentation have been established in cardiac (Chen et al., 2011; Papanicolaou et al., 2011) and in skeletal muscle (Chen et al., 2010), but it remains elusive whether mitochondrial fusion actually occurs in skeletal muscle. Novel approaches used in simpler cellular models could be useful to elucidate the processes involved in the dark secrets and mysteries of SR/ER–mitochondria coupling in the muscle. For instance, synthetic SR/ER–mitochondria linkers will be useful to test the relevance of SR–mitochondria linkage in muscle cell physiology (Csordás et al., 2006; Kornmann et al., 2009; Csordás et al., 2010). To validate specific protein constituents of the SR/ER–mitochondria interface, genetic tags for proteins that can be detected by electron microscopy [e.g. mini Singlet Oxygen Generator (Shu et al., 2011)] are now available and might be useful.
Functional role of SR–mitochondria communication
Mutation of the ERMES complex, the putative ER–mitochondria tethering factor, is lethal in yeast. The mutant strain can be rescued by synthetic ER–mitochondria linkers, indicating that the physical linkage between ER and mitochondria is essential for yeast survival (Kornmann et al., 2009). In mammalian cells, destruction of the ER–mitochondrial linkage causes the specific loss of the IP3R-mediated Ca2+ transfer to the mitochondria and the ensuing stimulation of oxidative metabolism (Csordás et al., 2006). Specific roles of SR/ER–mitochondria associations have been identified in different cellular processes, including biosynthetic pathways, local signaling and membrane dynamics, as discussed below (Fig. 3A).
Biosynthetic pathways – phospholipid biosynthesis and protein transport
The term MAM was first used to describe a subcellular fraction that is associated with mitochondria and displays a high activity for several phospholipid biosynthetic enzymes previously ascribed to ER (Vance, 1990). This finding was complemented by the reconstruction of phosphatidylserine metabolism in a cell-free system that consisted of microsomes and mitochondria (Voelker, 1989) and by the subsequent demonstration of interorganellar phospholipid transfer between the ER and mitochondria (Ardail et al., 1993; Gaigg et al., 1995; for a recent review see Rowland and Voeltz, 2012). Evidence for a requirement of the ERMES complex in yeast in lipid transport has been presented (Kornmann et al., 2009), but was not reproduced in a subsequent study (Nguyen et al., 2012). It has also been suggested that there is protein transfer at the ER–mitochondria associations, in particular for proteins that are present in both the ER and OMM (e.g. Bcl-xL). Some evidence has been presented for the interorganellar transfer of the human cytomegalovirus protein vMIA (Bozidis et al., 2008), but a direct ER–mitochondria protein transfer remains to validated.
Local signaling
Cytoplasmic Ca2+ ([Ca2+]c) spikes and oscillations propagate to the mitochondrial matrix and stimulate oxidative energy production (Rizzuto et al., 1993; Hajnóczky et al., 1995; Jouaville et al., 1999). Constitutive Ca2+ release through IP3Rs has also been proposed to be essential for maintaining cellular bioenergetics (Cárdenas et al., 2010). However, the IP3R-mediated transient increases of global [Ca2+]c peak in the submicromolar range and would not activate the low-affinity mitochondrial uniporter. Instead, it was proposed that much higher and short-lasting local [Ca2+]c increases in the vicinity of IP3Rs stimulates Ca2+ uptake by adjacent mitochondria (Rizzuto et al., 1993; Rizzuto et al., 1998; Csordás et al., 1999). More recently, [Ca2+] has been quantified as ≥10 µM at the ER–mitochondria interface, directly confirming that mitochondria can sense high [Ca2+]c microdomains in the vicinity of the ER (Csordás et al., 2010; Giacomello et al., 2010). In further support for the role of the ER–mitochondria interface in Ca2+ signaling, mitochondrial Ca2+ uptake has been shown to modulate the local Ca2+ feedback regulation of IP3Rs (Jouaville et al., 1995; Simpson and Russell, 1996; Hajnóczky et al., 1999). Mitochondrial ATP production is relevant for locally supporting the activity of the Ca2+ ATPase SERCA and, in turn, the refilling of the ER/SR Ca2+ stores (Landolfi et al., 1998; De Marchi et al., 2011). In addition to stimulating oxidative ATP production, local Ca2+ delivery from the SR/ER to mitochondria can also control other Ca2+-sensitive mechanisms at the mitochondrial matrix and in the mitochondrial membranes. Of particular interest is that the IP3R-mediated local Ca2+ transfer can trigger apoptosis under conditions of cellular stress (Szalai et al., 1999; Pinton et al., 2001).
ROS are involved in both physiological signaling and induction of cell injury. Mitochondria generate ROS through the electron transport chain, whereas the oxidative folding machinery in the ER produces H2O2 and includes ERO1, which is localized to the MAM (Gilady et al., 2010; Anelli et al., 2012). ROS can change the activity of both ER and mitochondrial Ca2+ transport mechanisms, and ROS production itself is also affected by Ca2+ (Brookes et al., 2004). Thus, mutual local interactions between Ca2+ and ROS signaling are likely to occur and control various functions at the SR/ER–mitochondria associations (Csordás and Hajnóczky, 2009).
Many proteins that regulate cell survival or apoptosis are enriched in the SR/ER and OMM, and several show a dual localization. The SR/ER–mitochondria associations provide opportunity for interorganellar protein–protein interactions. For example, the Bcl-2 family member and ER membrane integral protein BAP31 interacts with FIS1 on the mitochondrial surface. Caspase-8-mediated cleavage of BAP31 yields p20BAP31, which stimulates local Ca2+ transfer to the mitochondria, and promotes mitochondrial fragmentation and cytochrome c release (Breckenridge et al., 2003; Iwasawa et al., 2011). The promyelocytic leukemia (PML) tumor suppressor also localizes to the ER and MAM and forms a complex with IP3R, Akt and PP2A to control Ca2+ transfer to the mitochondria and the induction of apoptosis via the phosphorylation state of the IP3R (Marchi et al., 2008; Giorgi et al., 2010). The GTPase Rab32 localizes to both the ER and OMM and controls the distribution and activity of several MAM proteins to influence the activation of mitochondrial apoptosis (Bui et al., 2010). Another type of local crosstalk is mediated by cytochrome c, which is released from the mitochondria to enhance IP3R-mediated Ca2+ release and, in turn, further stimulates mitochondrial membrane permeabilization and cytochrome c release (Boehning et al., 2003). Additional signaling proteins that are localized to MAM are the chaperone σ1 receptor, which can sense ER Ca2+ and regulate its ER–mitochondrial transfer (Hayashi and Su, 2007), the sorting protein PACS2, which localizes the ER chaperone calnexin to the MAM (Myhill et al., 2008) and controls cell fate (Simmen et al., 2005), and mitostatin, which modulates the ER–mitochondrial interface in an MFN2-dependent manner and regulates Ca2+ transfer and apoptosis (Cerqua et al., 2010). These results illustrate the presence of a complex protein array at the ER–mitochondrial interface, which utilizes direct protein–protein interactions, post-translational modification and modulation of Ca2+ transport to control cell survival and death.
Regarding lipids, the composition of the MAM resembles that of lipid rafts, supporting their relevance as signal integration domains (Area-Gomez et al., 2012). Ceramide and other sphingolipids are produced at the ER, mitochondria and also in the MAM (Bionda et al., 2004; Sano et al., 2009; Novgorodov et al., 2011). These lipids can evoke OMM permeabilization and apoptosis, either on their own (Siskind et al., 2002; Stiban et al., 2008) by stimulating the Bax–Bak pathway (Chipuk et al., 2012) or by interacting with Ca2+ (Szalai et al., 1999; Pinton et al., 2001; Sano et al., 2009). Thus, the local lipid dynamics converges with proteins and Ca2+ in the control of cell survival. In addition to the specific functions of individual MAM constituents, the MAM, as a complex entity, provides membrane components for autophagosome formation (Hailey et al., 2010) and marks the mitochondrial division planes to contract the mitochondria and allow dynamin-1-like protein (DRP1)-mediated fission (Friedman et al., 2011).
Functional relevance of the SR/ER–mitochondrial interface in muscle
Here, we focus on the local control of mitochondrial Ca2+ uptake under physiological conditions, the relevance of mitochondria to Ca2+ homeostasis, and Ca2+-induced stimulation of oxidative metabolism and local ROS signaling, as only little is known about other functions of the SR/ER–mitochondrial interface in the muscle.
Mitochondrial Ca2+ uptake in cardiac muscle
In the past decades, diverse techniques have been applied to address two main questions: (1) do cardiac muscle mitochondria show an increase in the mitochondrial Ca2+ concentration ([Ca2+]m) on a beat-to-beat basis, and (2) is there a local SR-to-mitochondria Ca2+ transfer? (Dedkova and Blatter, 2008). Several studies showed that there were only hardly detectable increases in [Ca2+]m in association with single [Ca2+]c transients. However, owing to their relatively slow decay, the small [Ca2+]c rises integrate in a manner that is dependent on the frequency of the [Ca2+]c transients (Sedova et al., 2006; Andrienko et al., 2009). By contrast, other studies have demonstrated beat-to-beat oscillations of [Ca2+]m in both adult (Trollinger et al., 1997; Ohata et al., 1998; Matlib et al., 1998) and neonatal cardiomyocytes (Robert et al., 2001b). More recent studies performed with genetically targeted Ca2+-sensitive proteins support beat-to-beat [Ca2+]m oscillations (Bell et al., 2006; Kettlewell et al., 2009; Lu et al., 2013), although the amount of Ca2+ taken up by the mitochondria during each spike remains to be determined.
Regarding the second question of whether there is a local SR-to-mitochondria Ca2+ transfer, it has been reported that RyR activation evokes a detectable [Ca2+]m rise in adult cardiomyocytes, even when the [Ca2+]c transients were suppressed by 1,2-bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA), providing evidence for local SR–mitochondrial Ca2+ transfer (Sharma et al., 2000). Similarly, in differentiated H9c2 cells, both depolarization and caffeine triggered a rise in [Ca2+]m that resisted cytoplasmic Ca2+ buffering with a slow chelator (Szalai et al., 2000). Furthermore, in the same model, fundamental SR Ca2+ release events (Ca2+ sparks) were shown to cause a miniature [Ca2+]m transient in the most adjacent single mitochondrion; these events were referred to as ‘Ca2+ marks’ (Pacher et al., 2002). On the basis of pharmacological evidence, Ca2+ marks depend on mitochondrial Ca2+ uptake and their decay phase is controlled by the mitochondrial Na+/Ca2+ exchanger (Pacher et al., 2002). Interestingly, transient mitochondrial depolarizations, often confined to a single mitochondrion and referred to as flickers, have also been described in cardiomyocytes. Flickering was blocked upon treatment with thapsigargin or ryanodine, indicating dependence on Ca2+ release from the SR (Duchen et al., 1998). As direct evidence for the exposure of cardiac mitochondria to high [Ca2+]c, microdomains or ‘Ca2+ hot spots’ on OMM were recorded in neonatal cardiomyocytes during [Ca2+]c oscillations (Drago et al., 2012). Furthermore, co-purification of SR with cardiac mitochondria allowed the demonstration of caffeine-induced local Ca2+ transfer from SR to the physically coupled mitochondria (García-Pérez et al., 2008). Finally, studies performed in H9c2 myoblasts and differentiated cells described a differentiation-dependent switch from IP3R–mitochondrial to RyR–mitochondrial local Ca2+ signaling (Yi et al., 2012). Thus, the opportunity for local RyR2–mitochondrial Ca2+ transfer occurring at the SR–mitochondrial associations has now been established (Fig. 3B).
Activation of oxidative metabolism by local RyR–mitochondrial Ca2+ transfer in cardiomyocytes
Early studies have shown the activity of Ca2+-sensitive matrix dehydrogenases (CSMDHs) in isolated heart mitochondria (McCormack and Denton, 1989) and that extramitochondrial Ca2+ increase can rapidly enhance the activities of CSMDH and ATP synthase to stimulate ATP production (Territo et al., 2001). In H9c2 cells, [Ca2+]m transients produced by local Ca2+ transfer from RyR to the mitochondria result in increased NAD(P)H fluorescence, reflecting CSMDH activation in vivo (Pacher et al., 2000; Szalai et al., 2000). More recently, using mitochondria-localized luciferase in cardiomyocytes, it has been shown that ATP levels increase when workload is increased by means of ionotropic stimulation with isoproterenol (Ginsburg and Bers, 2004). However, without stimulation, there are no fluctuations in ATP levels in paced rabbit myocytes (Bell et al., 2006). Collectively, these results suggest that local Ca2+ delivery to the mitochondria is a key to coupling the stimulation of energy metabolism with contraction in cardiac muscle. Mitochondrial Ca2+ uptake and ATP production might also exert some feedback effects on the neighboring RyR2s given that interference with mitochondrial activity affected Ca2+ spark generation in H9c2 cells (Pacher et al., 2002).
Mitochondria Ca2+ uptake in skeletal muscle
Expression of mitochondrial-matrix-targeted aequorin in skeletal muscle myotubes gave rise to [Ca2+]m transients upon RyR-mediated Ca2+ release that were stimulated by caffeine or plasma membrane depolarization (Brini et al., 1997). The RyR-mediated [Ca2+]m transients were enhanced by overexpression of VDAC, whereas the [Ca2+]m responses to bulk [Ca2+]c elevations were unaffected (Rapizzi et al., 2002). On the basis of these data, it was concluded that enhancement of the Ca2+ permeability by the overexpressed VDAC is needed to optimize the local transfer of the brief high-[Ca2+]c microdomains during RyR-mediated [Ca2+]m transients (Rapizzi et al., 2002). In vivo recording in skeletal muscle confirmed that mitochondria display increases in [Ca2+]m that closely follow [Ca2+]c transients under both single twitch and tetanic stimulation (Rudolf et al., 2004; Rogers et al., 2007). Another elegant study identified a subset of mitochondria that displayed a rise in [Ca2+]m upon caffeine stimulation even when global [Ca2+]c transients were buffered by BAPTA, further supporting a local SR–mitochondrial Ca2+ transfer (Shkryl and Shirokova, 2006). Furthermore, mitochondria were found to attenuate spontaneous [Ca2+]c sparks and transients in proportion to their abundance in fibers (Isaeva et al., 2005).
A recent electrophysiology study recorded the current through the mitochondrial Ca2+ uniporter in mitoplasts isolated from mitochondria of both skeletal and cardiac muscle (Fieni et al., 2012). Interestingly, the Ca2+ conductance of the uniporter was much higher in skeletal-muscle-derived than in cardiac-muscle-derived mitoplasts. Similarly, the mRNA levels of the putative pore-forming protein (MCU) and of an essential regulatory protein (MICU1) of the uniporter were also found to be higher in skeletal-muscle-derived mitoplasts than in those derived from cardiac muscle (De Stefani et al., 2011; Perocchi et al., 2010). In the context of the local Ca2+ transfer from SR to the mitochondria, it will be important to evaluate the distribution of the uniporters in the highly folded IMM and their alignment with the SR Ca2+ source and with the VDACs in the OMM.
Relevance of SR–mitochondrial Ca2+ transfer for oxidative metabolism in skeletal muscle
When [ATP] was measured using mitochondria-targeted luciferase it was found to be elevated during [Ca2+]m transients that are triggered by membrane depolarization in myotubes, confirming the competence of SR in transferring Ca2+ to mitochondria to stimulate oxidative metabolism (Jouaville et al., 1999). A major consumer of ATP is the Ca2+ transport machinery. Indeed, treatment with an uncoupler results in an increase in resting [Ca2+]c and a decrease in the amplitude of the [Ca2+]c transients that are induced by field stimulation in isolated smooth muscle (SM) fibers (Caputo and Bolanos, 2008). Thus, the RyR-mediated mitochondrial transfer of Ca2+ is relevant for the control of oxidative metabolism and likely allows a local feedback regulation of [Ca2+]c signaling through mitochondria, both in cardiac and skeletal muscle.
ROS as regulatory signaling factors in cardiac and skeletal muscle
The roles of mitochondria and of SR/ER-localized ROS producers, such as the electron transport chain and NAD(P)H oxidase (NOX) enzymes (reviewed in Duchen, 2004; Csordás and Hajnóczky, 2009) are depicted in Fig. 3C. In cardiomyocytes, mitochondrial ROS-induced ROS release has been described in response to light-induced ROS production and occurs through either opening of the mitochondrial permeability transition pore (PTP) (Zorov et al., 2000) or an IMM anion channel (Aon et al., 2003). Here, ROS waves do not depend on mitochondrial Ca2+ uptake. Spatially and temporally confined bursts of superoxide anion production (‘flashes’) by mitochondria have also been documented by means of a mitochondria-targeted circularly permuted YFP (cpYFP) and a superoxide-sensitive fluorophore, mitoSOX, in cardiomyocytes (Wang et al., 2008), skeletal muscle fibers (Pouvreau, 2010; Wei et al., 2011) and living animals (Fang et al., 2011). The reported flashes are not dependent on the transfer of Ca2+ from the SR to mitochondria. Nonetheless, the nature of flashes reported by cpYFP remains a subject of discussion, as they have also been attributed to pH fluctuations (Schwarzländer et al., 2012). Although the measurement of mitochondrial ROS in cells remains difficult, and, practically no measurement of SR ROS is available, recently introduced fluorescent protein-based H2O2 and glutathione reporters (such as HyPer, Grx1–roGFP) might facilitate reliable organelle-specific ROS measurements.
With regard to the specific targets of redox modifications, both RyR1 and RyR2 possess a number of reactive cysteine residues. Hyperactive cysteine residues that are susceptible to either S-nitrosylation or S-glutathionylations have been identified for RyR1 (Voss et al., 2004; Aracena-Parks et al., 2006). Modification of RyR1 by nitric oxide in a calmodulin-dependent manner has also been reported (Eu et al., 2000). Nitric oxide synthase was found localized to cardiac SR, and polynitrosylation was shown to induce progressive activation of RyR2 (Xu et al., 1998; Xu et al., 1999). SR/ER Ca2+ ATPase (SERCA) is another target of redox modifications. Oxidation of SERCA1 in skeletal muscle by peroxinitrite causes an attenuation of activity of the Ca2+ pump (Sharov et al., 2006). On the mitochondrial side, a vast body of evidence supports the activation of the PTP by ROS in cardiac muscle (Brookes et al., 2004; Halestrap et al., 2004). Thus, several key factors of the SR–mitochondrial Ca2+ transport are known to be subjected to redox modulation in cardiac and skeletal muscle. Additional redox targets are likely to be added to the list when the proteins that are redox-modulated in other tissues (e.g. VDAC; Madesh and Hajnóczky, 2001) or have just been identified (e.g. proteins forming the uniporter, such as MCU and MICU1) are tested in the muscle.
In cardiomyocytes, mitochondria-derived ROS have been implicated in the modulation of RyR2-mediated Ca2+ spark activity, supporting a local control of SR Ca2+ release by mitochondrial ROS (Yan et al., 2008; Zhou et al., 2011). The activity of NOX has also been linked to the regulation of RyR2-mediated Ca2+-induced Ca2+ release (Cherednichenko et al., 2004; Sánchez et al., 2005). In skeletal muscle, NOX has been localized at the TTs, a favorable position for it to perform redox modifications of RyR1 (Hidalgo et al., 2006). In addition, the NOX isoform 4 has been recently described as localizing to the SR and to co-immunoprecipitate with RyR1, thereby locally modulating its Ca2+ release activity (Sun et al., 2011). Taken together, the results described above indicate that ROS that are produced in the mitochondria and SR can exert local control of the Ca2+ transport by the SR. Owing to the spatial separation between RyR and mitochondria through the transverse tubules (TTs), SERCAs might become more accessible and thus more relevant at low levels of mitochondrial ROS production. However, although ROS production by the SR/ER has been established (Enyedi et al., 2010), its relevance for the regulation of mitochondrial Ca2+ transport needs further study.
Disease relevance of Ca2+ and ROS signaling at the SR/ER–mitochondria interface in cardiac and skeletal muscle
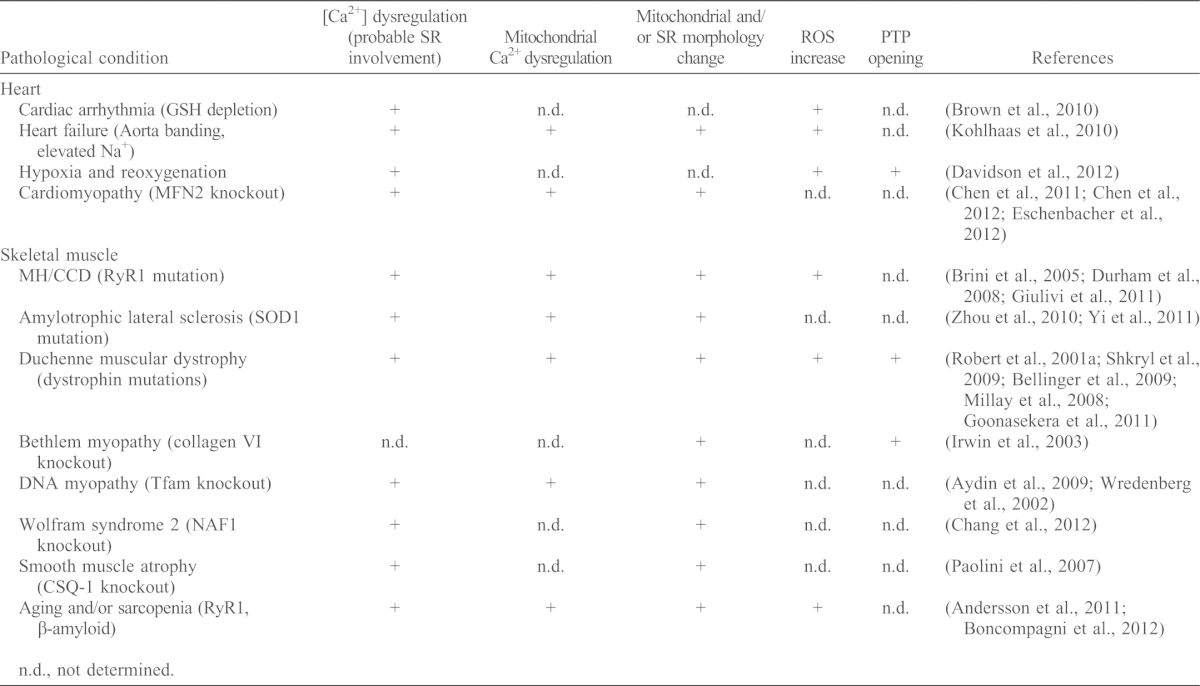
Despite the growing structural and functional evidence for SR–mitochondrial local communication, its relevance for muscle dysfunction has only recently been considered. This section reviews the scattered clues for the relevance of SR–mitochondria interactions in pathological models (summarized in Table 1).
Table 1. Cardiac and skeletal muscle disorders with a dysregulation of SR or mitochondria.
n.d., not determined.
Mitochondrial PTP opening as a consequence of hypoxia or reoxygenation has been recently described in perfused heart. Here, the PTP-opening-induced depolarization propagates from cell to cell and is preceded by Ca2+ and ROS waves that are sensitive to scavengers (Davidson et al., 2012). Ca2+ and ROS increases can be locally amplified by positive-feedback mechanisms at the SR–mitochondria associations (Fig. 4). In another line of investigation, conditional depletion of mitofusins has been reported to induce respiratory dysfunction of cardiomyocytes and progressive dilated cardiomyopathy (Chen et al., 2011). Depletion of MFN2, but not MFN1, causes the pacing-induced Ca2+ transients to become larger, which is accompanied by a decreased mitochondrial Ca2+ accumulation and oxidative response (Chen et al., 2012). As the SR–mitochondrial contact length is only decreased in the MFN2-deficient cells, cardiomyocyte dysfunction has been attributed to impaired SR–mitochondrial coupling rather than to a suppressed mitochondrial fusion activity (Chen et al., 2012).
Fig. 4.

Misregulation of SR/ER–mitochondrial communication as a potential source of muscle injury. (A) Under normal conditions, activation of RyR-mediated Ca2+ release is propagated to the mitochondria to activate oxidative metabolism. (B) Augmented Ca2+ release through increased RyR activity leads to a mitochondrial Ca2+ overload and the activation of the permeability transition pore (PTP). (C) Stress factors (e.g. ROS) sensitize (indicated by the red stars) either RyR-mediated Ca2+ release, or activation of the PTP by Ca2+. Prolonged PTP activation initiates mitochondrial membrane permeabilization, leading to dissipation of the mitochondrial membrane potential and release of apoptotic factors from the intermembrane space to the cytoplasm, which then can initiate cell death. MCU, mitochondrial Ca2+ uniporter.
The mitochondrial contribution to several genetic diseases has been tested in skeletal muscle tissue. For example, patients with malignant hyperthermia (MH) and central core disease (CCD) (Rosenberg et al., 2007; Treves et al., 2008) often show mutations in the RyR1, which leads to altered [Ca2+]c and [Ca2+]m transients (Brini et al., 2005). In the RyR1(Y522S) knock-in mouse model of MH, RyR1(Y522S) undergoes redox modifications due to elevated ROS and becomes leaky. These changes are accompanied by severely damaged and enlarged mitochondria (Durham et al., 2008). The mice present disrupted mitochondria early in development and muscles with extended contracture areas that lack SR or mitochondria (at two months of age) (Boncompagni et al., 2009). In another knock-in mouse model of MH, RyR1(R163C), bioenergetics defects, including elevated [Ca2+]m and ROS, and decreased oxidative phosphorylation have been reported (Giulivi et al., 2011). These results imply that there is a RyR1-mediated Ca2+ overload and subsequent mitochondrial injury in MH and CCD (Fig. 4). The altered Ca2+ homeostasis appears to facilitate ROS production, which, in turn, might exert important local feedback effects on RyR and other Ca2+ transport mechanisms.
Amylotrophic lateral sclerosis (ALS) is characterized by neuromuscular degeneration and muscle atrophy; 20% of the cases (familial ALS) are commonly associated with superoxide dismutase 1 (SOD1) mutations (Kunst, 2004). In skeletal muscle fibers derived from the SOD1(G93A) knock-in mouse model, depolarized mitochondria have been found in the area surrounding the neuromuscular junction (Zhou et al., 2010). Osmotic stress triggers Ca2+ release waves that are confined to the area of depolarized mitochondria. However, Ca2+ waves propagate throughout the muscle fibers upon inhibition of mitochondrial Ca2+ uptake by an uncoupler or Ru360 (Zhou et al., 2010). Furthermore, in the regions of depolarized mitochondria, [Ca2+]c signals that are associated with depolarization-induced excitation–contraction (EC) coupling are increased, whereas the amplitude of the [Ca2+]m transients is decreased (Yi et al., 2011). These data indicate that SOD1 mutations might cause mitochondrial injury that leads to a local dysregulation of EC coupling.
Studies of the mdx mouse model of Duchenne muscular dystrophy, which is characterized by loss-of-function mutations in the cytoskeleton-to-extracellular matrix-anchoring protein dystrophin, have shown elevated resting [Ca2+]c owing to leaky sarcolemma channels (Hopf et al., 1996). Elevated resting SR [Ca2+] and a larger decrease in response to membrane depolarization or caffeine, are associated with augmented [Ca2+]m transients in mdx skeletal muscle myotubes, demonstrating dysregulation of SR and mitochondrial Ca2+ signaling (Robert et al., 2001a). Furthermore, skeletal muscle fibers from mdx mice display elevated ROS levels that are produced by NOX, increased [Ca2+]c transients that lead to mitochondrial Ca2+ overload, and increased superoxide production (Shkryl et al., 2009). Another study also found nitrosylation of RyR1, dissociation of the stabilizing protein FKBP12 (calstabin) and a subsequent increase in spontaneous RyR1 activity and force reduction (Bellinger et al., 2009). The observed enhanced cell death in mdx skeletal muscle has been attributed to a mitochondrial Ca2+ overload and PTP opening (Millay et al., 2008). Furthermore, reduced SERCA activity has also been described in mdx muscles (Kargacin and Kargacin, 1996), and SERCA overexpression promotes an improvement of the dystrophic phenotype, including mitochondrial injury (Goonasekera et al., 2011). Although it is difficult to align these observations along a linear disease-causing pathway, they support the notion that enhanced SR–mitochondrial local Ca2+ transfer, Ca2+ overload-induced mitochondrial injury and ROS, which facilitate or induce these changes, all have an important role. In addition to the conditions described above, dysregulation of SR and mitochondrial Ca2+ signaling is implicated in several other genetic conditions and in aging (see Table 1). The cited examples also present a variety of adaptative or compensatory processes that might be associated with impaired function of the SR–mitochondrial interface.
A common pathogenic mechanism could be an amplification loop that is generated by the impairment of a component of the local Ca2+ or ROS signaling that triggers activation of the PTP and OMM permeabilization (Fig. 4). This amplification loop could be strengthened by means of ROS-induced ROS release, or Ca2+-induced Ca2+ release, that then propagate throughout the mitochondrial population (Ichas et al., 1997; Zorov et al., 2000; Pacher and Hajnóczky, 2001; Aon et al., 2003). A vast amount of evidence has already demonstrated the deleterious effects of mitochondrial Ca2+ overload, ROS generation and PTP opening during heart ischemia–reperfusion (Brookes et al., 2004; Halestrap et al., 2004; Csordás and Hajnóczky, 2009; Di Lisa et al., 2011; Rasola and Bernardi, 2011). However, a systematic evaluation of the same mechanism in other cardiac or skeletal muscle diseases has not yet been performed.
Concluding remarks
Ultrastructural studies combined with recent measurements of Ca2+ and ROS have provided evidence that the SR/ER is engaged in intimate structural and functional interactions with mitochondria in cardiac and skeletal muscle. In the muscle, but not in many other tissues, these interactions show a periodic spatial pattern and are likely to be relatively stable over time owing to the sarcomeric structure that is framed by the myofilaments. However, contractile activity might involve dynamic changes at the interface between SR/ER and mitochondria, which requires further studies. Furthermore, only a few clues are available with regard to the molecular composition of the SR/ER–mitochondrial interaction sites. At this point, several potential functions of the interface, such as phospholipid synthesis, apoptotic signaling, protein transfer and mitochondria fission, among others, remain untested in the muscle and, therefore, their participation in disease mechanisms is unclear. Identification of the proteins at the SR/ER–mitochondrial interface, for example, the tethers, will allow to specifically target the coupling and, in turn, might help to dissect its physiological and pathophysiological relevance. Novel technologies that enable the measurement of the functional changes at the specialized domains marking SR/ER–mitochondrial associations will help to elucidate the local transport and signaling mechanisms. Importantly, future studies also have to consider different types of skeletal muscle (e.g. fast twitch versus slow twitch) and the distinct properties of the different subsets of mitochondria in both cardiac and skeletal muscle. However, even on the basis of the current knowledge, it is safe to predict that the local coupling of mitochondria to the SR/ER will be found to be central to the function and dysfunction of the muscle, which is among the tissues most dependent on SR Ca2+ regulation and mitochondrial ATP production.
Acknowledgments
The authors thank David Weaver and Pal Pacher for acquiring some of the images shown in Fig. 1.
Footnotes
Funding
Our work in this area was supported by Pew Latin American Fellows Program in the Biomedical Sciences and Duchenne Parents Project to V.E.; and by the National Institutes of Health [grant number s DK051526 and RC2AA019416 to G.H.]. Deposited in PMC for release after 12 months.
References
- Andersson D. C., Betzenhauser M. J., Reiken S., Meli A. C., Umanskaya A., Xie W., Shiomi T., Zalk R., Lacampagne A., Marks A. R. (2011). Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 14, 196–207 10.1016/j.cmet.2011.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrienko T. N., Picht E., Bers D. M. (2009). Mitochondrial free calcium regulation during sarcoplasmic reticulum calcium release in rat cardiac myocytes. J. Mol. Cell. Cardiol. 46, 1027–1036 10.1016/j.yjmcc.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli T., Bergamelli L., Margittai E., Rimessi A., Fagioli C., Malgaroli A., Pinton P., Ripamonti M., Rizzuto R., Sitia R. (2012). Ero1α regulates Ca2+ fluxes at the endoplasmic reticulum-mitochondria interface (MAM). Antioxid. Redox Signal. 16, 1077–1087 10.1089/ars.2011.4004 [DOI] [PubMed] [Google Scholar]
- Anesti V., Scorrano L. (2006). The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta 1757, 692–699 10.1016/j.bbabio.2006.04.013 [DOI] [PubMed] [Google Scholar]
- Aon M. A., Cortassa S., Marbán E., O'Rourke B. (2003). Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 278, 44735–44744 10.1074/jbc.M302673200 [DOI] [PubMed] [Google Scholar]
- Aracena-Parks P., Goonasekera S. A., Gilman C. P., Dirksen R. T., Hidalgo C., Hamilton S. L. (2006). Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J. Biol. Chem. 281, 40354–40368 10.1074/jbc.M600876200 [DOI] [PubMed] [Google Scholar]
- Ardail D., Gasnier F., Lermé F., Simonot C., Louisot P., Gateau-Roesch O. (1993). Involvement of mitochondrial contact sites in the subcellular compartmentalization of phospholipid biosynthetic enzymes. J. Biol. Chem. 268, 25985–25992 [PubMed] [Google Scholar]
- Area-Gomez E., Del Carmen Lara Castillo M., Tambini M. D., Guardia-Laguarta C., de Groof A. J., Madra M., Ikenouchi J., Umeda M., Bird T. D., Sturley S. L. et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123 10.1038/emboj.2012.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin J., Andersson D. C., Hänninen S. L., Wredenberg A., Tavi P., Park C. B., Larsson N. G., Bruton J. D., Westerblad H. (2009). Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Hum. Mol. Genet. 18, 278–288 10.1093/hmg/ddn355 [DOI] [PubMed] [Google Scholar]
- Bell C. J., Bright N. A., Rutter G. A., Griffiths E. J. (2006). ATP regulation in adult rat cardiomyocytes: time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. J. Biol. Chem. 281, 28058–28067 10.1074/jbc.M604540200 [DOI] [PubMed] [Google Scholar]
- Bellinger A. M., Reiken S., Carlson C., Mongillo M., Liu X., Rothman L., Matecki S., Lacampagne A., Marks A. R. (2009). Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 15, 325–330 10.1038/nm.1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bionda C., Portoukalian J., Schmitt D., Rodriguez-Lafrasse C., Ardail D. (2004). Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J. 382, 527–533 10.1042/BJ20031819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning D., Patterson R. L., Sedaghat L., Glebova N. O., Kurosaki T., Snyder S. H. (2003). Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 5, 1051–1061 10.1038/ncb1063 [DOI] [PubMed] [Google Scholar]
- Boncompagni S., Rossi A. E., Micaroni M., Hamilton S. L., Dirksen R. T., Franzini-Armstrong C., Protasi F. (2009). Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc. Natl. Acad. Sci. USA 106, 21996–22001 10.1073/pnas.0911496106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompagni S., Moussa C. E., Levy E., Pezone M. J., Lopez J. R., Protasi F., Shtifman A. (2012). Mitochondrial dysfunction in skeletal muscle of amyloid precursor protein-overexpressing mice. J. Biol. Chem. 287, 20534–20544 10.1074/jbc.M112.359588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozidis P., Williamson C. D., Colberg-Poley A. M. (2008). Mitochondrial and secretory human cytomegalovirus UL37 proteins traffic into mitochondrion-associated membranes of human cells. J. Virol. 82, 2715–2726 10.1128/JVI.02456-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge D. G., Stojanovic M., Marcellus R. C., Shore G. C. (2003). Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 160, 1115–1127 10.1083/jcb.200212059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M., De Giorgi F., Murgia M., Marsault R., Massimino M. L., Cantini M., Rizzuto R., Pozzan T. (1997). Subcellular analysis of Ca2+ homeostasis in primary cultures of skeletal muscle myotubes. Mol. Biol. Cell 8, 129–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M., Manni S., Pierobon N., Du G. G., Sharma P., MacLennan D. H., Carafoli E. (2005). Ca2+ signaling in HEK-293 and skeletal muscle cells expressing recombinant ryanodine receptors harboring malignant hyperthermia and central core disease mutations. J. Biol. Chem. 280, 15380–15389 10.1074/jbc.M410421200 [DOI] [PubMed] [Google Scholar]
- Brookes P. S., Yoon Y., Robotham J. L., Anders M. W., Sheu S. S. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. 287, C817–C833 10.1152/ajpcell.00139.2004 [DOI] [PubMed] [Google Scholar]
- Brough D., Schell M. J., Irvine R. F. (2005). Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem. J. 392, 291–297 10.1042/BJ20050738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. A., Aon M. A., Frasier C. R., Sloan R. C., Maloney A. H., Anderson E. J., O'Rourke B. (2010). Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J. Mol. Cell. Cardiol. 48, 673–679 10.1016/j.yjmcc.2009.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui M., Gilady S. Y., Fitzsimmons R. E., Benson M. D., Lynes E. M., Gesson K., Alto N. M., Strack S., Scott J. D., Simmen T. (2010). Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J. Biol. Chem. 285, 31590–31602 10.1074/jbc.M110.101584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo C., Bolanos P. (2008). Effect of mitochondria poisoning by FCCP on Ca2+ signaling in mouse skeletal muscle fibers. Pflugers Arch. 455, 733–743 [DOI] [PubMed] [Google Scholar]
- Cárdenas C., Miller R. A., Smith I., Bui T., Molgó J., Müller M., Vais H., Cheung K. H., Yang J., Parker I. et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283 10.1016/j.cell.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqua C., Anesti V., Pyakurel A., Liu D., Naon D., Wiche G., Baffa R., Dimmer K. S., Scorrano L. (2010). Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep. 11, 854–860 10.1038/embor.2010.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D., Frank S., Rojo M. (2006). Mitochondrial dynamics in cell life and death. Cell Death Differ. 13, 680–684 10.1038/sj.cdd.4401857 [DOI] [PubMed] [Google Scholar]
- Chang N. C., Nguyen M., Bourdon J., Risse P. A., Martin J., Danialou G., Rizzuto R., Petrof B. J., Shore G. C. (2012). Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 21, 2277–2287 10.1093/hmg/dds048 [DOI] [PubMed] [Google Scholar]
- Chen H., Vermulst M., Wang Y. E., Chomyn A., Prolla T. A., McCaffery J. M., Chan D. C. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289 10.1016/j.cell.2010.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Liu Y., Dorn G. W., 2nd (2011). Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331 10.1161/CIRCRESAHA.111.258723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Csordás G., Jowdy C., Schneider T. G., Csordás N., Wang W., Liu Y., Kohlhaas M., Meiser M., Bergem S. et al. (2012). Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 111, 863–875 10.1161/CIRCRESAHA.112.266585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherednichenko G., Zima A. V., Feng W., Schaefer S., Blatter L. A., Pessah I. N. (2004). NADH oxidase activity of rat cardiac sarcoplasmic reticulum regulates calcium-induced calcium release. Circ. Res. 94, 478–486 10.1161/01.RES.0000115554.65513.7C [DOI] [PubMed] [Google Scholar]
- Chipuk J. E., McStay G. P., Bharti A., Kuwana T., Clarke C. J., Siskind L. J., Obeid L. M., Green D. R. (2012). Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 148, 988–1000 10.1016/j.cell.2012.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Hajnóczky G. (2009). SR/ER-mitochondrial local communication: calcium and ROS. Biochim. Biophys. Acta 1787, 1352–1362 10.1016/j.bbabio.2009.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Thomas A. P., Hajnóczky G. (1999). Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 18, 96–108 10.1093/emboj/18.1.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Renken C., Várnai P., Walter L., Weaver D., Buttle K. F., Balla T., Mannella C. A., Hajnóczky G. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 10.1083/jcb.200604016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G., Várnai P., Golenár T., Roy S., Purkins G., Schneider T. G., Balla T., Hajnóczky G. (2010). Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132 10.1016/j.molcel.2010.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S. M., Yellon D. M., Murphy M. P., Duchen M. R. (2012). Slow calcium waves and redox changes precede mitochondrial permeability transition pore opening in the intact heart during hypoxia and reoxygenation. Cardiovasc. Res. 93, 445–453 10.1093/cvr/cvr349 [DOI] [PubMed] [Google Scholar]
- de Brito O. M., Scorrano L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 10.1038/nature07534 [DOI] [PubMed] [Google Scholar]
- De Marchi U., Castelbou C., Demaurex N. (2011). Uncoupling protein 3 (UCP3) modulates the activity of Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing mitochondrial ATP production. J. Biol. Chem. 286, 32533–32541 10.1074/jbc.M110.216044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedkova E. N., Blatter L. A. (2008). Mitochondrial Ca2+ and the heart. Cell Calcium 44, 77–91 10.1016/j.ceca.2007.11.002 [DOI] [PubMed] [Google Scholar]
- Di Lisa F., Carpi A., Giorgio V., Bernardi P. (2011). The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim. Biophys. Acta 1813, 1316–1322 10.1016/j.bbamcr.2011.01.031 [DOI] [PubMed] [Google Scholar]
- Drago I., De Stefani D., Rizzuto R., Pozzan T. (2012). Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 109, 12986–12991 10.1073/pnas.1210718109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen M. R. (2004). Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol. Aspects Med. 25, 365–451 10.1016/j.mam.2004.03.001 [DOI] [PubMed] [Google Scholar]
- Duchen M. R., Leyssens A., Crompton M. (1998). Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J. Cell Biol. 142, 975–988 10.1083/jcb.142.4.975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhunty A. F. (2006). Excitation-contraction coupling from the 1950s into the new millennium. Clin. Exp. Pharmacol. Physiol. 33, 763–772 10.1111/j.1440-1681.2006.04441.x [DOI] [PubMed] [Google Scholar]
- Durham W. J., Aracena-Parks P., Long C., Rossi A. E., Goonasekera S. A., Boncompagni S., Galvan D. L., Gilman C. P., Baker M. R., Shirokova N. et al. (2008). RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell 133, 53–65 10.1016/j.cell.2008.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi B., Várnai P., Geiszt M. (2010). Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal. 13, 721–729 10.1089/ars.2009.2880 [DOI] [PubMed] [Google Scholar]
- Eschenbacher W. H., Song M., Chen Y., Bhandari P., Zhao P., Jowdy C. C., Engelhard J. T., Dorn G. W., 2nd (2012). Two rare human mitofusin 2 mutations alter mitochondrial dynamics and induce retinal and cardiac pathology in Drosophila. PLoS ONE 7, e44296 10.1371/journal.pone.0044296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eu J. P., Sun J., Xu L., Stamler J. S., Meissner G. (2000). The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell 102, 499–509 10.1016/S0092-8674(00)00054-4 [DOI] [PubMed] [Google Scholar]
- Fang H., Chen M., Ding Y., Shang W., Xu J., Zhang X., Zhang W., Li K., Xiao Y., Gao F. et al. (2011). Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res. 21, 1295–1304 10.1038/cr.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieni F., Lee S. B., Jan Y. N., Kirichok Y. (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 3, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson A., Ruusala A., Aspenström P. (2003). Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J. Biol. Chem. 278, 6495–6502 10.1074/jbc.M208609200 [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C., Boncompagni S. (2011). The evolution of the mitochondria-to-calcium release units relationship in vertebrate skeletal muscles. J. Biomed. Biotechnol. 2011, 830573 10.1155/2011/830573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C., Protasi F., Ramesh V. (1998). Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann. N. Y. Acad. Sci. 853, 20–30 10.1111/j.1749-6632.1998.tb08253.x [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C., Protasi F., Tijskens P. (2005). The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 1047, 76–85 10.1196/annals.1341.007 [DOI] [PubMed] [Google Scholar]
- Friedman J. R., Webster B. M., Mastronarde D. N., Verhey K. J., Voeltz G. K. (2010). ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 190, 363–375 10.1083/jcb.200911024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J. R., Lackner L. L., West M., DiBenedetto J. R., Nunnari J., Voeltz G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362 10.1126/science.1207385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M., Hayashi T. (2011). New insights into the role of mitochondria-associated endoplasmic reticulum membrane. Int. Rev. Cell Mol. Biol. 292, 73–117 [DOI] [PubMed] [Google Scholar]
- Gaigg B., Simbeni R., Hrastnik C., Paltauf F., Daum G. (1995). Characterization of a microsomal subfraction associated with mitochondria of the yeast, Saccharomyces cerevisiae. Involvement in synthesis and import of phospholipids into mitochondria. Biochim. Biophys. Acta 1234, 214–220 10.1016/0005-2736(94)00287-Y [DOI] [PubMed] [Google Scholar]
- García-Pérez C., Hajnóczky G., Csordás G. (2008). Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J. Biol. Chem. 283, 32771–32780 10.1074/jbc.M803385200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Pérez C., Schneider T. G., Hajnóczky G., Csordás G. (2011). Alignment of sarcoplasmic reticulum-mitochondrial junctions with mitochondrial contact points. Am. J. Physiol. 301, H1907–H1915 10.1152/ajpheart.00397.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez C., Roy S. S., Naghdi S., Lin X., Davies E., Hajnóczky G. (2012). Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc. Natl. Acad. Sci. USA 109, 4497–4502 10.1073/pnas.1118244109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomello M., Drago I., Bortolozzi M., Scorzeto M., Gianelle A., Pizzo P., Pozzan T. (2010). Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 38, 280–290 10.1016/j.molcel.2010.04.003 [DOI] [PubMed] [Google Scholar]
- Gilady S. Y., Bui M., Lynes E. M., Benson M. D., Watts R., Vance J. E., Simmen T. (2010). Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 15, 619–629 10.1007/s12192-010-0174-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg K. S., Bers D. M. (2004). Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J. Physiol. 556, 463–480 10.1113/jphysiol.2003.055384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C., Ito K., Lin H. K., Santangelo C., Wieckowski M. R., Lebiedzinska M., Bononi A., Bonora M., Duszynski J., Bernardi R. et al. (2010). PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330, 1247–1251 10.1126/science.1189157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulivi C., Ross-Inta C., Omanska-Klusek A., Napoli E., Sakaguchi D., Barrientos G., Allen P. D., Pessah I. N. (2011). Basal bioenergetic abnormalities in skeletal muscle from ryanodine receptor malignant hyperthermia-susceptible R163C knock-in mice. J. Biol. Chem. 286, 99–113 10.1074/jbc.M110.153247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz J. G., Genty H., St-Pierre P., Dang T., Joshi B., Sauvé R., Vogl W., Nabi I. R. (2007). Reversible interactions between smooth domains of the endoplasmic reticulum and mitochondria are regulated by physiological cytosolic Ca2+ levels. J. Cell Sci. 120, 3553–3564 10.1242/jcs.03486 [DOI] [PubMed] [Google Scholar]
- Goonasekera S. A., Lam C. K., Millay D. P., Sargent M. A., Hajjar R. J., Kranias E. G., Molkentin J. D. (2011). Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest. 121, 1044–1052 10.1172/JCI43844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey D. W., Rambold A. S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P. K., Lippincott-Schwartz J. (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667 10.1016/j.cell.2010.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnóczky G., Robb-Gaspers L. D., Seitz M. B., Thomas A. P. (1995). Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424 10.1016/0092-8674(95)90430-1 [DOI] [PubMed] [Google Scholar]
- Hajnóczky G., Hager R., Thomas A. P. (1999). Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+. J. Biol. Chem. 274, 14157–14162 10.1074/jbc.274.20.14157 [DOI] [PubMed] [Google Scholar]
- Halestrap A. P., Clarke S. J., Javadov S. A. (2004). Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc. Res. 61, 372–385 10.1016/S0008-6363(03)00533-9 [DOI] [PubMed] [Google Scholar]
- Hayashi T., Su T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610 10.1016/j.cell.2007.08.036 [DOI] [PubMed] [Google Scholar]
- Hayashi T., Martone M. E., Yu Z., Thor A., Doi M., Holst M. J., Ellisman M. H., Hoshijima M. (2009). Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J. Cell Sci. 122, 1005–1013 10.1242/jcs.028175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo C., Sánchez G., Barrientos G., Aracena-Parks P. (2006). A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. J. Biol. Chem. 281, 26473–26482 10.1074/jbc.M600451200 [DOI] [PubMed] [Google Scholar]
- Hopf F. W., Turner P. R., Denetclaw W. F., Jr, Reddy P., Steinhardt R. A. (1996). A critical evaluation of resting intracellular free calcium regulation in dystrophic mdx muscle. Am. J. Physiol. 271, C1325–C1339 [DOI] [PubMed] [Google Scholar]
- Ichas F., Jouaville L. S., Mazat J. P. (1997). Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89, 1145–1153 10.1016/S0092-8674(00)80301-3 [DOI] [PubMed] [Google Scholar]
- Irwin W. A., Bergamin N., Sabatelli P., Reggiani C., Megighian A., Merlini L., Braghetta P., Columbaro M., Volpin D., Bressan G. M. et al. (2003). Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 35, 367–371 10.1038/ng1270 [DOI] [PubMed] [Google Scholar]
- Isaeva E. V., Shkryl V. M., Shirokova N. (2005). Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J. Physiol. 565, 855–872 10.1113/jphysiol.2005.086280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasawa R., Mahul-Mellier A. L., Datler C., Pazarentzos E., Grimm S. (2011). Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 30, 556–568 10.1038/emboj.2010.346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville L. S., Ichas F., Holmuhamedov E. L., Camacho P., Lechleiter J. D. (1995). Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature 377, 438–441 10.1038/377438a0 [DOI] [PubMed] [Google Scholar]
- Jouaville L. S., Pinton P., Bastianutto C., Rutter G. A., Rizzuto R. (1999). Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 96, 13807–13812 10.1073/pnas.96.24.13807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargacin M. E., Kargacin G. J. (1996). The sarcoplasmic reticulum calcium pump is functionally altered in dystrophic muscle. Biochim. Biophys. Acta 1290, 4–8 10.1016/0304-4165(95)00180-8 [DOI] [PubMed] [Google Scholar]
- Kettlewell S., Cabrero P., Nicklin S. A., Dow J. A., Davies S., Smith G. L. (2009). Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. J. Mol. Cell. Cardiol. 46, 891–901 10.1016/j.yjmcc.2009.02.016 [DOI] [PubMed] [Google Scholar]
- Kohlhaas M., Liu T., Knopp A., Zeller T., Ong M. F., Böhm M., O'Rourke B., Maack C. (2010). Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121, 1606–1613 10.1161/CIRCULATIONAHA.109.914911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B., Currie E., Collins S. R., Schuldiner M., Nunnari J., Weissman J. S., Walter P. (2009). An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481 10.1126/science.1175088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B., Osman C., Walter P. (2011). The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc. Natl. Acad. Sci. USA 108, 14151–14156 10.1073/pnas.1111314108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst C. B. (2004). Complex genetics of amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 933–947 10.1086/426001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland C. G., Andersson S. G. (2000). Origin and evolution of the mitochondrial proteome. Microbiol. Mol. Biol. Rev. 64, 786–820 10.1128/MMBR.64.4.786-820.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolfi B., Curci S., Debellis L., Pozzan T., Hofer A. M. (1998). Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured In situ in intact cells. J. Cell Biol. 142, 1235–1243 10.1083/jcb.142.5.1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartigue L., Medina C., Schembri L., Chabert P., Zanese M., Tomasello F., Dalibart R., Thoraval D., Crouzet M., Ichas F. et al. (2008). An intracellular wave of cytochrome c propagates and precedes Bax redistribution during apoptosis. J. Cell Sci. 121, 3515–3523 10.1242/jcs.029587 [DOI] [PubMed] [Google Scholar]
- Liu X., Weaver D., Shirihai O., Hajnóczky G. (2009). Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 28, 3074–3089 10.1038/emboj.2009.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Ginsburg K. S., Kettlewell S., Bossuyt J., Smith G. L., Bers D. M. (2013). Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ. Res. 112, 424–431 10.1161/CIRCRESAHA.111.300501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko V., Ziman A., Lukyanenko A., Salnikov V., Lederer W. J. (2007). Functional groups of ryanodine receptors in rat ventricular cells. J. Physiol. 583, 251–269 10.1113/jphysiol.2007.136549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes E. M., Simmen T. (2011). Urban planning of the endoplasmic reticulum (ER): how diverse mechanisms segregate the many functions of the ER. Biochim. Biophys. Acta 1813, 1893–1905 10.1016/j.bbamcr.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madesh M., Hajnóczky G. (2001). VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 155, 1003–1015 10.1083/jcb.200105057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S., Rimessi A., Giorgi C., Baldini C., Ferroni L., Rizzuto R., Pinton P. (2008). Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem. Biophys. Res. Commun. 375, 501–505 10.1016/j.bbrc.2008.07.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlib M. A., Zhou Z., Knight S., Ahmed S., Choi K. M., Krause-Bauer J., Phillips R., Altschuld R., Katsube Y., Sperelakis N. et al. (1998). Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J. Biol. Chem. 273, 10223–10231 10.1074/jbc.273.17.10223 [DOI] [PubMed] [Google Scholar]
- McBride H. M., Neuspiel M., Wasiak S. (2006). Mitochondria: more than just a powerhouse. Curr. Biol. 16, R551–R560 [DOI] [PubMed] [Google Scholar]
- McCormack J. G., Denton R. M. (1989). The role of Ca2+ ions in the regulation of intramitochondrial metabolism and energy production in rat heart. Mol. Cell. Biochem. 89, 121–125 10.1007/BF00220763 [DOI] [PubMed] [Google Scholar]
- Millay D. P., Sargent M. A., Osinska H., Baines C. P., Barton E. R., Vuagniaux G., Sweeney H. L., Robbins J., Molkentin J. D. (2008). Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 14, 442–447 10.1038/nm1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner D. J., Mavroidis M., Weisleder N., Capetanaki Y. (2000). Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 150, 1283–1298 10.1083/jcb.150.6.1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min C. K., Yeom D. R., Lee K. E., Kwon H. K., Kang M., Kim Y. S., Park Z. Y., Jeon H., Kim H. (2012). Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem. J. 447, 371–379 10.1042/BJ20120705 [DOI] [PubMed] [Google Scholar]
- Mitchell R. D., Palade P., Fleischer S. (1983). Purification of morphologically intact triad structures from skeletal muscle. J. Cell Biol. 96, 1008–1016 10.1083/jcb.96.4.1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morré D. J., Merritt W. D., Lembi C. A. (1971). Connections between mitochondria and endoplasmic reticulum in rat liver and onion stem. Protoplasma 73, 43–49 10.1007/BF01286410 [DOI] [PubMed] [Google Scholar]
- Myhill N., Lynes E. M., Nanji J. A., Blagoveshchenskaya A. D., Fei H., Carmine Simmen K., Cooper T. J., Thomas G., Simmen T. (2008). The subcellular distribution of calnexin is mediated by PACS-2. Mol. Biol. Cell 19, 2777–2788 10.1091/mbc.E07-10-0995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. T., Lewandowska A., Choi J. Y., Markgraf D. F., Junker M., Bilgin M., Ejsing C. S., Voelker D. R., Rapoport T. A., Shaw J. M. (2012). Gem1 and ERMES do not directly affect phosphatidylserine transport from ER to mitochondria or mitochondrial inheritance. Traffic 13, 880–890 10.1111/j.1600-0854.2012.01352.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novgorodov S. A., Wu B. X., Gudz T. I., Bielawski J., Ovchinnikova T. V., Hannun Y. A., Obeid L. M. (2011). Novel pathway of ceramide production in mitochondria: thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA. J. Biol. Chem. 286, 25352–25362 10.1074/jbc.M110.214866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata H., Chacon E., Tesfai S. A., Harper I. S., Herman B., Lemasters J. J. (1998). Mitochondrial Ca2+ transients in cardiac myocytes during the excitation-contraction cycle: effects of pacing and hormonal stimulation. J. Bioenerg. Biomembr. 30, 207–222 10.1023/A:1020588618496 [DOI] [PubMed] [Google Scholar]
- Pacher P., Hajnóczky G. (2001). Propagation of the apoptotic signal by mitochondrial waves. EMBO J. 20, 4107–4121 10.1093/emboj/20.15.4107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P., Csordás P., Schneider T., Hajnóczky G. (2000). Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J. Physiol. 529, 553–564 10.1111/j.1469-7793.2000.00553.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P., Thomas A. P., Hajnóczky G. (2002). Ca2+ marks: miniature calcium signals in single mitochondria driven by ryanodine receptors. Proc. Natl. Acad. Sci. USA 99, 2380–2385 10.1073/pnas.032423699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolini C., Quarta M., Nori A., Boncompagni S., Canato M., Volpe P., Allen P. D., Reggiani C., Protasi F. (2007). Reorganized stores and impaired calcium handling in skeletal muscle of mice lacking calsequestrin-1. J. Physiol. 583, 767–784 10.1113/jphysiol.2007.138024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanicolaou K. N., Khairallah R. J., Ngoh G. A., Chikando A., Luptak I., O'Shea K. M., Riley D. D., Lugus J. J., Colucci W. S., Lederer W. J. et al. (2011). Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 31, 1309–1328 10.1128/MCB.00911-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F., Gohil V. M., Girgis H. S., Bao X. R., McCombs J. E., Palmer A. E., Mootha V. K. (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467, 291–296 10.1038/nature09358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P., Ferrari D., Rapizzi E., Di Virgilio F., Pozzan T., Rizzuto R. (2001). The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20, 2690–2701 10.1093/emboj/20.11.2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouvreau S. (2010). Superoxide flashes in mouse skeletal muscle are produced by discrete arrays of active mitochondria operating coherently. PLoS ONE 5, e13035 10.1371/journal.pone.0013035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapizzi E., Pinton P., Szabadkai G., Wieckowski M. R., Vandecasteele G., Baird G., Tuft R. A., Fogarty K. E., Rizzuto R. (2002). Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624 10.1083/jcb.200205091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A., Bernardi P. (2011). Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 50, 222–233 10.1016/j.ceca.2011.04.007 [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Brini M., Murgia M., Pozzan T. (1993). Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262, 744–747 10.1126/science.8235595 [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Pinton P., Carrington W., Fay F. S., Fogarty K. E., Lifshitz L. M., Tuft R. A., Pozzan T. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 10.1126/science.280.5370.1763 [DOI] [PubMed] [Google Scholar]
- Robert V., Massimino M. L., Tosello V., Marsault R., Cantini M., Sorrentino V., Pozzan T. (2001a). Alteration in calcium handling at the subcellular level in mdx myotubes. J. Biol. Chem. 276, 4647–4651 10.1074/jbc.M006337200 [DOI] [PubMed] [Google Scholar]
- Robert V., Gurlini P., Tosello V., Nagai T., Miyawaki A., Di Lisa F., Pozzan T. (2001b). Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 20, 4998–5007 10.1093/emboj/20.17.4998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers K. L., Picaud S., Roncali E., Boisgard R., Colasante C., Stinnakre J., Tavitian B., Brûlet P. (2007). Non-invasive in vivo imaging of calcium signaling in mice. PLoS ONE 2, e974 10.1371/journal.pone.0000974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg H., Davis M., James D., Pollock N., Stowell K. (2007). Malignant hyperthermia. Orphanet J. Rare Dis. 2, 21 10.1186/1750-1172-2-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland A. A., Voeltz G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625 10.1038/nrm3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf R., Mongillo M., Magalhães P. J., Pozzan T. (2004). In vivo monitoring of Ca(2+) uptake into mitochondria of mouse skeletal muscle during contraction. J. Cell Biol. 166, 527–536 10.1083/jcb.200403102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salnikov V., Lukyanenko Y. O., Lederer W. J., Lukyanenko V. (2009). Distribution of ryanodine receptors in rat ventricular myocytes. J. Muscle Res. Cell Motil. 30, 161–170 10.1007/s10974-009-9186-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez G., Pedrozo Z., Domenech R. J., Hidalgo C., Donoso P. (2005). Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J. Mol. Cell. Cardiol. 39, 982–991 10.1016/j.yjmcc.2005.08.010 [DOI] [PubMed] [Google Scholar]
- Sano R., Annunziata I., Patterson A., Moshiach S., Gomero E., Opferman J., Forte M., d'Azzo A. (2009). GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 36, 500–511 10.1016/j.molcel.2009.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton W. M., Hollenbeck P. J. (2012). The axonal transport of mitochondria. J. Cell Sci. 125, 2095–2104 10.1242/jcs.053850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzländer M., Murphy M. P., Duchen M. R., Logan D. C., Fricker M. D., Halestrap A. P., Müller F. L., Rizzuto R., Dick T. P., Meyer A. J. et al. (2012). Mitochondrial ‘flashes’: a radical concept repHined. Trends Cell Biol. 22, 503–508 10.1016/j.tcb.2012.07.007 [DOI] [PubMed] [Google Scholar]
- Sedova M., Dedkova E. N., Blatter L. A. (2006). Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am. J. Physiol. 291, C840–C850 10.1152/ajpcell.00619.2005 [DOI] [PubMed] [Google Scholar]
- Sharma V. K., Ramesh V., Franzini-Armstrong C., Sheu S. S. (2000). Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenerg. Biomembr. 32, 97–104 10.1023/A:1005520714221 [DOI] [PubMed] [Google Scholar]
- Sharov V. S., Dremina E. S., Galeva N. A., Williams T. D., Schöneich C. (2006). Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem. J. 394, 605–615 10.1042/BJ20051214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkryl V. M., Shirokova N. (2006). Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. J. Biol. Chem. 281, 1547–1554 10.1074/jbc.M505024200 [DOI] [PubMed] [Google Scholar]
- Shkryl V. M., Martins A. S., Ullrich N. D., Nowycky M. C., Niggli E., Shirokova N. (2009). Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Archiv. Eur. J. Physiol. 458, 915–928 [DOI] [PubMed] [Google Scholar]
- Shore G. C., Tata J. R. (1977). Two fractions of rough endoplasmic reticulum from rat liver. I. Recovery of rapidly sedimenting endoplasmic reticulum in association with mitochondria. J. Cell Biol. 72, 714–725 10.1083/jcb.72.3.714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X., Lev-Ram V., Deerinck T. J., Qi Y., Ramko E. B., Davidson M. W., Jin Y., Ellisman M. H., Tsien R. Y. (2011). A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 9, e1001041 10.1371/journal.pbio.1001041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmen T., Aslan J. E., Blagoveshchenskaya A. D., Thomas L., Wan L., Xiang Y., Feliciangeli S. F., Hung C. H., Crump C. M., Thomas G. (2005). PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 24, 717–729 10.1038/sj.emboj.7600559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson P. B., Russell J. T. (1996). Mitochondria support inositol 1,4,5-trisphosphate-mediated Ca2+ waves in cultured oligodendrocytes. J. Biol. Chem. 271, 33493–33501 10.1074/jbc.271.52.33493 [DOI] [PubMed] [Google Scholar]
- Siskind L. J., Kolesnick R. N., Colombini M. (2002). Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 277, 26796–26803 10.1074/jbc.M200754200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spät A., Szanda G., Csordás G., Hajnóczky G. (2008). High- and low-calcium-dependent mechanisms of mitochondrial calcium signalling. Cell Calcium 44, 51–63 10.1016/j.ceca.2007.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiban J., Caputo L., Colombini M. (2008). Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 49, 625–634 10.1194/jlr.M700480-JLR200 [DOI] [PubMed] [Google Scholar]
- Sun Q. A., Hess D. T., Nogueira L., Yong S., Bowles D. E., Eu J., Laurita K. R., Meissner G., Stamler J. S. (2011). Oxygen-coupled redox regulation of the skeletal muscle ryanodine receptor-Ca2+ release channel by NADPH oxidase 4. Proc. Natl. Acad. Sci. USA 108, 16098–16103 10.1073/pnas.1109546108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G., Bianchi K., Várnai P., De Stefani D., Wieckowski M. R., Cavagna D., Nagy A. I., Balla T., Rizzuto R. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911 10.1083/jcb.200608073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G., Krishnamurthy R., Hajnóczky G. (1999). Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 18, 6349–6361 10.1093/emboj/18.22.6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G., Csordás G., Hantash B. M., Thomas A. P., Hajnóczky G. (2000). Calcium signal transmission between ryanodine receptors and mitochondria. J. Biol. Chem. 275, 15305–15313 10.1074/jbc.275.20.15305 [DOI] [PubMed] [Google Scholar]
- Territo P. R., French S. A., Dunleavy M. C., Evans F. J., Balaban R. S. (2001). Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, AND light scattering. J. Biol. Chem. 276, 2586–2599 10.1074/jbc.M002923200 [DOI] [PubMed] [Google Scholar]
- Treves S., Jungbluth H., Muntoni F., Zorzato F. (2008). Congenital muscle disorders with cores: the ryanodine receptor calcium channel paradigm. Curr. Opin. Pharmacol. 8, 319–326 10.1016/j.coph.2008.01.005 [DOI] [PubMed] [Google Scholar]
- Trollinger D. R., Cascio W. E., Lemasters J. J. (1997). Selective loading of Rhod 2 into mitochondria shows mitochondrial Ca2+ transients during the contractile cycle in adult rabbit cardiac myocytes. Biochem. Biophys. Res. Commun. 236, 738–742 10.1006/bbrc.1997.7042 [DOI] [PubMed] [Google Scholar]
- Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G. et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 10.1038/sj.emboj.7601963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256 [PubMed] [Google Scholar]
- Voelker D. R. (1989). Reconstitution of phosphatidylserine import into rat liver mitochondria. J. Biol. Chem. 264, 8019–8025 [PubMed] [Google Scholar]
- Voss A. A., Lango J., Ernst-Russell M., Morin D., Pessah I. N. (2004). Identification of hyperreactive cysteines within ryanodine receptor type 1 by mass spectrometry. J. Biol. Chem. 279, 34514–34520 10.1074/jbc.M404290200 [DOI] [PubMed] [Google Scholar]
- Wang H. J., Guay G., Pogan L., Sauvé R., Nabi I. R. (2000). Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J. Cell Biol. 150, 1489–1498 10.1083/jcb.150.6.1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Fang H., Groom L., Cheng A., Zhang W., Liu J., Wang X., Li K., Han P., Zheng M. et al. (2008). Superoxide flashes in single mitochondria. Cell 134, 279–290 10.1016/j.cell.2008.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L., Salahura G., Boncompagni S., Kasischke K. A., Protasi F., Sheu S. S., Dirksen R. T. (2011). Mitochondrial superoxide flashes: metabolic biomarkers of skeletal muscle activity and disease. FASEB J. 25, 3068–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wredenberg A., Wibom R., Wilhelmsson H., Graff C., Wiener H. H., Burden S. J., Oldfors A., Westerblad H., Larsson N. G. (2002). Increased mitochondrial mass in mitochondrial myopathy mice. Proc. Natl. Acad. Sci. USA 99, 15066–15071 10.1073/pnas.232591499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Eu J. P., Meissner G., Stamler J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237 10.1126/science.279.5348.234 [DOI] [PubMed] [Google Scholar]
- Xu K. Y., Huso D. L., Dawson T. M., Bredt D. S., Becker L. C. (1999). Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA 96, 657–662 10.1073/pnas.96.2.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y., Liu J., Wei C., Li K., Xie W., Wang Y., Cheng H. (2008). Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 77, 432–441 10.1093/cvr/cvm047 [DOI] [PubMed] [Google Scholar]
- Yi M., Weaver D., Hajnóczky G. (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167, 661–672 10.1083/jcb.200406038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J., Ma C., Li Y., Weisleder N., Ríos E., Ma J., Zhou J. (2011). Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J. Biol. Chem. 286, 32436–32443 10.1074/jbc.M110.217711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M., Weaver D., Eisner V., Várnai P., Hunyady L., Ma J., Csordás G., Hajnóczky G. (2012). Switch from ER-mitochondrial to SR-mitochondrial calcium coupling during muscle differentiation. Cell Calcium 52, 355–365 10.1016/j.ceca.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Yi J., Fu R., Liu E., Siddique T., Ríos E., Deng H. X. (2010). Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 285, 705–712 10.1074/jbc.M109.041319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L., Aon M. A., Liu T., O'Rourke B. (2011). Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J. Mol. Cell. Cardiol. 51, 632–639 10.1016/j.yjmcc.2011.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov D. B., Filburn C. R., Klotz L. O., Zweier J. L., Sollott S. J. (2000). Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192, 1001–1014 10.1084/jem.192.7.1001 [DOI] [PMC free article] [PubMed] [Google Scholar]