

Background: This study aimed to elucidate the structure and function of HNTX-III.
Results: HNTX-III adopts the inhibitor cystine knot motif and inhibits Nav1.7 activation through binding to site 4 in the closed state.
Conclusion: HNTX-III is distinct from other β-toxins in its action mechanism, specificity, and affinity.
Significance: HNTX-III is a useful tool for studying toxin-VGSC interaction and a potential prototype analgesic.
Keywords: NMR, Patch Clamp Electrophysiology, Peptides, Sodium Channels, Toxins, Nav1.7, Inhibitor Cystine Knot Motif, Spider Venom
Abstract
In the present study, we investigated the structure and function of hainantoxin-III (HNTX-III), a 33-residue polypeptide from the venom of the spider Ornithoctonus hainana. It is a selective antagonist of neuronal tetrodotoxin-sensitive voltage-gated sodium channels. HNTX-III suppressed Nav1.7 current amplitude without significantly altering the activation, inactivation, and repriming kinetics. Short extreme depolarizations partially activated the toxin-bound channel, indicating voltage-dependent inhibition of HNTX-III. HNTX-III increased the deactivation of the Nav1.7 current after extreme depolarizations. The HNTX-III·Nav1.7 complex was gradually dissociated upon prolonged strong depolarizations in a voltage-dependent manner, and the unbound toxin rebound to Nav1.7 after a long repolarization. Moreover, analysis of chimeric channels showed that the DIIS3-S4 linker was critical for HNTX-III binding to Nav1.7. These data are consistent with HNTX-III interacting with Nav1.7 site 4 and trapping the domain II voltage sensor in the closed state. The solution structure of HNTX-III was determined by two-dimensional NMR and shown to possess an inhibitor cystine knot motif. Structural analysis indicated that certain basic, hydrophobic, and aromatic residues mainly localized in the C terminus may constitute an amphiphilic surface potentially involved in HNTX-III binding to Nav1.7. Taken together, our results show that HNTX-III is distinct from β-scorpion toxins and other β-spider toxins in its mechanism of action and binding specificity and affinity. The present findings contribute to our understanding of the mechanism of toxin-sodium channel interaction and provide a useful tool for the investigation of the structure and function of sodium channel isoforms and for the development of analgesics.
Introduction
Voltage-gated sodium channels (VGSCs)5 are essential for the initiation and propagation of action potentials in excitable tissues, such as nerves and muscles. Mutations in VGSC proteins have been associated with several diseases in humans (1–7). All VGSCs are composed of a pore-forming α-subunit and two auxiliary β-subunits. To date, nine α-subunits (Nav1.1–Nav1.9, also referred as channels) and four β-subunits (β1–β4) have been identified in mammals (1, 3). The nine isoforms of VGSCs are classified into two groups according to their sensitivity to tetrodoxin (TTX); Nav1.1–1.4, Nav1.6, and Nav1.7 are sensitive to nanomolar concentrations of TTX (TTX-S), whereas Nav1.5, Nav1.8, and Nav1.9 are resistant (TTX-R) (1, 2). Of the nine isoforms of VGSCs, Nav1.7–1.9 play specialized roles in nociceptive pathways and are considered as useful targets for the development of analgesic drugs (3–7).
Neurotoxins, including TTX, μ-conotoxins, α- and β-scorpion toxins, sea anemone toxins, and β-spider toxins, interact with VGSCs, blocking sodium currents or modulating the gating properties of these channels (8–13). To date, six different neurotoxin receptor sites (sites 1–6) have been identified on VGSCs. Different neurotoxins bind to one of the six receptor sites and therefore have distinct effects on VGSCs. Certain neurotoxins target VGSCs with high affinity (at nanomolar potency) and selectivity (subtype-selective) and have been used to isolate VGSCs and to explore their structure and function. Moreover, neurotoxins have been used to develop therapeutic drugs that selectively target certain VGSC isoforms (8–13). To date, more than 100 neurotoxins acting on VGSCs have been identified from venomous animals, such as scorpions, snails, spiders, and sea anemones. However, only a small proportion of toxins have been intensively investigated, which has limited their potential applications. Therefore, a comprehensive analysis of the structure and function of specific toxins affecting VGSCs is still necessary (8–13).
Spider venoms are a rich source of diverse bioactive peptides affecting VGSCs and are therefore potential candidates for therapeutic drug development (14, 15). However, compared with conotoxins and scorpion toxins, relatively few spider toxins with activity against VGSCs have been identified, and for most of them, the precise pattern of VGSC subtype selectivity either is unknown or has not been fully elucidated. The mechanisms by which spider toxins interact with VGSCs have only recently become the topic of research (9). Therefore, the identification and characterization of novel spider peptide toxins affecting VGSCs are expected to expand our understanding of the mechanisms of their interaction and provide new templates for drug development.
We previously identified a 33-residue peptide named hainantoxin-III (HNTX-III), isolated from the venom of the spider Ornithoctonus hainana. This peptide potently inhibited TTX-S VGSCs in rat dorsal root ganglion (DRG) cells, whereas it had no effect on TTX-R VGSCs. In our previous study, we showed that HNTX-III is a novel spider toxin, and its effect on mammalian neural Na+ channels is mediated by a mechanism different from that of other spider toxins targeting the neural receptor site 3, such as δ-aractoxins and μ-agatoxins (16). In the present study, we elucidated the solution structure and subtype selectivity of HNTX-III and showed that HNTX-III inhibits Nav1.7 activation in a manner similar to HWTX-IV, which binds to site 4 of Nav1.7, trapping the voltage sensor in the closed state (17–20). This mechanism of action is different from those of α- and β-scorpion toxins. Compared with many other β-spider toxins, HNTX-III possesses distinct sodium channel-inhibiting properties in the manner of channel gating modification and binding specificity and affinity.
EXPERIMENTAL PROCEDURES
Materials and Animals
Kunming albino mice and Sprague-Dawley rats were purchased from Xiangya School of Medicine, Central South University. All sequencing reagents were purchased from the Applied Biosystems (Foster City, CA) Division of PerkinElmer Life Sciences. The 3′- and 5′-RACE kits and TRIzol reagent were purchased from Invitrogen. Restriction enzymes, Taq DNA polymerase, and the pGEMT easy vector system were acquired from Promega (Madison, WI). All synthesis reagents were purchased from Chemassist Corp. Trifluoroacetic acid (TFA) and α-cyano-4-hydroxycinnamic acid were obtained from Sigma. All reagents were of analytical grade.
Toxin Purification
The venom was obtained by electrical stimulation of female spiders, and the freeze-dried crude venom was stored at −20 °C prior to analysis. Lyophilized venom, dissolved in double-distilled water, was purified by ion exchange chromatography and reverse-phase HPLC. Ion exchange chromatography was performed using a Waters Protein-Pak CM 8H column (10 × 100 mm) on a Waters 650 advanced protein purification system equipped with a model 486 detector. Fractions obtained from ion exchange chromatography were further fractionated using reverse phase HPLC on a Vydac C18 column (300 Å, 4.6 × 250 mm) on a Waters alliance 2690 HPLC system with a model 996 photodiode array detector. The disulfide connections of HNTX-III was determined by partial reduction according to the method described by Gray (21, 22).
Electrophysiological Assays
Whole cell clamp recordings of voltage-gated sodium currents were generated from rat DRG neurons that were acutely dissociated from 30-day old Sprague-Dawley rats and maintained in short term primary culture according to the methods described by Hu and Li (23). DRG cells with large diameter (around 50 picosiemens in slow capacitance) and those with a relatively small diameter (around 20 picosiemens for slow-capacitance) were selected for the study of TTX-S and TTX-R sodium currents, respectively. Meanwhile, TTX at a final concentration of 200 nmol/liter was used to separate the TTX-R sodium current from the TTX-S sodium current. A human Nav1.1–1.5 or Nav1.7 channel plasmid and a plasmid for green fluorescent protein were transiently transfected into human embryonic kidney 293 (HEK 293) cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. HEK 293 cells were grown under standard tissue culture conditions (5% CO2; 37 °C) in DMEM supplemented with 10% FBS. The β1-subunit was cotransfected with the Nav1.7 channel to increase the current density. Cells with green fluorescence were selected for whole-cell patch clamp recording at 36–72 h after transfection. Patch clamp experiments were performed at room temperature. Suction pipettes (2.0–3.0 megaohms) were made from borosilicate glass capillary tubes using a two-step vertical micropipette puller. The pipette solution contained 145 mm CsCl, 4 mm MgCl2·6H2O, 10 mm HEPES, 10 mm EGTA, 10 mm glucose, 2 mm ATP (pH 7.2). The external solution contained 145 mm NaCl, 2.5 mm KCl, 1.5 mm CaCl2, 1.2 mm MgCl2·6H2O, 10 mm HEPES, 10 mm glucose (pH 7.4). For the experiments described in the legend to Fig. 7, modified solutions were used to enhance outward sodium current amplitudes. The modified pipette solution contained 108 mm NaCl, 35 mm CsF, 1 mm EGTA, 2 mm KCl, and 10 mm HEPES, pH 7.3. The modified bathing solution contained 105 mm NaCl, 35 mm N-methyl-d-glucamine chloride, 5 mm CsCl, 2 mm KCl, 1 mm MgCl2, 1 mm CaCl2, and 10 mm HEPES, pH 7.3 (19). Experimental data were collected and analyzed using the program Pulse/Pulsefit version 8.0 (HEKA Electronics). Macroscopic sodium currents were filtered at 5 kHz and digitized at 20 kHz with an EPC-9 patch clamp amplifier (HEKA Electronics). Series resistance was kept near 5 megaohms and compensated to 65–70%; linear capacitative and leakage currents were digitally subtracted using the P/4 protocol.
FIGURE 7.

Solution structure of HNTX-III. A, superimposition of backbone heavy atoms (N, Cα, and C) for 20 converged structures of HNTX-III. B, Richardson style diagrams of the backbone fold of HNTX-III. The β-sheets, turns, and random coils are colored in yellow, blue, and green, respectively. The disulfide bridge linkage pattern (1–4, 2–5, 3–6) is shown in yellow. C and D, surface profiles of HNTX-III. Positively and negatively charged residues and hydrophobic residues are colored in blue, red, and orange, respectively.
NMR Spectroscopy of HNTX-III
The NMR sample was prepared by dissolving the HNTX-III in 450 μl of 20 mm deuterium sodium acetate buffer (H2O/D2O, 9:1, v/v) containing 0.002% NaN3 and 0.01 mm EDTA with a final concentration of 6 mm HNTX-III and a pH of 4.0. Sodium 3-(trimethylsilyl) propionate-2,2,3,3-d4 was added to a final concentration of 200 μm as an internal chemical shift reference. For experiments in D2O, the sample used in H2O experiments was lyophilized and then redissolved in 450 μl of 99.996% D2O (Cambridge Isotope Laboratories). All NMR spectra were observed on a 500-MHz Bruker DRX-500 spectrometer with a sample temperature of 298 K. Several sets of two-dimensional spectra were recorded in a phase-sensitive mode by the time-proportional phase increment method with standard pulse sequences and phase cycling. Total correlation spectroscopy spectra were obtained with a mixing time of 85 ms. NOESY spectra were recorded in D2O with a mixing time of 200 ms and in H2O with mixing times of 100, 200, and 400 ms. Solvent suppression was achieved according to the presaturation method. All two-dimensional measurements were recorded with 512–1024 frequency data points and were zero-filled to yield 1024–2048 data matrices, except in the high resolution double quantum-filled COSY spectrum. The double quantum-filled COSY spectrum was recorded with 2048–1024 data points in the t2 and t1 dimensions, respectively, and zero-filled to 4096–2048 points to measure the coupling constants. All spectra were processed and analyzed using Felix 98.0 (Biosym Technologies) software running on a Silicon Graphics O2 work station. Before Fourier transformation, the signal was multiplied by a sine bell or sine bell square window function with a π/2 phase shift.
Structure Calculations
Distance constraints were obtained from the intensities of the cross-peaks in NOESY spectra with a mixing time of 200 ms. All NOE data were classified into three distance ranges, 1.8–2.7, 1.8–3.5, and 1.8–5.0 Å, corresponding to strong, medium, and weak NOE values, respectively. Pseudo-atom corrections were applied to non-stereospecifically assigned methyl and methylene protons according to the method of Wüthrich (24). Ten dihedral angle restraints derived from 3JNH-CαH coupling constants were restrained to −120 ± 30° for 3JNH-CαH ≥ 8.80 Hz and −65 ± 25° for 3JNH-CαH ≤ 5.5 Hz. Three distance constraints were added to every disulfide bridge that was determined from NMR data. The corresponding distances were 2.02 ± 0.02, 2.99 ± 0.5, and 2.99 ± 0.5 Å for S(i)-S(j), S(i)-Cβ(j), and S(j)-Cβ(i), respectively. Structure calculations of HNTX-III were run on a Silicon Graphics work station using the standard protocol of the X-PLOR NIH-2.9.6 program (25).
RESULTS
Inhibitory Effect of HNTX-III on VGSC Isoforms Expressed on HEK 293 Cells
HNTX-III was purified through the combined use of ion exchange HPLC and reverse-phase HPLC as described in our previous study (16). The amino acid sequence of HNTX-III is GCKGFGDSCTPGKNECCPNYACSSKHKWCKVYL, and it contains an amidated C-terminal residue (26). The disulfide linkages of HNTX-III were determined to be Cys2-Cys17, Cys9-Cys22, and Cys16-Cys29 (known as the 1–4, 2–5, and 3–6 disulfide pattern) by tris(2-carboxyethyl)phosphine partial reduction and Edman degradation. Consistent with our previous study, HNTX-III purified in the present study was able to inhibit TTX-S Na+ currents in rat DRG cells, whereas it had no detectable effect on TTX-R Na+ currents (data not shown).
The inhibitory activity of HNTX-III on Nav1.1–1.5 and Nav1.7 channels transiently expressed on HEK 293 cells was examined next. The current of each channel was triggered by 50-ms depolarization potentials to −10 mV from the clamped voltage of −80 mV. Of the six VGSC isoforms, Nav1.1–1.3 and Nav1.7 currents were inhibited by 1 μm HNTX-III, and the inhibitory effect was dose-dependent (Fig. 1). The IC50 value was calculated to be ∼1.27 μm for Nav1.1, 275 nm for Nav1.2, 491 nm for Nav1.3, and 232 nm for Nav1.7. Because we failed to detect a current in Nav1.6 expressed in HEK 293 cells, the inhibitory effect of HNTX-III on Nav1.6 could not be examined in the present study. However, because 1 μm toxin completely suppressed TTX-S Na+ currents in rat DRG cells, which show high levels of Nav1.6 expression (27), we concluded that HNTX-III has inhibitory activity against Nav1.6 currents. On the other hand, no inhibitory effect was observed on Nav1.4 and Nav1.5 currents even in the presence of 10 μm HNTX-III (Fig. 1).
FIGURE 1.

Inhibitory effect of HNTX-III on six VGSC isoforms expressed in HEK 293 cells. HNTX-III at 1 μm potently inhibited Nav1.1–1.3 and Nav1.7 currents, respectively. No inhibitory effect was observed on Nav1.4 and Nav1.5 currents in the presence of 10 μm HNTX-III. Current traces before and after application of the toxin are shown in black and red, respectively. The inhibition of HNTX-III on Nav1.1–1.3 or Nav1.7 was dose-dependent. Each data point (mean ± S.E.), which was derived from 5–8 cells, shows current relative to control. Apparent IC50 values were ∼1.27 μm for Nav1.1, 270 nm for Nav1.2, 491 nm for Nav1.3, and 232 nm for Nav1.7. The inward Na+ currents were elicited by a 50-ms depolarization from a holding potential of −80 to −10 mV.
Voltage-dependent HNTX-III Inhibition of Nav1.7 Expressed on HEK 293 Cells
To investigate the mechanism by which HNTX-III inhibits the Nav1.7 current expressed on HEK 293 cells, the time-dependent inhibition of Nav1.7 by 1 μm HNTX-III (τon = 20.5 ± 0.3 s) was examined, as shown in Fig. 2A. The inhibition by HNTX-III was reversible upon washing, with a recovery of ∼80% of the control current within 7 min. The current-voltage (I-V) curves of Nav1.7 (Fig. 2B) showed that HNTX-III did not affect the initial activated voltage, the active voltage of peak inward current, and the reversal potential of the Nav1.7 current. The effects of HNTX-III on the activation and inactivation of Nav1.7 were analyzed. The half-activation voltage and half-inactivation voltage of Nav1.7 after treatment with 1 μm HNTX-III were 42.8 and 90.8 mV, respectively, compared with 39.6 and 87.0 mV, respectively, in the control group (Fig. 2C), indicating that HNTX-III inhibited the peak current of Nav1.7 without significantly changing its activation and inactivation kinetics. Furthermore, we examined the effect of HNTX-III on the repriming kinetics (recovery from inactivation) of Nav1.7 (28). As shown in Fig. 2D, the Nav1.7 current was gradually recovered with a progressive increase in the recovery times to the recovery potential of −80 mV before and after application of 1 μm HNTX-III. Consistent with results showing that HNTX-III does not alter the inactivation kinetics of Nav1.7, HNTX-III had no effect on the rate of recovery from inactivation (27.2 ± 0.8 ms for the control and 25.2 ± 1.1 ms for HNTX-III-treated samples) (Fig. 2E). This was contrary to other spider toxins, such as δ-actratoxins and β-spider toxins, which modulate the gating behaviors of VGSCs (9, 29–31). These findings suggest that HNTX-III, similar to TTX, seems to act as a pore blocker in the physiologically relevant voltage range of −80 to 60 mV. In our previous study, we found that HNTX-III caused a hyperpolarizing shift of ∼10 mV in the half-inactivation voltage of rat DRG TTX-S VGSCs and significantly decreased the recovery rate from inactivation, which is different from the results obtained with Nav1.7 expressed in HEK 293 cells. This difference might be explained as follows. 1) Rat DRG cells express multiple TTX-S VGSC isoforms, including Nav1.1, Nav1.2, Nav1.6, and Nav1.7. The effect on rat DRG TTX-S VGSCs described above could be attributed to HNTX-III acting on TTX-S VGSC isoforms other than Nav1.7. 2) Nav1.7 channels expressed on these two cell types have different regulatory proteins, such as β-subunits, which may modulate the action of HNTX-III on Nav1.7, similar to the effect of β-subunits on modulating the inhibition of Nav1.8 by the gating modifier μO-conotoxin MrVIB (32).
FIGURE 2.

The inhibition of HNTX-III on Nav1.7 expressed in HEK 293 cells. A, time course of inhibition of Nav1.7 by 1 μm HNTX-III (τon = 20.5 ± 0.3 s). The inhibition could be reversed by washing. B, no evident changes in the current-voltage relationship for Nav1.7 were observed in the presence of 1 μm HNTX-III. C, HNTX-III did not significantly alter the steady-state activation and inactivation of Nav1.7. Data are plotted as a fraction of the maximum conductance. The voltage dependence of steady-state inactivation was estimated using a standard double-pulse protocol, in which a 20-ms depolarizing test potential of 0 mV followed a 500-ms prepulse at potentials that ranged from −130 to −10 mV with a 10-mV increment. Cells were held at −100 mV. D, representative current traces were recorded from cells expressing Nav1.7 before (left) and after (right) the application of 1 μm HNTX-III, indicating the rate of recovery from inactivation (repriming) at −80 mV. The cells were prepulsed to −20 mV for 20 ms to inactivate Nav1.7 current and then returned to the recovery potential (−80 mV) for increasing recovery durations prior to a test pulse to 0 mV. The times labeled for each trace correspond to the recovery duration for that trace. E, kinetics of current recovery from fast inactivation before (τ = 27.2 ± 0.8 ms) and after (τ = 25.2 ± 1.1 ms) the application of HNTX-III (n = 5). Error bars, S.E.
Additional experiments were performed to clarify whether HNTX-III acts as a pore blocker or a gating modifier. It is known that when gating modifier toxins cause complete inhibition of all inward currents, outward currents can still be observed (33–35). In the present study, cells were depolarized from −100 mV to potentials ranging from −60 to 160 mV in 10-mV increments, and the depolarization duration was 1 ms. In the modified solutions described in our previous study (19), we found that the Nav1.7 current was reversed at ∼0 mV, and the amplitude of the outward current elicited with strong depolarizations was increased (Fig. 3A). In the presence of 5 μm HNTX-III, the inward current was completely inhibited, indicating that the toxin-bound Nav1.7 channels expressed on HEK 293 cells could not be activated in response to moderate depolarizations that evoke inward currents through the unblocked channel. Nevertheless, the toxin-bound channel could still be opened by large depolarizations at voltages above 40 mV. The outward current increased gradually with increasing depolarization potentials, with a current amplitude of ∼27% of the current amplitude elicited before toxin application (Fig. 3, A and B). On the contrary, TTX, a pore blocker, completely blocked both inward and outward currents with depolarization potentials from −60 to 160 mV (Fig. 3B). Furthermore, the IC50 values were ∼232 nm, 415 nm, and 1.6 μm for Nav1.7 currents evoked with depolarizations of −10, +50 mV, and +100 mV, respectively (Fig. 3C). These data indicated that HNTX-III inhibition of Nav1.7 was voltage-dependent or state-dependent.
FIGURE 3.

Voltage-dependent inhibition of HNTX-III on Nav1.7 expressed in HEK 293 cells. A, short extreme depolarizations partially activated toxin-bound Nav1.7 channel. The currents were recorded from HEK 293 cells expressing Nav1.7 channel before (left) and after (right) toxin application (for about 10 min of incubation). The depolarizing voltage steps ranged from −60 to 160 mV. B, current-voltage plot of peak current amplitude obtained from five cells expressing Nav1.7 channel before and after toxin application. Current amplitudes were normalized to the amplitude recorded at −100 mV before toxin application. C, the dose-dependent inhibition of HNTX-III on Nav1.7 currents evoked by different depolarizing potentials (−10, +50, and +100 mV) from a holding potential of −80 mV. Each data point (mean ± S.E. (error bars)), which was derived from 5–8 cells, shows current relative to control.
HNTX-III Binds to the DIIS3-S4 Linker of Nav1.7 Expressed on HEK 293 Cells
Considering our previous results with HWTX-IV, the present results indicated that HNTX-III might be a site 4 toxin acting on the DIIS3-S4 linker of Nav1.7. To clarify this hypothesis, we constructed chimeric channels of Nav1.5 or Nav1.7 (Fig. 4A). Because the toxin has no inhibitory activity against Nav1.5, we assumed that HNTX-III would not affect a Nav1.7 chimeric channel in which the DIIS3-S4 linker (-LADVEGLSV-) of Nav1.7 was replaced with the corresponding linker (-LSRMGNLSV-) of Nav1.5, whereas it should inhibit the Nav1.5 chimeric channel in which the DIIS3-S4 linker of Nav1.5 was replaced with the corresponding linker of Nav1.7 (Fig. 4A). As shown in Fig. 4, 10 μm HNTX-III could only reduce ∼9% of the amplitude current of the Nav1.7 chimeric channel (Fig. 4, B and D). On the other hand, 1 μm HNTX-III could inhibit more than 50% of the amplitude current of the Nav1.5 chimeric channel, and the IC50 value was calculated to be 975 nm (Fig. 4, C and D). These data indicate that the DIIS3-S4 linker might be critical for HNTX-III binding to Nav1.7. The lack of an effect of HNTX-III on the activation and inactivation kinetics was suggestive of a toxin that might trap the domain II voltage sensor of Nav1.7 in its resting conformation.
FIGURE 4.
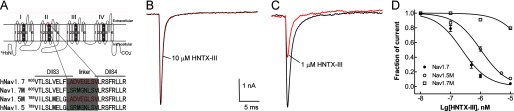
HNTX-III binds to the DIIS3-S4 linker of Nav1.7. A, strategy for chimeric channel construction. Top, schematic diagram of VGSC α-subunit. The voltage sensor (segment 4) of each domain is shaded in gray and marked with ++. The DIIS3-S4 linker is labeled in red. Below, sequence alignment of DIIS3-S4 of Nav1.5, Nav1.7, and chimeric channels. The linker is shaded in gray. Nav1.7 M, Nav1.7 chimeric channel in which the DIIS3-S4 linker of Nav1.7 was replaced by the corresponding linker of Nav1.5; Nav1.5 M, Nav1.5 chimeric channel in which the DIIS3-S4 linker of Nav1.5 was replaced by the corresponding linker of Nav1.7. B, 10 μm HNTX-III can only reduce less than 10% current amplitude of the Nav1.7 chimeric channel. C, 1 μm HNTX-III can inhibit more than 50% current amplitude of the Nav1.5 chimeric channel. The channels were expressed in HEK 293 cells, and Na+ current was evoked by a 50-ms depolarization from a holding potential of −80 mV to −10 mV. D, apparent IC50 values were ∼231 nm for Nav1.7 and 975 nm for the Nav1.5 chimeric channel, as calculated from the dose-dependent inhibition curves. Note that the IC50 value of HNTX-III against the Nav1.7 chimeric channel (Nav1.7M) could not be calculated because even the highest concentration used had a very low inhibitory effect on this channel. Error bars, S.E.
Kinetics of Dissociation of HNTX-III from Nav1.7
Binding of HWTX-IV was reversed by prolonged strong depolarizations that activate the voltage sensor (19). Therefore, we examined whether prolonged strong depolarizations could reverse the inhibitory effect of HNTX-III on Nav1.7 by using the protocol described by Wang et al. (36) with some modifications (Fig. 5A). As shown in Fig. 5B, in the absence of HNTX-III, a current of ∼10 nA was induced by the test pulse, indicating the recovery from inactivation. In the presence of 2 μm HNTX-III, a progressively longer strong depolarization (to 100 mV) led to an increase in the fraction of sodium current recovered from inhibition by HNTX-III. Depolarizations lasting 100 and 600 ms resulted in ∼30% and complete recovery of the Na+ current, respectively. These data indicate that HNTX-III dissociated from Nav1.7 in response to prolonged strong depolarizations. As shown in Fig. 5C, increased depolarization potentials (the depolarization time was set at 400 ms) were correlated with increased dissociation. Fig. 5D shows the time course of dissociation of the toxin after strong depolarizations in the presence of 2 μm HNTX-III, from which the dissociation time constant (τ) was calculated to be 189 ± 5 ms at 100 mV, 323 ± 10 ms at 80 mV, or 566 ± 13 ms at 60 mV. These results indicated that the rate of HNTX-III dissociation was voltage-dependent, and a stronger depolarization was correlated with a higher rate of dissociation (Fig. 5E). The dissociation was also concentration-dependent, with a lower rate of dissociation in the presence of higher concentrations of HNTX-III. In the presence of 5 μm HNTX-III, the dissociation time constant (τ) was calculated to be 294 ± 7 ms at 100 mV, 476 ± 12 ms at 80 mV, or 733 ± 17 ms at 60 mV (Fig. 5E).
FIGURE 5.

Kinetics of dissociation and reassociation of HNTX-III·Nav1.7 complex. A, diagram showing the protocol for dissociation analysis. Cells expressing Nav1.7 channels were incubated in HNTX-III for 3 min at a holding potential of −100 mV to allow binding. The rate of toxin dissociation was determined with the illustrated pulse paradigm by stepping to a depolarizing pulse of 100, 80, or 60 mV for 10–1110 ms at increments of 100 ms, returning to −100 mV for 200 ms to allow recovery from fast inactivation, and then assessing the effect of the depolarizing pulse with a 50-ms test pulse to −10 mV (test). B, in the presence of 2 μm HNTX-III, progressively prolonged strong depolarization (to 100 mV) led to a greater degree of Nav1.7 current recovery from HNTX-III inhibition. C, in the presence of 2 μm HNTX-III, Nav1.7 current recovery increased in correlation with the increment of depolarization potentials (the depolarization duration time was set as 400 ms). D, time course of dissociation of 2 μm HNTX-III from Nav1.7 at 100 mV (τ = 189 ± 5 ms, n = 3), 80 mV (τ = 323 ± 10 ms, n = 3), and 60 mV (τ = 566 ± 13 ms, n = 4). E, time constants of dissociation as a function of potentials for Nav1.7 in the presence of 2 and 5 μm HNTX-III. Note that the dissociation was voltage-dependent and dose-dependent. F, diagram showing the protocol for reassociation analysis. The Nav1.7 current was first activated by a 50-ms depolarization from −100 to −10 mV, followed by a 600-ms strong depolarization to 100 mV to cause toxin dissociation, progressively longer hyperpolarizing pulses to allow toxin rebinding, and a final test depolarization to −10 mV to assess HNTX-III rebinding. G, gradual rebinding of HNTX-III to Nav1.7 occurred after progressively prolonged repolarizations with complete reassociation at 55 s. H, time course of reassociation of 2 μm HNTX-III with Nav1.7 at −100 mV (τ = 22.3 ± 2.4 s) and −80 mV (τ = 21.0 ± 2.1 s). I, time constants of reassociation as a function of potential for Nav1.7 in the presence of 2 and 5 μm HNTX-III. Error bars, S.E.
Kinetics of Reassociation of HNTX-III with Nav1.7
The rates of reassociation of unbound HNTX-III with Nav1.7 were also assessed using a protocol described by Wang et al. (36) (Fig. 5F). In agreement with the data described above, HNTX-III completely dissociated upon the 600-ms depolarization to 100 mV, as indicated by the complete recovery of the sodium current (Fig. 5G). A gradual rebinding of HNTX-III to Nav1.7 occurred after longer repolarizations with complete reassociation at 55 s (Fig. 5, G and H). In the presence of 2 μm HNTX-III, the reassociation time constant was 22.3 ± 2.4 s at −100 mV and 21.0 ± 2.1 s at −80 mV (Fig. 5, H and I), indicating that toxin reassociation was independent of membrane potential. In the presence of 5 μm HNTX-III, the reassociation time constant was 18.7 ± 3.1 s at −100 mV and 17.9 ± 2.6 s at −80 mV (Fig. 5, H and I), with no significant difference between the two concentrations.
Kinetics of Deactivation of Nav1.7 in the Presence HNTX-III
HNTX-III dissociated from Nav1.7 upon prolonged strong depolarizations and reassociated with the channel after long repolarizations. This behavior could be explained by state-dependent interaction of HNTX-III with Nav1.7, by which HNTX-III binding to Nav1.7 would be dependent on the configuration of the channel. Another explanation is based on an electric field effect, by which HNTX-III is positively charged at pH 7.4 and the dissociation and reassociation of the toxin to Nav1.7 occurs by electric repulsion and attraction in an electric field. Our data described above supported the former mechanism. Furthermore, additional experiments were conducted to analyze the current deactivation kinetics as shown for hanatoxin and Kv2.1 (37). As indicated above, short extreme depolarizations activated the Nav1.7 channel in the presence of 5 μm HNTX-III, which eliminated the inward current of Nav1.7. If extreme depolarization caused the activation of toxin-bound channel, then the deactivation kinetics (the rate at which current decays or channels close in response to repolarization of the membrane potential) following the activating pulses would be different from the kinetics in the absence of the toxin. However, if the toxin must dissociate from the channel before current activation, the deactivation kinetics following extreme depolarizations should be the same in the presence and absence of the toxin (19, 37). Therefore, we determined the time course for Nav1.7 current deactivation following a 0.2-ms depolarization to 250 mV (Fig. 6). In the presence of 5 μm HNTX-III, the outward current amplitude evoked by the short depolarizing pulse was ∼50% of that activated before the toxin application (Fig. 6A). The relative amplitude (the peak current in the presence of HNTX-III was normalized to that before toxin application) of tail current evoked by the repolarizing pulse following the depolarizing pulse was also reduced in the presence of HNTX-III (Fig. 6B), indicating a more rapid closure of the Nav1.7 channel during the repolarizing transition. The time course for the decay of the tail current was also much faster in the presence of HNTX-III (Fig. 6, B (inset) and C). The significantly faster deactivation of the Nav1.7 current in the presence of 5 μm HNTX-III could be explained as follows. The toxin-bound channel activated by an extreme depolarization would be deactivated or returned to its resting state more rapidly if the toxin bound with greater affinity to the channel in the resting configuration. The gradual dissociation of HNTX-III in response to prolonged strong depolarizations could be the result of prolonged activation maintaining the channel in a configuration with lower toxin affinity, which promotes toxin dissociation. Taken together, our results indicate that HNTX-III, similar to HWTX-IV, should bind to the Nav1.7 voltage sensor in the closed state, inhibiting its activation.
FIGURE 6.

HNTX-III promotes the deactivation of Nav1.7 current following an extreme depolarization. A, current traces were recorded HEK 293 cells expressing Nav1.7 in control before and after application of 5 μm HNTX-III. Nav1.7 current was activated with a 0.2-ms depolarization to 250 mV followed by a repolarization to −40 mV to elicit the tail current. B, the currents shown in A were normalized to the maximum current evoked during the depolarization. Note that the relative amplitude of the tail current became smaller in the presence of HNTX-III. The inset shows just the tail currents after normalization of the maximum tail current amplitude. Note that the decay of the tail current was faster in the presence of HNTX-III compared with that of control. C, time constants for tail current decay was determined at repolarizing pulses ranging from −20 to −80 mV before and after application of 1 μm HNTX-III (n = 5). Data are expressed as mean ± S.E. (error bars).
The Solution Structure of HNTX-III
Sequence-specific resonance assignments were performed according to the standard procedures established by Wüthrich (24). All of the backbone protons and more than 95% of the side chain protons were identified. The two Pro residues (Pro11 and Pro18) of HNTX-III were assigned clearly by the strong sequential Hα–Hδ cross-peaks for Xaa–Pro, which also indicated the presence of trans-peptide bonds for these residues (38).
The structure of HNTX-III was determined using 451 intramolecular distance constraints, six dihedral constraints, and nine distance constraints derived from the three disulfide bridges. Altogether, the final experimental set corresponded to 10 restraints per residue on average. A family of 20 accepted structures with lower energies and better Ramachandran plots was selected to represent the three-dimensional solution structure of HNTX-III (Protein Data Bank code 2JTB). The structures have no distance violations greater than 0.2 Å and no dihedral violations greater than 2.0°. They have favorable non-bonded contacts, as evidenced by the low values of the mean Lennard-Jones potentials, and good covalent geometry, as indicated by the small deviations from ideal bond lengths and bond angles. Analysis of the structures in PROCHECK showed that 80.2% of non-Pro, non-Gly residues lie in the most favored regions of the Ramachandran plot with a further 19.8% in additionally allowed regions. The best fit superposition of the backbone atoms (N, Cα, and C) for the 20 converged structures of HNTX-III resulted in an average root mean square deviation with respect to the mean structure values of 0.55 ± 0.09 Å for backbone atoms and 1.19 ± 0.12 Å for all heavy atoms.
Fig. 7A shows the best fit superposition of the backbone atoms (N, Cα, and C) for the 20 converged structures of HNTX-III. Analysis of the 20 converged structures indicated that the molecular structure of HNTX-III contains a short triple-stranded anti-parallel β-sheet formed by the strands Gly7-Cys9, Ala21-Ser23, and Lys27-Lys30 (Fig. 7B). The strong sequential dαN and weak dNN, interstrand NOE connectivities, chemical shift index, large 3JNH-CαH coupling constants, and slowly exchanging amide protons confirmed the β-sheet structure (39). The turns in HNTX-III were also identified using a standard definition that states that the distance between Cα(i) and Cα(i + 3) should be less than 7 Å and that the characteristic NOE connectivities of backbone protons for the corresponding turn segments are present. These analyses led to the identification of three β-turns: Gly4–Asp7 (type II), Thr10–Lys13 (type II), and Ser24–Lys27 (type I) (Fig. 7B).
HNTX-III adopts an inhibitor cystine knot motif commonly observed in other toxic and inhibitory peptides (40, 41). The cystine knot in HNTX-III is formed by three disulfide bridges between Cys2 and Cys17, Cys9 and Cys22, and Cys16 and Cys29, with the Cys16-Cys29 disulfide bridge crossing a 10-residue ring formed by the intervening polypeptide backbone and the Cys2-Cys19 and Cys9-Cys22 disulfide bridges (Fig. 7B). The inhibitor cystine knot toxins have diverse bioactivities, which are derived from structural differences caused by the amino acid residues located on the loops between two cysteine residues (40, 41). A significant advantage of this motif is that it imparts to peptides extraordinary stability, even in human serum (14). It is believed that HNTX-III has this advantage. The molecular surface analysis showed that the basic residues (Lys3, Lys13, Lys27, His26, and Lys25) are mainly distributed on one side, forming a basic patch on the surface of HNTX-III, whereas most hydrophobic residues (Phe5, Pro18, Tyr20, Ala21, Val31, Tyr32, and Leu33) are clustered on the other side, forming a hydrophobic patch (Fig. 7, C and D).
DISCUSSION
HNTX-III Is a Selective Antagonist of Neuronal TTX-S VGSCs
In the present study, the specific inhibitory effect of HNTX-III on VGSCs was examined in HEK 293 cells transiently expressing VGSC isoforms. Our previous study showed that the TTX-R isoforms Nav1.8 and Nav1.9 are resistant to inhibition by HNTX-III (16). Of the six VGSC isoforms tested, Nav1.1–1.3 and Nav1.7 but not Nav1.4 (a skeletal TTX-S VGSC) and Nav1.5 (a cardiac TTX-R VGSC) are inhibited by HNTX-III, suggesting that HNTX-III is a selective antagonist of neuronal TTX-S VGSCs.
HNTX-III Inhibits Nav1.7 Opening by Binding to Site 4 and Trapping the Voltage Sensor in the Closed State
Our data suggest that HNTX-III acts as a gating modifier inhibiting Nav1.7 opening by binding to site 4 in the closed state. First, the inhibition of Nav1.7 by HNTX-III was voltage-dependent. Higher concentrations (IC50 = 2.5 μm) of HNTX-III were required to inhibit the Nav1.7 current evoked with stronger depolarization (100 mV). A saturating concentration of HNTX-III (5 μm) could completely suppress all inward Nav1.7 currents elicited with moderate depolarizations, but upon short extreme depolarizations (above 40 mV), a fraction of outward currents (∼27% of the current amplitude elicited before toxin application at 160 mV) were still observed, indicating the activation of the toxin-bound channel. Second, our data on chimeric channels showed that the DIIS3-S4 linker plays an important role in HNTX-III binding to Nav1.7, providing direct evidence that HNTX-III is a gating modifier acting on site 4. Third, we further demonstrated that the toxin-bound channel could deactivate or return to a closed configuration more rapidly than the free channel in the absence of HNTX-III, as revealed by the deactivation kinetics analysis. These data suggest that HNTX-III preferentially binds to Nav1.7 in the closed state with high affinity and stabilizes the voltage sensor. Consequently, extreme depolarizations should be required to overcome the energy of the interaction of HNTX-III with the channel in the resting state, driving it into the activated conformation. Fourth, the inhibition could be reversed by prolonged strong depolarizations. HNTX-III dissociated more rapidly at higher depolarized potentials. We propose that prolonged strong depolarization may push the outward movement of the voltage sensor from the inward position even in the presence of bound HNTX-III. This would cause a conformational change of the binding site, resulting in the dissociation of the HNTX-III·Nav1.7 complex. Additionally, HNTX-III was able to gradually rebind to Nav1.7 after longer repolarizations that caused a return to the closed state from the inactivated state. These data suggest that HNTX-III and HWTX-IV share a common mechanism of inhibition of Nav1.7 by binding to the S3-S4 linker in domain II of Nav1.7 in the closed state and consequently trapping the voltage sensor in its inward position. Thus, these toxins prevent the outward movement of the voltage sensor upon membrane depolarization and keep Nav1.7 from opening, inhibiting Na+ conductance (17–19).
HNTX-III Is Distinct from Other β-Toxins Acting on VGSCs
To date, more than 100 peptide neurotoxins affecting VGSCs have been identified from venomous animals (42, 43). These neurotoxins are known as μ-conotoxins, α- and β-scorpion toxins, sea anemone toxins, and δ- and β-spider toxins. They have been shown to bind to the four extracellular receptor sites (sites 1, 3, 4, and 6) (8–13, 44, 45). Although β-scorpion toxins and β-spider toxins target the same site 4, they have rather different mechanisms of action. β-Scorpion toxins shift the voltage dependence of channel activation in the hyperpolarizing direction and reduce the peak current amplitude, which is the direct result of toxin binding to site 4 in the activated state, trapping the voltage sensor in its outward position (11–13, 46). On the contrary, previous studies and our current results indicate that HNTX-III and many other β-spider toxins (namely HWTX-I; HWTX-IV; HNTX-I; HNTX-IV; CcoTx1, -2, and -3; PaurTx3; ProTx-I; and ProTx-II) can target site 4 in the closed state, trapping the voltage sensor in its inward position (17–19, 35, 47–53). However, it is important to note that HNTX-III and its analog toxins (HWTX-I, HWTX-IV, HNTX-I, and HNTX-III) differ from many other β-spider toxins (e.g. CcoTx1, -2, and -3; PaurTx3; ProTx-I; and ProTx-II) in their mechanism of channel gating modification. The latter toxins shift the voltage-dependent activation to more depolarized potentials (35, 51–53), whereas the former ones cannot significantly change the activation kinetics (17–19, 47–50).
Compared with HNTX-I, HNTX-IV, HWTX-I, and HWTX-IV, the VGSC-inhibiting properties of HNTX-III are distinct. First, these toxins show differences in their selectivity for VGSC isoforms. HWTX-I, HWTX-IV, and HNTX-IV display slightly higher selectivity for Nav1.7 compared with Nav1.2 and Nav1.3, and they also show weak inhibitory activity against Nav1.4 and Nav1.5 (17–20, 47–50). HNTX-I is a weak antagonist of Nav1.2 (47). HNTX-III is a selective inhibitor of neuronal TTX-S VGSCs with similar selectivity for Nav1.2, -1.3, and -1.7. Second, the inhibition of Nav1.7 by HNTX-III is reversible upon washing, but no reversibility was observed for HNTX-IV and HWTX-IV (19, 20, 48). Additionally, compared with HWTX-IV and HNTX-IV, HNTX-III is much less toxic against animals. The intraperitoneal LD50 of HNTX-III in mice was determined to be ∼5 mg/kg body weight, whereas those of HWTX-IV and HNTX-IV are ∼0.2 mg/kg body weight, as determined in our previous studies (20, 48), indicating the lower toxicity of HNTX-III. Although HNTX-III belongs to the β-spider toxin family by binding to the same “macro” site 4 of VGSCs, it has distinct VGSC-inhibiting properties in the manner of channel gating modification and binding specificity and affinity, which could be related to its binding to different “micro” sites of VGSCs. Actually, multiple sites contributing to β-toxin interaction have been identified (8–13). Recently, Zhang et al. (54) found that apart from the residues in DIIS3-S4 of Nav1.2, five residues in the DIII SS2-S6 loop are also important for Css IV binding. Therefore, HNTX-III may be a useful probe for exploring novel toxin binding sites affecting the gating properties of VGSCs.
The Structure-Function Relationship of HNTX-III
Sodium channel toxins are known to have different amino acid sequences and structural scaffolds. However, extensive research on the structure-function relationships of certain toxins has highlighted certain common features of the bioactive surface. Multiple amino acid residues, particularly charged, hydrophobic, and aromatic residues, are commonly involved in forming a big and amphiphilic bioactive surface, which is necessary to determine the high affinity and selectivity for binding to VGSC isoforms (9, 55, 56). Analysis of the molecular surfaces of HNTX-I, HNTX-III, and HWTX-IV highlighted their amphiphilic properties, in which the basic residues (e.g. Lys25, His26, Lys27, and Lys30 in HNTX-III) form a positively charged patch, whereas the N- and C-terminal hydrophobic and aromatic residues (e.g. Phe5, Trp28, Val31, Tyr32, and Leu33 in HNTX-III) may make up a continuous hydrophobic surface that is separated from the positively charged surface (20, 47, 49) (Fig. 8). The side chains of the residues in both surfaces project outward from the molecular surface in the same orientation, suggesting that together they could form the bioactive surface of the three toxins. The residues in the conserved motif (K/R)X(K/R)WCK among the five toxins (HNTX-I, HNTX-III, HNTX-IV, HWTX-I, and HWTX-IV) form the positively charged surface. Our previous studies indicated that Lys27, His28, Arg29, and Lys32 in this motif of HNTX-IV are critical residues for its inhibitory activity on TTX-S VGSCs in rat DRG cells (49, 57), confirming that this basic surface may interact directly with the acidic residues in site 4. However, subtle structural differences may contribute to the inhibitory activities of these toxins. For example, the acidic residue Asp26 (corresponding to His26 in HNTX-III and Thr28 in HWTX-IV) is localized in the positively charged patch of HNTX-I and thus interferes with its binding to the channels. This may explain the weak inhibitory activity of HNTX-I on Nav1.2 (47). These data suggest that the amphiphilic surface consisting of basic, hydrophobic, and aromatic residues is critical for toxin binding to VGSCs. A comparative analysis will provide valuable information for our understanding of the mechanisms underlying the interaction between β-spider toxins (HNTX-III and analogous toxins) and isoforms of VGSCs in future studies.
FIGURE 8.
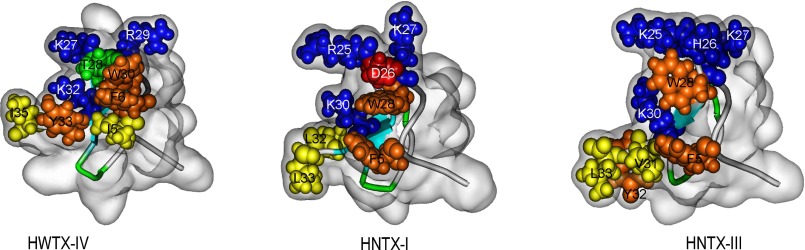
Structures and bioactive surfaces of HWTX-IV, HNTX-I and HNTX-III. The structures of HWTX-IV (Protein Data Bank code 1MB6), HNTX-I (Protein Data Bank code 1NIX), and HNTX-III (Protein Data Bank code 2JTB) were determined by two-dimensional NMR. The schematic backbone structures were covered by the semitransparent molecular surface of the toxins. Residues composing the bioactive surfaces are space-filled and colored as follows: basic (blue), acidic (red), aromatic (orange), hydrophobic (yellow), and hydrophilic (green).
In summary, HNTX-III is a potent and specific antagonist of neuronal TTX-S VGSCs. Its structure possesses a stable and compact inhibitor cystine knot motif. It inhibits the activation of Nav1.7 by trapping the domain II voltage sensor in the closed state, an action mechanism different from that of β-scorpion toxins. Moreover, compared with many other β-spider toxins, HNTX-III is distinct with regard to gating modification and binding specificity and affinity. These distinct properties of HNTX-III make it a valuable molecular tool for exploring the mechanisms of toxin-VGSC interaction, which may help identify novel binding sites on VGSCs. This information is essential for explaining the different selectivity of toxins to VGSC isoforms in addition to providing new insights into the structure and function of VGSCs. Moreover, HNTX-III might be a potential prototype analgesic.
Acknowledgments
We thank Dr. Mingqiang Rong for technical assistance. We also thank Jason Mulcahey (LC Sciences) for proofreading and correcting the English.
This work was supported in part by National Basic Research Program of China (973 Program) Grants 2010CB529801 and 2012CB22305 (to S. L.) and National Science Foundation Projects 31070699 and 31170746 (to Z. L.) and 31100764 (to M. D.).
The atomic coordinates and structure factors (code 2JTB) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- VGSC
- voltage-gated sodium channel
- TTX
- tetrodoxin
- TTX-S
- TTX-sensitive
- TTX-R
- TTX-resistant
- HNTX
- hainantoxin
- HWTX
- huwentoxin.
REFERENCES
- 1. Catterall W. A. (2012) Voltage-gated sodium channels at 60. Structure, function, and pathophysiology. J. Physiol. 590, 2577–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Catterall W. A. (2000) From ionic currents to molecular mechanisms. The structure and function of voltage-gated sodium channels. Neuron 26, 13–25 [DOI] [PubMed] [Google Scholar]
- 3. Dib-Hajj S. D., Cummins T. R., Black J. A., Waxman S. G. (2010) Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347 [DOI] [PubMed] [Google Scholar]
- 4. Yu F. H., Catterall W. A. (2003) Overview of the voltage-gated sodium channel family. Genome Biol. 4, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dib-Hajj S. D., Black J. A., Waxman S. G. (2009) Voltage-gated sodium channels. Therapeutic targets for pain. Pain Med. 10, 1260–1269 [DOI] [PubMed] [Google Scholar]
- 6. Wang W., Gu J., Li Y. Q., Tao Y. X. (2011) Are voltage-gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol. Pain 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dubin A. E., Patapoutian A. (2010) Nociceptors. The sensors of the pain pathway. J. Clin. Invest. 120, 3760–3772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cestèle S., Catterall W. A. (2000) Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 82, 883–892 [DOI] [PubMed] [Google Scholar]
- 9. Bosmans F., Swartz K. J. (2010) Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 31, 175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee C. H., Ruben P. C. (2008) Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels 2, 407–412 [DOI] [PubMed] [Google Scholar]
- 11. Billen B., Bosmans F., Tytgat J. (2008) Animal peptides targeting voltage-activated sodium channels. Curr. Pharm. Des. 14, 2492–2502 [DOI] [PubMed] [Google Scholar]
- 12. Catterall W. A., Cestèle S., Yarov-Yarovoy V., Yu F. H., Konoki K., Scheuer T. (2007) Voltage-gated ion channels and gating modifier toxins. Toxicon 49, 124–141 [DOI] [PubMed] [Google Scholar]
- 13. Stevens M., Peigneur S., Tytgat J. (2011) Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. King G. F. (2011) Venoms as a platform for human drugs. Translating toxins into therapeutics. Expert Opin. Biol. Ther. 11, 1469–1484 [DOI] [PubMed] [Google Scholar]
- 15. Escoubas P., King G. F. (2009) Venomics as a drug discovery platform. Expert Rev. Proteomics 6, 221–224 [DOI] [PubMed] [Google Scholar]
- 16. Xiao Y., Liang S. (2003) Inhibition of neuronal tetrodotoxin-sensitive Na+ channels by two spider toxins. Hainantoxin-III and hainantoxin-IV. Eur. J. Pharmacol. 477, 1–7 [DOI] [PubMed] [Google Scholar]
- 17. Xiao Y., Jackson J. O., 2nd, Liang S., Cummins T. R. (2011) Common molecular determinants of tarantula huwentoxin-IV inhibition of Na+ channel voltage sensors in domains II and IV. J. Biol. Chem. 286, 27301–27310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiao Y., Blumenthal K., Jackson J. O., 2nd, Liang S., Cummins T. R. (2010) The tarantulatoxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol. Pharmacol. 78, 1124–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao Y., Bingham J. P., Zhu W., Moczydlowski E., Liang S., Cummins T. R. (2008) Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain II voltage sensor in the closed configuration. J. Biol. Chem. 283, 27300–27313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng K., Shu Q., Liu Z., Liang S. (2002) Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena. J. Biol. Chem. 277, 47564–47571 [DOI] [PubMed] [Google Scholar]
- 21. Gray W. R. (1993) Disulfide structures of highly bridged peptides. A new strategy for analysis. Protein Sci. 2, 1732–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gray W. R. (1993) Echistatin disulfide bridges. Selective reduction and linkage assignment. Protein Sci. 2, 1749–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu H. Z., Li Z. W. (1997) Modulation by adenosine of GABA-activated current in rat dorsal root ganglion neurons. J. Physiol. 501, 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wüthrich K. (1986) NMR of Protein and Nucleic Acids, John Wiley & Sons, Inc., New York [Google Scholar]
- 25. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 26. Tang X., Zhang Y., Hu W., Xu D., Tao H., Yang X., Li Y., Jiang L., Liang S. (2010) Molecular diversification of peptide toxins from the tarantula Haplopelma hainanum (Ornithoctonus hainana) venom based on transcriptomic, peptidomic, and genomic analyses. J. Proteome Res. 9, 2550–2564 [DOI] [PubMed] [Google Scholar]
- 27. Ekberg J., Adams D. J. (2006) Neuronal voltage-gated sodium channel subtypes. Key roles in inflammatory and neuropathic pain. Int. J. Biochem. Cell Biol. 38, 2005–2010 [DOI] [PubMed] [Google Scholar]
- 28. Herzog R. I., Cummins T. R., Ghassemi F., Dib-Hajj S. D., Waxman S. G. (2003) Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McDonough S. I., Lampe R. A., Keith R. A., Bean B. P. (1997) Voltage-dependent inhibition of N- and P-type calcium channels by the peptide toxin ω-grammotoxin-SIA. Mol. Pharmacol. 52, 1095–1104 [DOI] [PubMed] [Google Scholar]
- 30. Yamaji N., Little M. J, Nishio H., Billen B., Villegas E., Nishiuchi Y., Tytgat J., Nicholson G. M., Corzo G. (2009) Synthesis, solution structure, and phylum selectivity of a spider δ-toxin that slows inactivation of specific voltage-gated sodium channel subtypes. J. Biol. Chem. 284, 24568–24582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Corzo G., Sabo J. K., Bosmans F., Billen B., Villegas E., Tytgat J., Norton R. S. (2007) Solution structure and alanine scan of a spider toxin that affects the activation of mammalian voltage-gated sodium channels. J. Biol. Chem. 282, 4643–4652 [DOI] [PubMed] [Google Scholar]
- 32. Wilson M. J., Zhang M. M, Azam L., Olivera B. M, Bulaj G., Yoshikami D. (2011) Navβ subunits modulate the inhibition of Nav1.8 by the analgesic gating modifier μO-conotoxin MrVIB. J. Pharmacol. Exp. Ther. 338, 687–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Billen B., Vassilevski A., Nikolsky A., Debaveye S., Tytgat J., Grishin E. (2010) Unique bell-shaped voltage-dependent modulation of Na+ channel gating by novel insect-selective toxins from the spider Agelena orientalis. J. Biol. Chem. 285, 18545–18554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bourinet E., Stotz S. C., Spaetgens R. L., Dayanithi G., Lemos J., Nargeot J., Zamponi G. W. (2001) Interaction of SNX482 with domains III and IV inhibits activation gating of α(1E) (Cav2.3) calcium channels. Biophys. J. 81, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bosmans F., Rash L., Zhu S., Diochot S., Lazdunski M., Escoubas P., Tytgat J. (2006) Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol. Pharmacol. 69, 419–429 [DOI] [PubMed] [Google Scholar]
- 36. Wang J., Yarov-Yarovoy V., Kahn R., Gordon D., Gurevitz M., Scheuer T., Catterall W. A. (2011) Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 108, 15426–15431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Phillips L. R., Milescu M., Li-Smerin Y., Mindell J. A., Kim J. I., Swartz K. J. (2005) Voltage-sensor activation with a tarantula toxin as cargo. Nature 436, 857–860 [DOI] [PubMed] [Google Scholar]
- 38. Liu Z., Zhu Q., Hu W., Liang S. (2013) Sequence-specific 1H-NMR assignment and determination of the secondary structure of Hainantoxin-III from the spider Ornithoctonus hainana. Protein Pept. Lett. 20, 761–766 [DOI] [PubMed] [Google Scholar]
- 39. Wishart D. S., Sykes B. D., Richards F. M. (1992) The chemical shift index. A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry 31, 1647–1651 [DOI] [PubMed] [Google Scholar]
- 40. Pallaghy P. K., Nielsen K. J., Craik D. J., Norton R. S. (1994) A common structural motif incorporating a cystine knot and a triple-stranded β-sheet in toxic and inhibitory polypeptides. Protein Sci. 3, 1833–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Craik D. J., Daly N. L., Waine C. (2001) The cystine knot motif in toxins and implications for drug design. Toxicon 39, 43–60 [DOI] [PubMed] [Google Scholar]
- 42. He Q. Y., He Q. Z., Deng X. C., Yao L., Meng E., Liu Z. H., Liang S. P. (2008) ATDB. A uni-database platform for animal toxins. Nucleic Acids Res. 36, D293–D297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. He Q., Han W., He Q., Huo L., Zhang J., Lin Y., Chen P., Liang S. (2010) ATDB 2.0. A database integrated toxin-ion channel interaction data. Toxicon 56, 644–647 [DOI] [PubMed] [Google Scholar]
- 44. Wilson M. J., Yoshikami D., Azam L., Gajewiak J., Olivera B. M., Bulaj G., Zhang M. M. (2011) μ-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl. Acad. Sci. U.S.A. 108, 10302–10307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Choudhary G., Aliste M. P., Tieleman D. P., French R. J., Dudley S. C., Jr. (2007) Docking of μ-conotoxin GIIIA in the sodium channel outer vestibule. Channels 1, 344–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang J. Z., Yarov-Yarovoy V., Scheuer T., Karbat I., Cohen L., Gordon D., Gurevitz M., Catterall W. A. (2011) Structure-function map of the receptor site for β-scorpion toxins in domain II of voltage-gated sodium channels. J. Biol. Chem. 286, 33641–33651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li D., Xiao Y., Hu W., Xie J., Bosmans F., Tytgat J., Liang S. (2003) Function and solution structure of hainantoxin-I, a novel insect sodium channel inhibitor from the Chinese bird spider Selenocosmia hainana. FEBS Lett. 555, 616–622 [DOI] [PubMed] [Google Scholar]
- 48. Liu Z., Dai J., Chen Z., Hu W., Xiao Y., Liang S. (2003) Isolation and characterization of hainantoxin-IV, a novel antagonist of tetrodotoxin-sensitive sodium channels from the Chinese bird spider Selenocosmia hainana. Cell Mol. Life Sci. 60, 972–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li D., Xiao Y., Xu X., Xiong X., Lu S., Liu Z., Zhu Q., Wang M., Gu X., Liang S. (2004) Structure-activity relationships of hainantoxin-IV and structure determination of active and inactive sodium channel blockers. J. Biol. Chem. 279, 37734–37740 [DOI] [PubMed] [Google Scholar]
- 50. Wang M., Rong M., Xiao Y., Liang S. (2012) The effects of huwentoxin-I on the voltage-gated sodium channels of rat hippocampal and cockroach dorsal unpaired median neurons. Peptides 34, 19–25 [DOI] [PubMed] [Google Scholar]
- 51. Sokolov S., Kraus R. L., Scheuer T., Catterall W. A. (2008) Inhibition of sodium channel gating by trapping the domain II voltage sensor with protoxin II. Mol. Pharmacol. 73, 1020–1028 [DOI] [PubMed] [Google Scholar]
- 52. Smith J. J., Cummins T. R., Alphy S., Blumenthal K. M. (2007) Molecular interactions of the gating modifier toxin ProTx-II with NaV 1.5. Implied existence of a novel toxin binding site coupled to activation. J. Biol. Chem. 282, 12687–12697 [DOI] [PubMed] [Google Scholar]
- 53. Priest B. T., Blumenthal K. M., Smith J. J., Warren V. A., Smith M. M. (2007) ProTx-I and ProTx-II. Gating modifiers of voltage-gated sodium channels. Toxicon 49, 194–201 [DOI] [PubMed] [Google Scholar]
- 54. Zhang J. Z., Yarov-Yarovoy V., Scheuer T., Karbat I., Cohen L., Gordon D., Gurevitz M., Catterall W. A. (2012) Mapping the interaction site for a β-scorpion toxin in the pore module of domain III of voltage-gated Na+ channels. J. Biol. Chem. 287, 30719–30728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kahn R., Karbat I., Ilan N., Cohen L., Sokolov S., Catterall W. A., Gordon D., Gurevitz M. (2009) Molecular requirements for recognition of brain voltage-gated sodium channels by scorpion α-toxins. J. Biol. Chem. 284, 20684–20691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cohen L., Karbat I., Gilles N., Ilan N., Benveniste M., Gordon D., Gurevitz M. (2005) Common features in the functional surface of scorpion β-toxins and elements that confer specificity for insect and mammalian voltage-gated sodium channels. J. Biol. Chem. 280, 5045–5053 [DOI] [PubMed] [Google Scholar]
- 57. Liu Y., Li D., Wu Z., Li J., Nie D., Xiang Y., Liu Z. (2012) A positively charged surface patch is important for hainantoxin-IV binding to voltage-gated sodium channels. J. Pept. Sci. 18, 643–649 [DOI] [PubMed] [Google Scholar]