

Background: Host innate immunity is against virus infection and replication.
Results: Toll-like receptor 3 activation leads to enhanced expression of a key Kaposi's sarcoma-associated herpesvirus (KSHV) protein.
Conclusion: KSHV uses host Toll-like receptor pathway to augment its critical gene expression.
Significance: A virus may usurp host innate immunity for its own benefits.
Keywords: Innate Immunity, Toll-like Receptors (TLR), Translation Control, TRIF, Tumor Viruses
Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is a human γ-herpesvirus. KSHV replication and transcription activator (RTA) is necessary and sufficient for KSHV reactivation from latency. Toll-like receptors (TLRs) recognize pathogen-associated molecular patterns, act through adaptors, and initiate innate and adaptive immune responses against pathogens. Toll/interleukin-1-receptor domain containing adaptor protein inducing interferon-β (TRIF) is an adaptor associated with TLR3 and TLR4 signaling, and is closely related to antiviral signaling to activate type I interferon (IFN) production. We previously found that KSHV RTA degrades TRIF indirectly and blocks TLR3 pathways. In this report, we find that TRIF, as well as TLR3 activation, enhances KSHV RTA protein expression. The C-terminal region of the RTA is involved in the responding TRIF-mediated enhancement. The degradation of TRIF and the enhancement of RTA expression are using two different pathways. The enhancement by TLR-TRIF is at least partially via promoting translational efficiency of RTA mRNA. Finally, the receptor-interacting protein 1 (RIP1) may be involved in TRIF-mediated enhancement of RTA expression, but not in the RTA-mediated degradation of TRIF. Therefore, the activation of TLR-TRIF pathway enhances KSHV RTA protein expression, and KSHV RTA in turn degrades TRIF to block innate immunity. The putative KSHV-TLR-adaptor-interacting loop may be a critical element to evade and usurp host innate immunity in KSHV life-cycle.
Introduction
Toll-like receptors (TLRs)4 are a family of evolutionarily conserved receptors that recognize molecular patterns unique to pathogens and activate host innate immunity against the pathogen (1, 2). One of the major products from TLR activation is production of inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin 1 (IL1), and type I interferons (IFN). IFN is key component to mount a proper and robust immune response to a viral infection (3, 4). Toll-IL-1 receptor (TIR) domain-containing adaptor inducing IFN-β (TRIF), also called TIR domain-containing adaptor molecule-1 (TICAM-1), is an adaptor protein involved in signal transduction during the activation of TLR3 and 4 leading to activation of nuclear factor κB (NF-κB) and type I IFN (5–7). TLR3 seems to be dependent on TRIF for its downstream cascades (5, 8).
Kaposi's Sarcoma-Associated Herpesvirus (KSHV), also known as human herpesvirus 8, is a member of the human γ-herpesviruses family. KSHV is believed to be an etiological factor for Kaposi's Sarcoma (KS), and associated with several other B lymphocytes malignancies such as primary effusion lymphoma and multicentric Castleman's disease. As other herpesviruses, KSHV consists of two distinct phases: latent and lytic replication during its life cycle. During latency, the virus establishes persistent infection and only a small subset of genes are typically expressed. Under conditions of lytic replication, all the viral genes are activated in cascade mode and new viruses are packaged and released from cells (9–12). KSHV replication and transcription activator (RTA) is an immediate early gene and highly conserved among γ-herpesviruses (13–15). RTA is apparently necessary and sufficient for the switch from KSHV latency to lytic replication (12, 16). Beyond functioning in initiating viral lytic replication, RTA is involved in the induction of cellular IL6 (17), degradation of TRIF, IFN regulatory factor 7 (IRF7), K-RBP, and Hey1 through proteasome pathway (18–21), and blockage of p53-mediated apoptosis by competing for binding to CBP (22). RTA interacts with other factors to modulate its transcription potential and other cellular activities (22–26).
KSHV mainly infects endothelial cells and B lymphocytes, and those cells express multiple TLRs (27–30). TLR4 is identified as an important barrier against KSHV infection, and KSHV has developed a mechanism to rapidly suppress TLR4 expression (31). KSHV infection activates TLR3 and TL9 pathways, and TLR7 signaling may lead to lytic replication of KSHV (31–34). Also, murine γ-herpesvirus 68 (MHV68) is a herpesvirus with significant similarities to KSHV. Activation of the TLR3/4 pathway potently inhibits the replication of MHV68 (35). However, other reports suggest that TLR3 activation increases MHV68 viral replication in vivo (36).
Previously, we found that KSHV RTA degrades TRIF protein through a proteasome pathway (21). In this report, we have found that TRIF up-regulates the expression of RTA protein. The enhanced RTA protein expression by TRIF is at the translational efficiency of its mRNA. The downstream target of TRIF, receptor-interacting protein 1 (RIP1), may be involved in the process. Those data strongly suggest that KSHV usurps host innate immune system for its own benefits.
MATERIALS AND METHODS
Plasmids, Antibodies, and dsRNA
Expression plasmids of KSHV RTA and its mutant (RTA-K152E), EBV RTA, pcDNA3.1-myc-TRIF, TRIF mutants (TRIF-Del, TRIF-N, and TRIF-C) were described previously (21, 37–42). RTA-ΔC plasmid (aa 1–527) was a gift from Dr. Charles Wood (43). Human FLAG-tagged-RIP1 expression plasmid was obtained from Dr. Ning Shunbin. RTA antibody was described (44). Pan-luc, K14A-luc, and β-galactosidase expression plasmids were all described (37). Tubulin (T6557) and FLAG antibody (F1804) were obtained from Sigma. The antibodies for Myc (SC-40), IRF-1 (SC-497), and GAPDH (SC-47724) were from Santa Cruz Biotechnology. TRIF antibody was from Cell Signaling (Cat. 4596). EBV-R antibody was from Argene (8C12). Poly (I:C) (double-stranded RNA) was purchased from InvivoGen (tlrl-pic) and used at 10 μg/ml.
Cell Culture
293T is a human fibroblast line. WT11(clone 9) is a TLR-3-expressing cell line (45) (gift from Dr. Ganes Sen). RIP1(−/−) and RIP1(+/+) lines were obtained from Dr. Ning Shunbin (46). Those cells were grown in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 1% penicillin-streptomycin (PS) at 37 °C in 5% CO2 incubation. 400 μg/ml G418 (Invitrogen) was used to maintain the TLR-3 expression in WT11 cells. BC3 is a KSHV-positive PEL line and were maintained in RPMI 1640 plus 10% FBS.
Transient Transfection
Effectene (Qiagen) was used for the transfection of 293T or WT11–9 cell lines following the manufacturer's recommendation. For BC3 cells, electroporation (320 V; 925 microfarads) was used for transfection along with other plasmids. One day after the transfection, the cells were used for the treatment of various concentrations of sodium butyrate. One day later, transfected cells were incubated with Dynabeads-CD4 beads for 20–30 min at room temperature with gentle rotation. CD4-positive cells were isolated by placing the test tubes in a magnetic separation device (Dynal magnet). The CD4-positive cells were washed three times in phosphate-buffered saline and used to prepare cell lysates.
Protein Stability Assays
Protein biosynthesis inhibitor, cycloheximide, was purchased from Sigma (C4859) and used at 50–100 μg/ml. Cells were transfected in 10-cm dishes, and transfected cells were split 4–6 h after transfection into a 6-well plate. Next day, the cells were treated with cycloheximide for the indicated period, and cell lysates were made and used for Western blot analysis.
Western Blot Analysis, RNA Extraction, and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Standard Western blot analysis was performed as described (47–49). Total RNA was isolated from cells using TRIzol extraction. cDNA was synthesized using 100 ng of RNA, oligo dT priming, and SuperScriptTM reverse transcriptase (Invitrogen). LGH4930 (5′-TTCGCCTGTTAGACGAAGC-3′) and LGH4929 (5′-GATTCGCAAGCTTCAGTCTCGGAAGTAATTACG-3′) primers were used for detection of RTA expression, and actin1 (5-TTCTACAATGAG CTGCGTGT-3′) and actin2 (5′-GCCAGACAGCACTGTGTTGG-3′) primers were used for actin control.
Translational Efficiency Assays
293T cells were transfected with various plasmids. 24 h after transfection, cells were washed, cultured in methionine-free DMEM (GIBCO) supplemented with 10% of dialyzed FBS for 30 min, and then labeled with of [35S]methionine (Perkin Elmer, 100 μCi/ml) for 30 min. Cells were lysed in 1% Nonidet P-40 buffer (100 mm Tris, pH 8.0, 1 mm EDTA, 100 mm NaCl, 1% Nonidet P-40, 1 mm PMSF plus one tablet of protease inhibitor (Roche) per 10 ml) for 30 min on ice. Supernatants were immunoprecipitated with FLAG antibody. Proteins were transferred to a PVDF membrane and allowing to air dry. The membrane was exposed overnight to a Kodak film to capture the 35S signal. The membrane was re-hydrated in 100% methanol, and Western blot was carried out with RTA antibody. The signal strengths were enumerated by the use of the Bio-Rad Software Quantity One (version 4.6.7).
Transient Transfection and Reporter Assays
Effectene (Qiagen) was used for the transfection of 293T cells. The luciferase assays were performed using the assay kit from Promega according to the manufacturer's recommendation.
RESULTS
TRIF Increases Steady State Levels of RTA Protein
To examine an effect of TRIF on RTA expression, equal amounts of RTA along with various amounts of TRIF plasmids were transfected into 293T cells. As shown in Fig. 1A, TRIF protein expression is gradually decreased as reported (21). However, the steady state of RTA protein expression was increased upon TRIF expression. Of note, the expression of TRIF could be detected with longer exposure and more lysates (data not shown). Epstein-Barr virus (EBV), the prototype of human γ-herpesvirus, has a RTA homologue (EBV-R). We further tested if EBV-R was regulated by TRIF expression with similar approach. EBV-R protein expression was not increased by TRIF expression, and EBV-R did not degrade TRIF (Fig. 1B). In addition, the same phenomenon can be observed in other human cell lines, such as MCF7 and U2OS (data not shown). Because RTA degrades TRIF, whether the same pathway was used by TRIF to enhance the expression of RTA was examined. RTA-K152E weakly degraded TRIF, but the expression of the mutant was also enhanced by TRIF (Fig. 1C). Furthermore, we tested if TRIF enhanced the RTA-mediated transactivation of reporter gene constructs. As shown in Fig. 1D, TRIF increased the activation of Pan as well as K14 promoter reporter constructs, probably through the enhancing the expression of RTA. Therefore, TRIF activation increased steady state levels of KSHV RTA protein, and the pathways for TRIF degradation and RTA enhancement seem to be different.
FIGURE 1.

TRIF increases the expression of RTA. A, 293T cells were transfected with cDNA, RTA (0.1 μg), and various TRIF plasmid (0.01, 0.05, 0.1 μg). The cell lysates were collected 1 day later, and the expression was examined by Western blot. B, same as A, but TRIF (0.1 μg) and EBV-R (0.05, 0.1 μg) were used. C, 293T cells were transfected with various plasmids as shown on the top. RTA (0.1 μg) and RTA-K152E (0.05 μg) were used for transfection. The images in the same box indicate that they are derived from the same membranes. The identity of proteins is as shown. D, 293T cells were transfected with various reporter constructs along with the CMV-β-gal, TRIF, or RTA expression plasmid as shown on the top. Both Pan and K14 promoters are well-known for their responsiveness to RTA. Luciferase activity was normalized to β-galactosidase activity. The fold activation of each promoter construct is shown with standard deviation. One representative of three independent experiments is shown.
TLR3 Activation Enhances RTA Expression
TRIF is required for TLR-3 signaling (5, 8). Whether the activation of TLR-3 increases RTA protein expression in TLR-3-expressing cells (WT11) was tested (45). The RTA expression plasmid was transfected into the cells, and the cells were treated with dsRNA for the activation of TLR3. The activation of TLR3 led to the enhancement of the RTA expression (Fig. 2A). This enhancement was specific for KSHV, as EBV-R could not be enhanced by TLR3 activation (Fig. 2A). Therefore, TLR3 signal specifically enhanced KSHV RTA expression.
FIGURE 2.
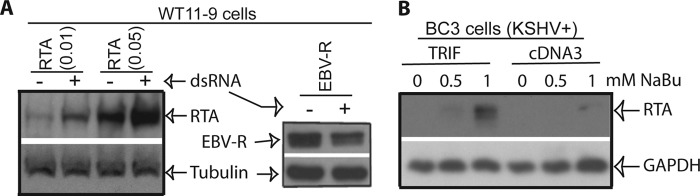
Activation of TLR-3 increases RTA expression. A, various plasmids were transfected into TLR-3-expressing cells (WT11–9) as shown on the top. After 4–6 h of transfection, the cells were washed once and then treated with(+) or without(−) dsRNA (10 μg/ml). The cells were collected after overnight treatment, and cell lysates were used for detection of RTA or EBV-R, respectively. The images in the same box indicate they are derived from the same membranes. The identities of the proteins are shown. B, BC3 cells were transfected with pcDNA3 or TRIF along with CD4 plasmids. Cells were treated with sodium butyrate (NaBu) for 24 h. The transfected cells were enriched, and the expressions of endogenous proteins were analyzed. Images in the same box indicate they derived from the same membrane. The identities of the proteins are shown.
TRIF Enhances Endogenous RTA Expression
To examine whether TRIF could enhance RTA under physiological conditions, KSHV latently infected PEL cell line, BC3, were transfected with various expression plasmids along with CD4 expression plasmids. BC3 was selected due to its transfection efficiency and relative tight control of the KSHV lytic replication. The cells were treated with sodium butyrate, and the transfected cells were enriched by CD4-magnetic bead selection as described before (50–54). The concentrations of sodium butyrate were adjusted to the levels that just barely induced the lytic replication of KSHV. Following the transfection of TRIF, the expression of RTA was clearly increased (Fig. 2B). Therefore, TRIF enhanced of RTA expression from viral genome in BC3 cells with chemical inductions.
TRIF Increases the Translation Efficiency of RTA mRNA
To examine the mechanism by which TRIF enhances the expression of RTA, whether the RNA translational efficiency was involved was examined. Basically, the cells were transfected with various plasmids, pulse labeled with [S35]methionine for 30 min, and cell lysates were used for immunoprecipitation with FLAG antibody, followed by Western blot analysis with RTA antibody. The newly synthesized proteins were metabolically labeled with [S35]methionine, and the relative translational efficiency was measured by the ratio of the newly synthesized proteins versus total proteins. As shown in Fig. 3, A and B, the translation efficiency of RTA mRNA was enhanced at least 50% in the presence of TRIF. In addition, TRIF did not have an effect on the half-life of RTA protein in the 24 h time period, during which RTA expression was enhanced (Fig. 3C). And the protein synthesis inhibitor was working properly because IRF1 has anticipated half-life (55). Next, the mRNA levels of RTA were not changed dramatically in the presence of TRIF (Fig. 3D). All those data collectively supports that notion that TRIF increased the translation efficiency of RTA mRNA, which consequently increased the levels of RTA proteins.
FIGURE 3.

TRIF increases the translation efficiency of RTA mRNA. A, 293T cells were transfected with cDNA, Flag-RTA (0.1 μg), and TRIF plasmid (0.05, 0.1 μg). The cells were labeled with [S35]methionine for 30 min, and the cells lysates were used for immunoprecipitation with FLAG antibody. The immunoprecipitates were separated and transferred onto Immobilon membranes. S35-labeled protein (newly synthesized) were measured. The membranes were then used for Western blots with RTA-antibody. The relative translation efficiency was calculated as newly-synthesized protein (S35-labeled) versus total proteins. Bottom numbers, the relative translation efficiency of RTA. B, average of relative translation efficiency of RTA from three independent experiments is as shown. Standard deviations are also shown. C, 293T cells were transfected with RTA or TRIF plus RTA plasmids. Cycloheximide (CHX) was added. Cell lysates were made at various time points after treatment (in hours). Western blot was performed to examine the protein stability. The images in the same box indicate that they are derived from the same membranes. D, different RTA-expression plasmids were transfected with various amounts of TRIF plasmids. Total RNA were isolated, and RTA mRNA levels were detected by semi-quantitative RT-PCR. Actin levels were used as a control. One representative of several independent experiments is shown.
Multiple Regions of TRIF Stimulate RTA Expression
To narrow down the region(s) of TRIF for the enhancement of RTA expression, we have used several mutants as shown in Fig. 4A (21). Those deletion mutants were transfected into 293T cells, and whether the TRIF mutants were enhancing the RTA expression was examined. As shown in Fig. 4B, all mutants were able to enhance the RTA expression. In addition, all those TRIF mutants were degraded by RTA as reported (21). These data suggest that the multiple regions of TRIF targeted RTA for its enhancement.
FIGURE 4.
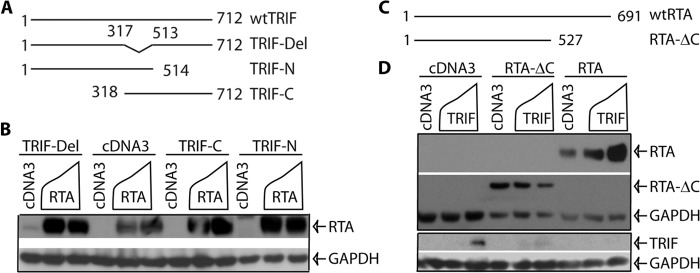
Domain analysis of TRIF-mediated enhancement of RTA. A, schematic diagram of TRIF mutant constructs. The number denotes amino acid positions. The drawing is not to scale. B, multiple regions of TRIF mediate the enhancement of RTA expression. 293T cells were transfected with vector pcDNA3, TRIF mutants (0.1 μg), and RTA (0.2 μg) expression plasmids in various combinations as shown on the top of the panel. Total DNA for transfection was maintained the same with the use of vector DNA. Cell lysates were made 1 day later, and Western blot analysis was performed. RTA and GAPDH antibodies were used. The identity of proteins is as shown. C, schematic diagram of RTA mutant constructs. The number denotes the amino acid positions. The drawing is not on scale. D, the C-terminal region of RTA is required for TRIF-mediated enhancement. 293T cells were transfected with cDNA, RTA-ΔC (aa1–527), RTA, and TRIF plasmids in various combinations. The expression of targets was examined by Western blot. RTA-ΔC was determined by FLAG antibody, and the RTA was determined by RTA-Ab. The images in the same box indicate they are derived from the same membranes. The identity of proteins is as shown.
A RTA Deletion Mutant Failed to Be Enhanced by TRIF
To determine the region of RTA responsive to TRIF expression, a serial of mutants had been examined, and one mutant in particular (RTA-ΔC) was different from others (Fig. 4C). TRIF failed to enhance the expression of the RTA-ΔC mutant, however, TRIF significantly enhanced the expression of wtRTA in the same assay (Fig. 4D). The expression of TRIF is also shown. Therefore, the C-terminal region of RTA was responsible for the enhancement of RTA expression by TRIF.
Receptor Interacting Protein-1 (RIP1) May Be Involved in the Enhancement of RTA Expression
Because we failed to detect physical interactions between TRIF and RTA (21), it is very likely that TRIF degradation and RTA expression enhancements occur indirectly. To examine the cellular factors involved in the degradation and enhancement, we used specific cell lines that lack critical components for TLR3 signaling, especially the downstream mediator of TRIF. RIP1 is critically involved in the TRIF-mediated NF-κB activation (56). In the RIP1(−/−) mouse embryonic fibroblast (MEF) line, TRIF failed to increase the expression of RTA; however, the degradation of TRIF was still exist (Fig. 5A). In the corresponding RIP1(+/+) MEFs, TRIF was still enhancing the expression of RTA (Fig. 5B). In addition, overexpression for human RIP1 alone was sufficient to increase the RTA expression in human 293T cells. RTA did not obviously degrade RIP1 because the relative ratios of RIP1 to tubulin were similar (Fig. 5C). All those data collectively suggested that RIP1 might be involved in TRIF-mediated enhancement of RTA expression.
FIGURE 5.
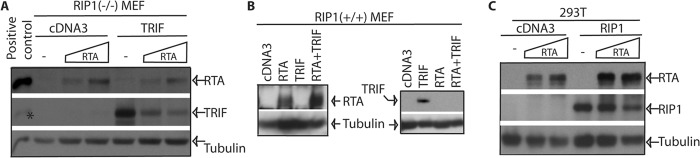
RIP1 may be involved in the enhancement of RTA expression. A and B, RIP1(−/−) MEFs (panel A) or RIP1(+/+) cells (panel B) were transfected with cDNA, TRIF, and RTA plasmid as shown on the top. Cells were collected 1 day later, and expression of target proteins was examined by Western blot with the RTA antibody first. The membranes were then stripped and probed with other antibodies. The identity of proteins is as shown.*: residual signals for RTA protein. The images in the same box indicate they are derived from the same membranes. C, 293T cells were transfected with cDNA, RIP1, and RTA plasmids. One day after the transfection, the cells were collected for Western blot analysis. The images in the same box indicate they are derived from the same membranes.
DISCUSSION
Innate immunity is important to control viral infection, and a successful counteraction of innate signaling may be a necessity for the survival of a virus in vivo. It has been shown that KSHV encodes several genes to counteract the innate system. KSHV uses at least two viral gene products to nullify the function of IRF7, a master gene for type I IFN production (20, 47). Latency-associated nuclear antigen blocks the activation of IFN through IRF-3 (48). ORF10 blocks the IFN signaling in KSHV-infected cells (49). On the other hands, it has been shown that virus may use molecules involved in the innate immunity for its own benefits. KSHV uses TLR7 signaling pathway for its reactivation process (33), EBV uses the same signaling pathway for its viral protein expression (57). MHV68 uses cellular Mavs adaptor molecular pathway to modify its gene products for the maximum replication (58). MyD88 may be involved in the establishment of MHV68 latency in vivo (59).
In this report, we studied the effects of TRIF on the regulation of KSHV. We find that: 1) TRIF specifically increases steady state levels of KSHV RTA protein and the increased RTA was functional (Fig. 1); 2) TLR3 activation leads to higher levels of steady-state RTA protein expression (Fig. 2A). Because TRIF is required for TLR3 signaling, the results suggest that the endogenous TRIF activation would contribute to RTA expression; 3) the ectopic expression of TRIF leads to enhance expression of RTA from the viral genome upon induction of lytic replication (Fig. 2B). All those data collectively indicates that TLR-TRIF pathway may enhance the expression of RTA under native environments. Therefore, there is a potential regulatory loop: the activation of TRIF leads to enhanced KSHV RTA protein expression, and RTA degrades TRIF to block innate immunity induced by TLR activations (Fig. 6).
FIGURE 6.
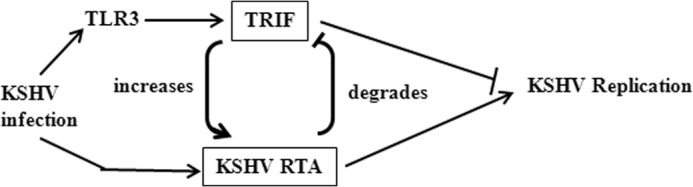
Schematic diagram of the TRIF and RTA interaction. Upon KSHV infection of a permissive cell, the TLR3 pathway is activated, and RTA is also expressed. RTA is able to degrade the critical adaptor molecule, TRIF, for TLR3 signaling, and consequently blocks TLR3-mediated innate immunity directed against KSHV. At the same time, the virus is able to use the TLR3-TRIF pathway to enhance the expression of RTA, and to expedite the degradation process of TRIF, and furthermore to block the TLR3-mediated inhibitory effects on KSHV replication.
We have extensively addressed the biological function of the regulatory loop. We thought the loop might suggest that TLR3 was a positive regulator of KSHV lytic replication as RTA is a key modulator of lytic replication. We have concentrated on the detection of TLR3 agonist (dsRNA) on the effect of KSHV for the following reasons: (A) KSHV infection activates TLR3 pathway. (B) TRIF is specifically involved in the TLR3 pathway; and (C) TLR4 already shown to be a negative regulator of KSHV (31). We had examined: (A) if TLR3 enhances spontaneous KSHV replication; (B) whether TLR3 activation plus chemical treatments lead to greater KSHV lytic replication; (C) If TLR3 activation at different times of lytic replication leads to KSHV replication enhancements. (D) Whether overexpression of TRIF in KSHV-infected cells, enhances the KSHV lytic replication. Of note, virus production in the supernatant was used as readout for the detection of viral replication, and all our data suggest that TLR3 activation and TRIF per se were negative regulators of KSHV lytic replication (data not shown). Therefore, the only evidence that TLR3 may enhance viral replication is from murine γ-herpesvirus 68 (MHV68), a murine KSHV homologue. It is reported that TLR3 activation increases MHV68 virus titers in vivo (36). However, other report argues against this notion (35). Based on all the results, it is apparent that TRIF-mediated RTA enhancement will facilitate the degradation of the TRIF itself, and thus blocks the inhibitory effects of TLR3 (Fig. 6).
TRIF is a multifunctional adaptor protein, mediating activation of several transcription factors including NF-κB (60), and trigger apoptosis. Our work has been added another novel function of TRIF, i.e. to enhance a viral gene expression for the benefits of viruses. The mechanism of the enhancement is mainly by promoting translational efficiency of RTA mRNA (Fig. 3), although other mechanisms, such as transcriptional control as well as protein stability, might also present. Furthermore, RIP1, which is a downstream molecule of TRIF activation pathway, might be involved in the TRIF-mediated enhancement of RTA expression (Fig. 5). However, the results seem to contradict to the fact that TRIF interacts with RIP1 using its C-terminal region. It is clear that the capability of TRIF to enhance RTA expression varies among different cell lines. In 293T cells, TRIF seems to have highest efficiency (Fig. 1 and data not shown). In addition, different TRIF fragments also have different efficiency for enhancing RTA expression. We summon that the different cell lines may be responsible for the apparent discrepancy. As RIP1 expression in 293T cells enhancing RTA expression (Fig. 5), RIP1 seems to be one of the mediators involved in the enhancement of RTA.
So far the exact responsive domain(s) for RTA to respond to TLR3/TRIF activation is not completely identified (Fig. 4). We narrowed the region down to the C-terminal (Fig. 4). Based on the results in 293T cells, a fine mapping of the RTA domain responsive to TRIF had been proven to be difficult (data not shown). RTAΔC is unique that it did not respond to TRIF. We suspect that multiple regions of RTA may response to different regions of TRIF for the enhancements.
The detailed molecular mechanism for the enhancement of translation efficiency of RTA mRNA is currently unknown. It is known that TRIF signaling stimulates translation efficiency of TNF-α mRNA via prolonged activation of protein kinase MK2 in macrophages (61). Also, TLR-TRIF signaling has been shown to suppress endoplasmic reticulum stress-induced translation inhibition through activation of eIF2B (62, 63). Whether a similar mechanism is also used for TRIF-mediated RTA enhancement is under investigation. The specific enhancement of KSHV RTA may have a unique mechanism because the responsive sequences are located in the C terminus of the molecule (Fig. 4).
We propose the following scenario upon primary KSHV infection: KSHV infection of cells would lead to the activation of TLR3, which may result in IFN production as well as the apoptosis of the infected cells for the suppression of viral replication. KSHV RTA is an immediate early gene that expressed upon viral infection, and RTA may block TLR3-mediated innate immunity by degrading TRIF. Furthermore, the activation of TLR3 pathway leads to the enhancement of RTA protein expression, which would accelerate the degradation process of TRIF, which would attenuate the TLR3 signaling (Fig. 6). In addition, RTA may indirectly affect TLR4 signaling by activating vIRF1 that inhibits TLR4 mRNA expression. Because TLR4 is involved in the KSHV pathogenesis in KS and exerts innate immunity against KSHV (31), and TRIF is involved in TLR3, 4 signaling (60), the putative regulatory loop may play an important role in KSHV life cycle.
In sum, we have identified a novel method that KSHV uses for its gene expression. Together with our previous findings, a regulatory loop is apparent: KSHV degrades TRIF, a TLR adaptor, to block TLR-mediated antiviral effects; and TRIF enhances expression of a KSHV critical protein to block TLR3-mediated repressive effects. This report provides evidence that a virus not only blocks the host innate immunity, but also harnesses an immune pathway for the benefits of virus infection.
Acknowledgments
We thank Shunbin Ning, Charles Wood, Rongtuan Lin, and Ganes Sen for providing various reagents for this work and Dr. Clinton Jones for critical reading of the manuscript.
This work was supported in part by National Institutes of Health Grants CA138213, RR15635, and Grant W81XWH-12-1-0225 (to L. Z.) from the Department of Defense-Army Medical Research.
- TLR
- Toll-like receptor
- KSHV
- Kaposi's sarcoma-associated herpesvirus
- RTA
- replication and transcription activator
- TRIF
- Toll/interleukin-1-receptor domain containing adaptor protein inducing interferon-β
- TIR
- Toll-IL-1 receptor
- RIP
- receptor-interacting protein.
REFERENCES
- 1. Kawai T., Akira S. (2007) TLR signaling. Semin Immunol. 19, 24–32 [DOI] [PubMed] [Google Scholar]
- 2. Takeda K., Akira S. (2007) Toll-like receptor. Curr. Protoc. Immunol. Chapter 14, Unit 14 12 [DOI] [PubMed] [Google Scholar]
- 3. Samuel C. E. (2001) Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sen G. (2001) Viruses and interferons. Annu. Rev. Microbiol. 55, 255–281 [DOI] [PubMed] [Google Scholar]
- 5. Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K., Akira S. (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643 [DOI] [PubMed] [Google Scholar]
- 6. Yamamoto M., Sato S., Hemmi H., Uematsu S., Hoshino K., Kaisho T., Takeuchi O., Takeda K., Akira S. (2003) TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat. Immunol. 4, 1144–1150 [DOI] [PubMed] [Google Scholar]
- 7. Hoebe K., Du X., Georgel P., Janssen E., Tabeta K., Kim S. O., Goode J., Lin P., Mann N., Mudd S., Crozat K., Sovath S., Han J., Beutler B. (2003) Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748 [DOI] [PubMed] [Google Scholar]
- 8. Sancho-Shimizu V., Pérez de Diego R., Lorenzo L., Halwani R., Alangari A., Israelsson E., Fabrega S., Cardon A., Maluenda J., Tatematsu M., Mahvelati F., Herman M., Ciancanelli M., Guo Y., AlSum Z., Alkhamis N., Al-Makadma A. S., Ghadiri A., Boucherit S., Plancoulaine S., Picard C., Rozenberg F., Tardieu M., Lebon P., Jouanguy E., Rezaei N., Seya T., Matsumoto M., Chaussabel D., Puel A., Zhang S. Y., Abel L., Al-Muhsen S., Casanova J. L. (2011) Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Invest. 121, 4889–4902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moore P., Chang Y. (2001) in Virology (Knipe D. M., ed), pp. 2803–2833, Lippincott Williams and Wilkins, Philadelphia [Google Scholar]
- 10. Moore P. S., Chang Y. (1995) Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N. Engl. J. Med. 332, 1181–1185 [DOI] [PubMed] [Google Scholar]
- 11. Moore P. S., Chang Y. (1998) Kaposi's sarcoma-associated herpesvirus-encoded oncogenes and oncogenesis. J. Natl. Cancer Inst. Monogr. 23, 65–71 [DOI] [PubMed] [Google Scholar]
- 12. West J. T., Wood C. (2003) The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 22, 5150–5163 [DOI] [PubMed] [Google Scholar]
- 13. Sarid R., Flore O., Bohenzky R. A., Chang Y., Moore P. S. (1998) Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72, 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun R., Lin S. F., Staskus K., Gradoville L., Grogan E., Haase A., Miller G. (1999) Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 73, 2232–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu F. X., Cusano T., Yuan Y. (1999) Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73, 5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dourmishev L. A., Dourmishev A. L., Palmeri D., Schwartz R. A., Lukac D. M. (2003) Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol. Biol. Rev. 67, 175–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng H., Chu J. T., Rettig M. B., Martinez-Maza O., Sun R. (2002) Rta of the human herpesvirus 8/Kaposi sarcoma-associated herpesvirus up-regulates human interleukin-6 gene expression. Blood 100, 1919–1921 [DOI] [PubMed] [Google Scholar]
- 18. Gould F., Harrison S. M., Hewitt E. W., Whitehouse A. (2009) Kaposi's sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 83, 6727–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Z., Yan Z., Wood C. (2008) Kaposi's sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 82, 3590–3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu Y., Wang S. E., Hayward G. S. (2005) The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22, 59–70 [DOI] [PubMed] [Google Scholar]
- 21. Ahmad H., Gubbels R., Ehlers E., Meyer F., Waterbury T., Lin R., Zhang L. (2011) Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing β-interferon (TRIF). J. Biol. Chem. 286, 7865–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gwack Y., Hwang S., Byun H., Lim C., Kim J. W., Choi E. J., Choe J. (2001) Kaposi's sarcoma-associated herpesvirus open reading frame 50 represses p53-induced transcriptional activity and apoptosis. J. Virol. 75, 6245–6248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang S., Liu S., Wu M. H., Geng Y., Wood C. (2001) Identification of a cellular protein that interacts and synergizes with the RTA (ORF50) protein of Kaposi's sarcoma-associated herpesvirus in transcriptional activation. J. Virol. 75, 11961–11973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gwack Y., Byun H., Hwang S., Lim C., Choe J. (2001) CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J. Virol. 75, 1909–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gwack Y., Hwang S., Lim C., Won Y. S., Lee C. H., Choe J. (2002) Kaposi's Sarcoma-associated herpesvirus open reading frame 50 stimulates the transcriptional activity of STAT3. J. Biol. Chem. 277, 6438–6442 [DOI] [PubMed] [Google Scholar]
- 26. Liao W., Tang Y., Lin S. F., Kung H. J., Giam C. Z. (2003) K-bZIP of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J. Virol. 77, 3809–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fitzner N., Clauberg S., Essmann F., Liebmann J., Kolb-Bachofen V. (2008) Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin. Vaccine Immunol. 15, 138–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bourke E., Bosisio D., Golay J., Polentarutti N., Mantovani A. (2003) The toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood 102, 956–963 [DOI] [PubMed] [Google Scholar]
- 29. Zarember K. A., Godowski P. J. (2002) Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 168, 554–561 [DOI] [PubMed] [Google Scholar]
- 30. Muzio M., Bosisio D., Polentarutti N., D'Amico G., Stoppacciaro A., Mancinelli R., van't Veer C., Penton-Rol G., Ruco L. P., Allavena P., Mantovani A. (2000) Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J. Immunol. 164, 5998–6004 [DOI] [PubMed] [Google Scholar]
- 31. Lagos D., Vart R. J., Gratrix F., Westrop S. J., Emuss V., Wong P. P., Robey R., Imami N., Bower M., Gotch F., Boshoff C. (2008) Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe 4, 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. West J., Damania B. (2008) Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus during primary infection. J. Virol. 82, 5440–5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gregory S. M., West J. A., Dillon P. J., Hilscher C., Dittmer D. P., Damania B. (2009) Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. U.S.A. 106, 11725–11730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. West J. A., Gregory S. M., Sivaraman V., Su L., Damania B. (2011) Activation of plasmacytoid dendritic cells by Kaposi's sarcoma-associated herpesvirus. J. Virol. 85, 895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doyle S., Vaidya S., O'Connell R., Dadgostar H., Dempsey P., Wu T., Rao G., Sun R., Haberland M., Modlin R., Cheng G. (2002) IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17, 251–263 [DOI] [PubMed] [Google Scholar]
- 36. Gargano L. M., Forrest J. C., Speck S. H. (2009) Signaling through Toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. J. Virol. 83, 1474–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang J., Wang J., Wood C., Xu D., Zhang L. (2005) Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 replication and transcription activator regulates viral and cellular genes via interferon-stimulated response elements. J. Virol. 79, 5640–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin R., Yang L., Nakhaei P., Sun Q., Sharif-Askari E., Julkunen I., Hiscott J. (2006) Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J. Biol. Chem. 281, 2095–2103 [DOI] [PubMed] [Google Scholar]
- 39. Zhang L., Pagano J. S. (1997) IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell Biol. 17, 5748–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jiang Y., Xu D., Zhao Y., Zhang L. (2008) Mutual inhibition between Kaposi's sarcoma-associated herpesvirus and Epstein-Barr virus lytic replication initiators in dually-infected primary effusion lymphoma. PLoS ONE 3, e1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao T., Yang L., Sun Q., Arguello M., Ballard D. W., Hiscott J., Lin R. (2007) The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nat. Immunol. 8, 592–600 [DOI] [PubMed] [Google Scholar]
- 42. Grandvaux N., Servant M. J., tenOever B., Sen G. C., Balachandran S., Barber G. N., Lin R., Hiscott J. (2002) Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 76, 5532–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wen H. J., Yang Z., Zhou Y., Wood C. (2010) Enhancement of autophagy during lytic replication by the Kaposi's sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 84, 7448–7458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu D., Coleman T., Zhang J., Fagot A., Kotalik C., Zhao L., Trivedi P., Jones C., Zhang L. (2007) Epstein-Barr virus inhibits Kaposi's sarcoma-associated herpesvirus lytic replication in primary effusion lymphomas. J. Virol. 81, 6068–6078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sarkar S. N., Smith H. L., Rowe T. M., Sen G. C. (2003) Double-stranded RNA signaling by Toll-like receptor 3 requires specific tyrosine residues in its cytoplasmic domain. J. Biol. Chem. 278, 4393–4396 [DOI] [PubMed] [Google Scholar]
- 46. Huye L. E., Ning S., Kelliher M., Pagano J. S. (2007) Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol. Cell Biol. 27, 2910–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang L., Pagano J. S. (1999) Interferon regulatory factor 2 represses the Epstein-Barr virus BamHI Q latency promoter in type III latency. Mol. Cell Biol. 19, 3216–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang L., Pagano J. S. (2000) Interferon regulatory factor 7 is induced by Epstein-Barr virus latent membrane protein 1. J. Virol. 74, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang L., Zhang J., Lambert Q., Der C. J., Del Valle L., Miklossy J., Khalili K., Zhou Y., Pagano J. S. (2004) Interferon regulatory factor 7 is associated with Epstein-Barr virus-transformed central nervous system lymphoma and has oncogenic properties. J. Virol. 78, 12987–12995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu D., Brumm K., Zhang L. (2006) The latent membrane protein 1 of Epstein-Barr virus (EBV) primes EBV latency cells for type I interferon production. J. Biol. Chem. 281, 9163–9169 [DOI] [PubMed] [Google Scholar]
- 51. Xu D., Zhao L., Del Valle L., Miklossy J., Zhang L. (2008) Interferon regulatory factor 4 is involved in Epstein-Barr virus-mediated transformation of human B lymphocytes. J. Virol. 82, 6251–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang J., Das S. C., Kotalik C., Pattnaik A. K., Zhang L. (2004) The latent membrane protein 1 of Epstein-Barr virus establishes an antiviral state via induction of interferon-stimulated genes. J. Biol. Chem. 279, 46335–46342 [DOI] [PubMed] [Google Scholar]
- 53. Zhang L., Hong K., Zhang J., Pagano J. (2004) Multiple signal transducers and activators of transcription are induced by EBV LMP-1. Virology 323, 141–152 [DOI] [PubMed] [Google Scholar]
- 54. Zhang L., Wu L. H., Hong K., Pagano J. S. (2001) Intracellular signaling molecules activated by Epstein-Barr virus for induction of interferon regulatory factor 7. J. Virol. 75, 12393–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harada H., Kitagawa M., Tanaka N., Yamamoto H., Harada K., Ishihara M., Taniguchi T. (1993) Anti-oncogenic and oncogenic potentials of interferon regulatory factors-1 and -2. Science 259, 971–974 [DOI] [PubMed] [Google Scholar]
- 56. Meylan E., Burns K., Hofmann K., Blancheteau V., Martinon F., Kelliher M., Tschopp J. (2004) RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κB activation. Nat. Immunol. 5, 503–507 [DOI] [PubMed] [Google Scholar]
- 57. Valente R. M., Ehlers E., Xu D., Ahmad H., Steadman A., Blasnitz L., Zhou Y., Kastanek L., Meng B., Zhang L. (2012) Toll-like receptor 7 stimulates the expression of Epstein-Barr virus latent membrane protein 1. PLoS One 7, e43317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dong X., Feng H., Sun Q., Li H., Wu T. T., Sun R., Tibbetts S. A., Chen Z. J., Feng P. (2010) Murine gamma-herpesvirus 68 hijacks MAVS and IKKβ to initiate lytic replication. PLoS Pathog 6, e1001001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gargano L. M., Moser J. M., Speck S. H. (2008) Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J. Virol. 82, 3853–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. O'Neill L. A., Bowie A. G. (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 61. Gais P., Tiedje C., Altmayr F., Gaestel M., Weighardt H., Holzmann B. (2010) TRIF signaling stimulates translation of TNF-α mRNA via prolonged activation of MK2. J. Immunol. 184, 5842–5848 [DOI] [PubMed] [Google Scholar]
- 62. Woo C. W., Kutzler L., Kimball S. R., Tabas I. (2012) Toll-like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat. Cell Biol. 14, 192–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Woo C. W., Cui D., Arellano J., Dorweiler B., Harding H., Fitzgerald K. A., Ron D., Tabas I. (2009) Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat. Cell Biol. 11, 1473–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]