


Abstract
Effective drug discovery and optimization can be accelerated by techniques capable of deconvoluting the complexities often present in targeted biological systems. We report a single-molecule approach to study the binding of an alternative splicing regulator, muscleblind-like 1 protein (MBNL1), to (CUG)n = 4,6 and the effect of small molecules on this interaction. Expanded CUG repeats (CUGexp) are the causative agent of myotonic dystrophy type 1 by sequestering MBNL1. MBNL1 is able to bind to the (CUG)n–inhibitor complex, indicating that the inhibition is not a straightforward competitive process. A simple ligand, highly selective for CUGexp, was used to design a new dimeric ligand that binds to (CUG)n almost 50-fold more tightly and is more effective in destabilizing MBNL1–(CUG)4. The single-molecule method and the analysis framework might be extended to the study of other biomolecular interactions.
INTRODUCTION
Myotonic dystrophy type 1 (DM1) is a triplet-repeat disease originating from a progressive expansion of the CTG repeat (CTGexp) in the 3′-untranslated region of the DMPK gene (1). The expanded DMPK gene produces a toxic RNA transcript (CUGexp containing RNA) that does not exit the nucleus but associates with proteins. One of these proteins, muscleblind-like 1 protein (MBNL1), is an important regulator of alternative splicing (2). Sequestration of MBNL1 in nuclear foci leads to multiple mis-spliced pre-mRNAs, incorrect protein levels and ultimately the disease (3). In a mouse model of DM1, a morpholino antisense oligonucleotide (ASO) (1), a 2′-O-(2-methoxyethyl) ASO (4) and a d-amino acid hexapeptide (ABP1) (5) reversed this process, rescued normal splicing and reversed the phenotype in mice thereby validating CUGexp as a therapeutic target. Because no drugs are currently available to treat DM1, there is intense interest in finding small molecules that may function in a manner similar to the morpholino antisense oligonucleotides, but avoid the limitations inherent in the antisense therapeutic approach (6). Pentamidine (7), benzo[g]quinolone heterocycle derivatives (8), a Hoechst derivative (H1) (9) and modularly assembled Hoechst 33258 (10) are examples of bioactive CUGexp binders that are able to restore MBNL1 function in DM1 cell and animal models.
Central to the discovery of new and improved therapeutic agents for DM1 is to understand how small molecules bind CUGexp and the mechanism by which they block the MBNL1 binding. Previously, we and others have analyzed the MBNL1–CUGexp-inhibitor system using a competitive inhibition model and have used bulk solution techniques to analyze the equilibrium binding. However, these bulk solution methods usually require large sample volumes and have low sensitivity. Most importantly, these techniques do not provide the kinetic information that can be vital for accelerating drug discovery and development (11) and sometimes do not give a full thermodynamic picture. Surface-based biosensors provide kinetic information, directly in real-time with a fast response and high sensitivity, by measuring the interaction between an immobilized macromolecule and its soluble binding partner. The localized surface plasmon resonance (LSPR) technique, in particular, is developing rapidly into a powerful method (12). Nonetheless, the SPR techniques are model dependent and can have difficulties distinguishing different analytes, for example, a protein from an inhibitor (13).
The lack of a suitable method for studying the MBNL1–CUGexp interaction and its inhibition by small molecules means that the overall complexation process remains largely unexplored. The same holds true for many other systems where it is desirable to disrupt off-pathway protein–RNA interactions without affecting the biologically important function of the protein. To aid our drug discovery program and provide a potentially general approach to study RNA–protein inhibition, we developed a single-molecule method to analyze the MBNL1–CUGexp-inhibitor system. The major advantage of our model-independent approach is the ability to detect and measure the individual binding events in real-time under equilibrium conditions (14). Analyzing the distributions of bound and free states gives the most reliable on and off rates and a full picture of the inhibition process (15,16).
Herein, we describe the use of total internal reflection fluorescence (TIRF) microscopy to study, at the single-molecule level, the inhibition of MBNL1 binding to CUG repeat by known small molecule inhibitor 1 (Figure 1). The synthesis of dimeric inhibitor ligand 2, based on structure 1, is described and evaluated using the TIRF method. Ligand 2 was shown to be bioactive in DM1 cell models (data will be published elsewhere). Both compounds are found to bind RNA at the same time as MBNL1, indicating that the inhibition of MBNL1–CUGexp interaction does not follow a simple competitive mechanism. Rather than competitive steric blocking of the MBNL1 complexation site on CUG repeat, it seems the ligands are accelerating the dissociation of the protein.
Figure 1.
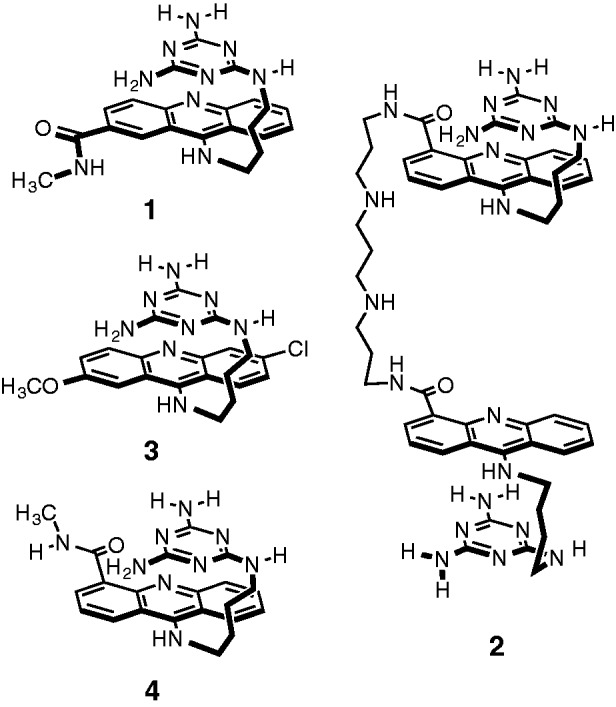
Compounds 1–4 used in this study.
MATERIALS AND METHODS
Instrumentation and chemicals
All reagents were purchased from commercial suppliers and were used without further purification. Anhydrous solvents were obtained from an anhydrous solvent dispensing system. For all reactions using anhydrous solvents, glassware was oven-dried, cooled under vacuum and then purged with dry nitrogen; all reactions were conducted under dry nitrogen. Purified compounds were further dried under high vacuum (0.01–0.05 Torr) or lyophilized using a Labconco lyophilizer. Yields refer to purified and spectroscopically pure compounds. Nuclear magnetic resonance (NMR) spectra were recorded at 23°C on either Varian Unity 500 or Varian Unity Inova 500NB, operating at 500 and 125 MHz for 1H and 13C acquisitions, respectively. NMR spectra were processed using MestReNova software. Chemical shifts are reported in ppm and referenced to the corresponding residual nuclei in the following deuterated solvents: CDCl3 (7.26 ppm 1H, 77.16 ppm 13C); dimethyl sulfoxide (DMSO) (2.50 ppm 1H, 39.52 ppm 13C); D2O (4.79 ppm 1H); and CD3OD (3.31 ppm 1H, 50.41 ppm 13C). Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), sext (sextet), dd (doublet of doublets), ddd (doublet of doublet of doublets), td (triplet of doublets), dt (doublet of triplets), m (multiplet) and b (broad). Integration is provided, and coupling constants, J, are reported in Hertz (Hz). Electrospray ionization (ESI) mass spectra were recorded using the Quattro or ZMD mass spectrometer. High-resolution mass spectra (HRMS) were obtained at the University of Illinois mass spectrometry facility. All compounds described herein gave NMR and mass spectral data in accord with their structures. Ligands 1 and 2 gave an HRMS within 5 ppm of calculated values.
9 -((4 -((4,6-Diamino-1,3,5-triazin-2-yl)amino)butyl)amino)-N-methylacridine-2-carboxamide (1). A round-bottom flask, equipped with a stir bar, was charged with S5 (290 mg, 1.07 mmol, 1 eq) and S1 (233 mg, 1.18 mmol, 1.1 eq) (compounds S1 and S5 and synthetic scheme are shown in Supplementary Figure S1). N,N-diisopropylethylamine (DIPEA) (305 mg, 2.36 mmol, 2.2 eq) and anhydrous N,N-dimethylformamide (DMF) (20 ml) were added. The solution was heated at 70°C for 5 h. The solvent was removed by rotary evaporation, and the product was purified via flash chromatography (basic alumina; DCM:methanol:NH4OH, 95:4.9:0.1 to 85:14:1) to yield a yellow solid (377 mg, 0.76 mmol, 71%). 1H NMR (500 MHz, methanol-d4) δ 8.91 (s, 1H), 8.33 (d, J = 8.6 Hz, 1H), 8.02 (dd, J = 9.1, 1.9 Hz, 1H), 7.85 (t, J = 7.6 Hz, 2H), 7.75–7.68 (m, 1H), 7.42–7.35 (m, 1H), 4.03 (t, J = 7.2 Hz, 2H), 3.37–3.33 (m, 2H), 3.00 (s, 3H), 1.93 (p, J = 7.4 Hz, 2H), 1.70 (p, J = 7.2 Hz, 2H); m/z HRMS (ESI) calculated for [M + H]+: 432.2260; found 432.2267.
N,N′-((Propane-1,3-diylbis(azanediyl))bis(propane-3,1-diyl))bis(9 -((4 -((4,6-diamino-1,3,5-triazin-2-yl)amino)butyl)amino)acridine-4-carboxamide) (2). A round-bottom flask, equipped with a stir bar, was charged with S12 (310 mg, 0.261 mmol, 1 eq). (compound S12 and synthetic scheme are shown in Supplementary Figures S2 and S3). trifluoroacetic acid (TFA) (30 ml) and anhydrous DCM (70 ml) was added, and the mixture was stirred at room temperature for 2 h. The solvents were removed to afford a yellow solid (437 mg, 0.261 mmol, 100%). 1H NMR (500 MHz, deuterium oxide) δ 8.34 (d, J = 8.3 Hz, 2H), 8.17 (d, J = 8.3 Hz, 2H), 8.11 (d, J = 7.4 Hz, 2H), 7.78 (t, J = 7.7 Hz, 2H), 7.52 (d, J = 8.3 Hz, 2H), 7.48–7.39 (m, 4H), 4.07 (t, J = 6.8 Hz, 4H), 3.58 (t, J = 6.7 Hz, 4H), 3.37–3.29 (m, 4H), 3.26 (t, J = 5.9 Hz, 8H), 2.31 (s, 2H), 2.14–2.08 (m, 4H), 1.98–1.91 (m, 4H), 1.71–1.65 (m, 4H); elemental analysis, calculated for C63H70F18N20O14: C, 45.22%; H, 4.22%; F, 20.44%; N, 16.74%; found: C, 45.15%; H, 4.20%; F, 20.09%; N, 16.46%, m/z HRMS (ESI) calculated for [M + H]+: 989.5599; found 989.5590.
MBNL1N plasmid and RNA
The expression vector pGEX-6 p-1/MBNL1N was obtained from Maurice S. Swanson (University of Florida, College of Medicine, Gainesville, FL, USA) (17). MBNL1N comprises the four zinc-finger motifs of MBNL1, the RNA-binding module of MBNL1 (17). It contains a 6xHis tag at the C-terminus and the Glutatione S-transferase (GST) tag at the N-terminus. MBNL1N binds RNA with similar affinity as the full-length MBNL1, but it does not form oligomers characteristic of the full-length protein (17). It is referred to as MBNL1 throughout this article for the sake of simplicity. All the oligonucleotides were purchased from Integrated DNA Technology and were high-performance liquid chromatography purified. The sequences and modifications for RNA constructs used in this study are shown in Supplementary Note S5.
MBNL1N protein expression and purification
Using BL21-CodonPlus(DE3)-RP competent cells (Stratagene), the expression of MBNL1N protein was induced with 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) at OD600 0.6 in Lysogeny Broth (LB) media with ampicillin for 2 h at 37°C. Bacterial cells were collected by centrifugation and were then resuspended in a lysis buffer containing 25 mM Tris–Cl (pH = 8), 0.5 M NaCl, 10 mM imidazole, 2 mM beta-mercaptoethanol (BME), 5% glycerol, 0.1% Triton X-100, 2 mg/ml lysozyme, 0.1 mM phenylmethanesulfonylfluoride (PMSF), 1 µM pepstatin and 1 µM leupeptin and sonicated six times for 15 s each. The cell pellet was centrifuged, and the clarified lysate was collected and filtered through a 45-µm Millex Filter (Millipore). To purify MBNL1N, Ni-Nitrilotriacetic acid (NTA) agarose (QIAGEN) was incubated with the lysate for 1 h at 4°C and washed with a washing buffer containing 25 mM Tris–Cl (pH = 8), 0.5 M NaCl, 20 mM imidazole and 0.1% Triton X-100, followed by elution with elution buffer of 25 mM Tris–Cl (pH = 8), 0.5 M NaCl, 250 mM imidazole and 0.1% Triton X-100. The eluate containing the GST fusion 6xHis-MBNL1N was dialyzed against phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4, pH 7.4), for SPR studies. The molecular weight was confirmed by Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry, and the concentration was determined by Bradford assay.
Preparation of Cy3-MBNL1 protein for TIRFM study
The GST fusion protein was incubated with Glutathione Sepharose 4B (GE Healthcare) for 1 h at 4°C. After washing with a buffer containing 25 mM Tris–Cl (pH = 8), 300 mM NaCl, 5 mM BME and 0.1% Triton X-100, the beads were collected and incubated with PreScission Protease (GE Healthcare) overnight at 4°C. After being cleaved from the beads, the protein was collected in the flow-through of the column. Fluorescent labeling of MBNL1 was performed by coupling Cy3 mono-reactive NHS esters (GE Healthcare) to the N-terminal amine group at pH 7.0 (18–20). MBNL1 was mixed with a 12.5-fold molar excess of the Cy3 mono-reactive NHS ester in potassium phosphate buffer (62 mM K2HPO4, 38 mM KH2PO4, pH 7.05, 100 mM NaCl and 1 mM dithiothreitol (DTT)) for 10 min at room temperature. The reaction mixture was incubated for 12 h at 4°C. The labeling reaction was terminated by the addition of 50 mM Tris–HCl, pH 7.5. Cy3-labeled MBNL1 was separated from the free dye using PD SpinTrap G-25 column (GE healthcare). The ratio of dye incorporated per protein molecule was determined to be 1.1 mol Cy3 per 1 mol MBNL1.
Surface plasmon resonance analysis
Detailed experimental procedure can be found in the Supplementary Methods.
Steady-state fluorescence-based–binding assays
To determine the equilibrium parameters for binding of 1 and 2 to CUGexp, we followed quenching of TAMRA in TAMRA-(CUG)6 at various ligand concentrations. The assays were performed using a Cary Eclipse Fluorescence Spectrophotometer (Varian). TAMRA-(CUG)6 was excited at 560 nm, and its emission was recorded at 590 nm. Stoichiometric titrations were carried out at 20°C in PBS, 1× buffer. The baseline fluorescence was recorded before addition of 20 nM TAMRA-(CUG)6. Increase in the fluorescence was recorded and attributed to the fluorescence of TAMRA-(CUG)6. On addition of each aliquot of 1 or 2, the fluorescence signal was allowed to equilibrate, recorded for 5 min and averaged. Titration was continued at a series of increasing final concentrations of the ligand until the fluorescence was completely quenched. Fluorescence intensities at different concentrations of 1 (1.95. 3.91, 7.81, 15.6, 31.4, 62.8, 94.2, 125.6, 157, 500 and 1000 µM) and 2 (39, 90, 210, 300, 390, 480, 570, 660, 750, 840 and 1250 nM) were fit to the following equation using Kaleidagraph software:
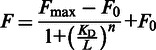 |
where KD is the dissociation binding constant, [L] is the ligand concentration, n is the Hill coefficient and F0 and Fmax are the fluorescence intensity of free and fully bound RNA, respectively. In the control experiment, ligands 1 and 2 had no effect on the fluorescence intensity of the free TAMRA.
Reaction conditions for the single-molecule assay
Biotinylated (CUG)4 or (CUG)12 were immobilized on a quartz surface, which was coated with polyethylene glycol to eliminate non-specific surface adsorption of proteins. The immobilization was mediated by biotin–neutravidin interaction between biotinylated Cy5-labeled (CUG)4, neutravidin and biotinylated polymer (polyethylene glycol (PEG), MW = 5000). Standard PBS buffer contained the oxygen scavenging system consisting of 1 mg/ml glucose oxidase, d-glucose (0.4%, w/v), 2-mercaptoethanol (1%, v/v) and 0.04 mg/ml catalase. Immobilization of 50 pM of each oligonucleotide allowed for detection of 100–600 individual molecules per slide. Cy3-labeled MBNL1 was then added and incubated for 5 min at 25°C in the standard PBS, 1× buffer in the absence or presence of the indicated concentrations of 1 and 2. Sample excitation and data recording were initiated after all components of the MBNL1– (CUG)4-inhibitor system were allowed to equilibrate in the TIRFM sample chamber. The presence of ligands had no effect on the fluorescence intensity of the Cy3-labeled MBNL1.
Single-molecule data acquisition and analysis
TIRFM was used to excite fluorophores present near the surface, within the evanescent field. Cy3 fluorescence was excited by a DPSS laser (532 nm, 75 mW), whereas diode laser (641 nm, 100 mW) was used for direct Cy5 excitation. The fluorescence signals originated from the Cy3 and Cy5 dyes were collected by a water immersion ×60 objective, separated by a 630 nm dichroic mirror, passed through a 550 nm long-pass filter to block out laser scattering and detected by EMCCD camera with a time resolution of 100 ms. Single-molecule fluorescence trajectories were extracted from the recorded video file by IDL software. The collected trajectories were visualized using an in-house MATLAB program and were validated for the presence of the Cy5 signal by visual inspection of the acquired data. The Cy3 excitation regions of all individual trajectories were fit to the two-state model using the QuB software. This fitting generated the dwell times in the bound (ON) and free (OFF) states of the immobilized RNA molecules, which were binned and plotted as the histograms. The resulting ON and OFF event distributions were globally fit to exponential decay functions or to models described by Equations (1) and (2) using Prism 4.0 software to obtain the kinetic parameters.
RESULTS
Design and synthesis of small molecules
Our previous studies validated 3 (Figure 1) as a selective CUG binder, which was able to inhibit the MBNL1–CUG interaction with an apparent IC50 = 52 ± 20 µM (21). Because the ultimate RNA target is CUGexp, a logical way to increase the selectivity and potency of this lead compound is to use the multivalent effect (22). Disney and co-workers (23) successfully applied this approach to the Hoechst 33258 ligand, which was known to bind CUG with modest selectivity. Because the rationally designed ligand 3 already exhibited a high affinity to (CUG)4 and showed excellent selectivity, we initially pursued a dimeric ligand.
The covalent linkage of ligand 3 to itself or other compounds required a functional derivative. The three possible sites for covalent modification of 3 are the acridine ring, the triaminotriazine recognition unit or the linking chain. The most expeditious synthetic approach used an acridine ring containing a carboxylic acid group to interconnect two ligands. It was found that the chloro- and methoxy-groups in 3 could be replaced with a 2- or 4-carboxamido group (see 1 and 4 in Figure 1, respectively) without altering its affinity for CUG or its inhibition of the MBNL1–CUG complex. Limited by the absence of structural data on the ligand–CUGexp complex, we designed and synthesized a small library of dimeric ligands that could potentially bind to consecutive CUG sites. This study, which will be published elsewhere, led us to 2 as the optimized dimeric ligand used in the current study (Figure 1). Ligand 2 was designed to target two consecutive UU mismatches, with the N,N′-bis(3-aminopropyl)-1,3-propanediamine linker spanning two central GC base pairs. Although the binding mode was not firmly established for 3, it was designed to act as a ‘stacked intercalator’ with the acridine and triaminotriazine rings π-stacked while the intercalator sits between the GC base pair and the U–triaminotriazine–U base triplet (21). A recent combined experimental and computational study provides support for that binding model in the major groove of the CUG RNA (24). By positioning the linking chain and triaminotriazine recognition units on opposite sides of the acridine unit, the bivalent complex likely requires a threading mechanism for binding (25). This design element was intentional with the goal of increasing the binding affinity through a higher residence time (26,27).
Establishing a TIRFM-based assay to monitor the MBNL1–(CUG)n interaction
To investigate binding of MBNL1 to (CUG)n and the effect of 1 and our bioactive dimeric ligand, 2, on MBNL1–(CUG)n interaction, we developed a TIRFM-based single-molecule–binding assay (28,29). A stem–loop (5′-bio-GCUGCUGUGCGCUGCUG-3′) containing two CUG pairs separated by a tetraloop (underlined), (CUG)4, was used as an RNA substrate to analyze the MBNL1 binding. This RNA construct has been previously established as the shortest CUG repeat to which MBNL1 binds with a similar affinity as to the long CUG repeats (30). The predicted stem–loop structure of (CUG)4 has been confirmed by melting studies (Tm = 60.6 ± 0.6) (30,31). Although (CUG)4 is much shorter than typical (CUG)n > 50 structure, it forms a hairpin structure characteristic of (CUG)exp; thus, it is the shortest validated CUG repeat model that can be used in biochemical analysis. Moreover, it contains only one MBNL1-binding site per RNA molecule, which simplifies analysis of the association/dissociation events. Validation of the single-site binding is described in the next section later in the text.
We followed the binding of Cy3-labeled MBNL1 to, and its dissociation from, individual (CUG)4 RNA molecules. Biotinylated and Cy5-labeled (CUG)4 was immobilized on a PEG-coated surface of the TIRFM flow chamber via biotin–neutravidin interaction (Figure 2a, left). First, locations of the surface-tethered (CUG)4 molecules were identified by illuminating the slide with red laser (641 nm), which selectively excited the Cy5 fluorophore present on (CUG)4. Then, the laser was switched to green (532 nm) to monitor binding of Cy3-labeled MBNL1 to (CUG)4 (Figure 2a, right). We followed MBNL1–(CUG)4 interaction in real-time, with a time resolution of 100 ms. In this experimental scheme, the surface-tethered (CUG)4 and Cy3-labeled MBNL1 are at equilibrium during the length of the measurement. TIR-generated evanescent field excites only the Cy3-labeled MBNL1 molecules that reside near the surface longer than two frames of the camera, which only occurs when MBNL1 is bound to the surface-tethered (CUG)4. This mode of excitation eliminates the background fluorescence of Cy3-MBNL1 in solution and thereby allows monitoring MBNL1–(CUG)4 interactions over a broad range of MBNL1 concentrations.
Figure 2.

Single-molecule analysis of MBNL1 binding to (CUG)4. (a) (CUG)4 was immobilized on a PEG-coated surface of the slide via biotin–neutravidin interaction. The experiment was carried out in two steps: (i) positions of (CUG)4 molecules on the slide were triangulated by exciting Cy5 label of (CUG)4 with a red laser and (ii) MBNL1–(CUG)4 interaction was monitored using green TIR illumination. On binding to (CUG)4, Cy3 label on MBNL1 is excited, as it is sequestered within the evanescent field. (b) Representative Cy3 (green) and Cy5 (red) fluorescence intensity trajectories from an individual (CUG)4 molecule. Arrows 1 and 2 depict periods of red and green excitation, respectively. ON and OFF events are indicated on the right.
Each event of MBNL1 binding to the surface-tethered (CUG)4 was observed as the appearance of a Cy3 signal at the location where (CUG)4 resided. Conversely, dissociation of MBNL1 from (CUG)4 resulted in the disappearance of the Cy3 signal. Fluorescence trajectories (Figure 2b) showed multiple events of a two-state (ON and OFF) association and dissociation process. The presence of only two states indicates that each observed event corresponded to binding of a single MBNL1 to a single (CUG)4. To confirm that potential non-specific interaction of MBNL1 with the surface does not interfere with our analysis, we substituted Cy5-labeled (CUG)4 with Cy5-labeled streptavidin. Only a few fluorescence trajectories showing both Cy5 and Cy3 signals were observed in the presence of 300 pM Cy3-MBNL1 [∼5% of what was observed in the presence of Cy5-labeled (CUG)4], confirming that the vast majority of binding events described earlier in the text resulted from specific MBNL1–(CUG)4 interaction. Analysis of the Cy3-MBNL1–(CUG)4 interaction in the presence of unlabeled MBNL1 confirmed that MBNL1-binding properties were not affected by the Cy3 conjugation (Supplementary Figure S9).
The Cy3 (green laser excitation) regions of the trajectories were fit to a two-state model using QuB software (32) (Supplementary Figure S4) yielding dwell times in the OFF (dissociated) and ON (bound) states. We determined kinetic parameters of MBNL1 binding to the surface-tethered (CUG)4 by globally analyzing distributions of ON and OFF dwell times in the presence of different concentrations of MBNL1 protein. All protein concentrations (50, 100, 150, 200, 300 and 400 pM) were used to determine the dissociation rate constant, whereas the association rate constant was determined using only 50–200 pM range of concentrations, which gave a linear dependence of the observed association rate on MBNL1 concentration. Figure 3a and b show individually fit ON and OFF time distributions obtained in the presence of 50 and 200 pM MBNL1, respectively. Global analyses of the dwell distributions yielded the dissociation rate constant for MBNL1–(CUG)4 complex, k−1 = 1.44 ± 0.3 s−1, and association rate constant, k1 = (4.6 ± 0.2) × 108 s−1 M−1, from which we calculated the equilibrium dissociation constant, KD1 = k−1/k1 = 3.1 ± 0.1 nM.
Figure 3.

(a) Distributions of all ON (left) and OFF (right) event durations detected in the 50 trajectories of individual (CUG)4 molecules are fit to single exponentials to yield k−1 ([MBNL1] = 50 pM) and von ([MBNL1] = 200 pM), respectively. (b) Effect of MBNL1 concentration on the k−1 and von of the observed events is shown. k−1 (dissociation rate constant) was independent of [MBNL1], suggesting the absence of functional cooperation between monomers. Initially, von (association rate) increased with increasing [MBNL1]. It was saturated at the MBNL1 concentrations >200 pM.
Stoichiometry of the MBNL1–(CUG)n interaction
Biotinylated (CUG)12 can simultaneously accommodate multiple Cy3-labeled MBNL1 molecules. Its Cy3 trajectories contained five states characterized by different Cy3 intensities (Figure 4a). An intensity histogram showed that there are three major Cy3 intensity populations consistent with the binding of one (∼196 a.u.), two (∼332 a.u.) and three (∼458 a.u.) MBNL1 proteins to the (CUG)12. A small fourth peak (∼632 a.u.) may represent more than one Cy3 label present on a fraction of MBNL1 molecules (Cy3 labeling efficiency of MBNL1 was 110%). Stoichiometry determination assay using SPR confirmed a stoichiometric ratio of MBNL1:(CUG)12 close to 3:1 (Supplementary Figure S10).
Figure 4.

Each (CUG)4 can accommodate a single-MBNL1 protein, whereas three MBNL1 proteins can be simultaneously bound to (CUG)12. (a) Schematic representation of (CUG)12 (left). (b) Schematic representation of (CUG)4. Representative fluorescence intensity trajectories for (MBNL1)3–(CUG)12 (a) and MBNL1–(CUG)4 (b) are shown in the middle (light green line shows the raw data collected at 100 ms time resolution, dark green line shows intensity averaged for nine data points). The fluorescence intensity histogram compiled from the trajectory is shown on the right (aqua) and is fit to five Gaussian peaks (black lines), corresponding to stepwise increase in (CUG)12 occupancy by MBNL1 (a), whereas only a single 1:1 binding state is observed for MBNL1–(CUG)4 (b).
Revealing the mechanism of the MBNL1–(CUG)n interaction inhibition by 1 and 2
We then carried out MBNL1–(CUG)4-binding studies in the presence of increasing concentrations of 1 and 2. Before starting the recording, the Cy3-labeled MBNL1, and the ligands were incubated for at least 5 min in the TIRFM reaction chamber to ensure that all components of the system are at equilibrium. The difference in the dwell-time distributions for the MBNL1–(CUG)4 complex in the absence and presence of 1 and 2, and therefore the difference in the apparent koff of the MBNL1–(CUG)4 complex, suggested that both 1 and 2 do not act as simple competitive inhibitors, which should only affect the on-rate (Supplementary Figures S5–S8). Based on the proposed modes of (CUG)4 binding by the ligand and MBNL1, it is possible that both can coexist on the same (CUG)4 molecule: although the ligand is expected to interact with the U–U mismatch (21), MBNL1 specifically binds to consecutive GC nucleotides (33). The observed inhibition likely stems from the different affinity of the MBNL1 protein for naked and ligand-bound (CUG)4. Most of this effect originates from an increased off-rate of the (CUG)4–MBNL1–ligand complex compared with (CUG)4–MBNL1. This situation can be described by a closed scheme of linked equilibria (Table 1a).
Table 1.
Parameters from global fitting of the dwell-time distributions to the closed (a) and open (b) schemes of linked equilibria describing state interconversion in the (CUG)4–MBNL1–ligand system
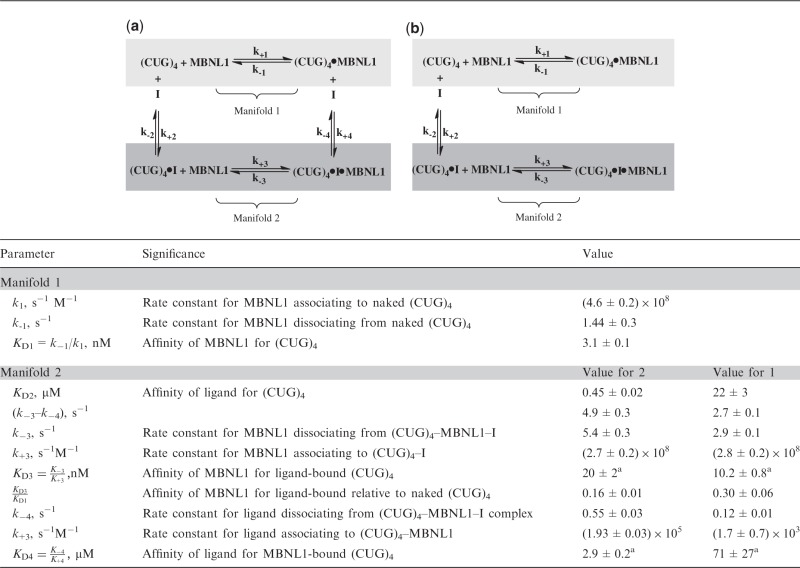 |
ki is the association rate constant, k−i is the dissociation rate constant and KDi = k−i/ki is the equilibrium dissociation constant for the respective steps in the scheme. I, inhibitor (1 or 2).
Unless indicated, the errors shown are standard errors from fitting the data.
aErrors were calculated as 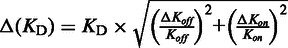
Assumptions derived from our experimental design and conditions (described in detail in the Supplementary Note S1) allowed us to globally fit distributions of the time intervals between binding events, obtained at a range of ligand concentrations, to Equation (1). Equation (1) assumes a simplified open scheme (Table 1b) and contains a double exponential
| (1) |
where the decay constants correspond to kinetic association constants for MBNL1 binding to (CUG)4, k1, and (CUG)4–I complex, k3, respectively. The two exponentials are weighted by the abundance of each manifold under given conditions, which are defined by the equilibrium between free and ligand-bound (CUG)4. The experiments were carried out in the presence of 0, 4, 20, 100 and 200 µM of ligand 1 or in the presence of 0, 0.5, 1, 2, 4, 6 and 8 µM of ligand 2.
The closed scheme in Table 1a was used to analyze the ON time distributions. These distributions were globally fit to Equation (2).
| (2) |
Const × KD2 is the weight of manifold (1), whereas Const × [I] is the weight of manifold (2) adjusted by the number of observed events.
These analyses yielded KD2, k4 and (k−3−k−4). Parameters k−3 and k−4 are linked as (k−3−k−4) and, therefore, cannot be determined individually by fitting but can be calculated from the linked equilibria as 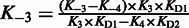 .
.
The results are summarized in the Table 1.
Indicative of its function as a bivalent inhibitor, the binding affinity of 2 for (CUG)4 was almost 50-fold higher than that of 1. Because neither 1 nor 2 behave as typical competitive inhibitors, but instead allow formation of the (CUG)4–MBNL1–ligand complex, the traditionally defined KI is replaced by KD2, which represents the affinity of the ligand for (CUG)4. We can, however, define an apparent IC50 as the concentration of ligand at which 50% of MBNL1 is free (Supplementary Note S2). It is notable, however, that this apparent IC50 depends on the concentration of the MBNL1 and (CUG)4 (Figure 5) making each inhibitor to be effective in a relatively narrow concentration range.
Figure 5.
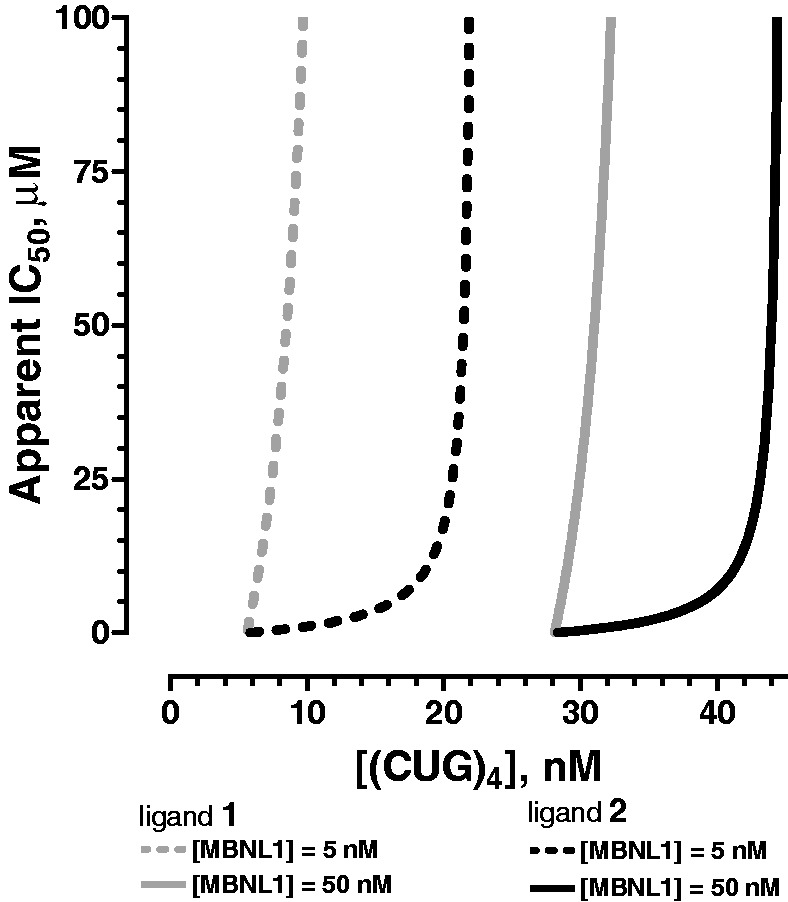
Theoretical curves for apparent IC50 values for 1 (gray) and 2 (black) at [MBNL1] = 5 (dashed lines) and 50 nM (solid lines). The concentration of ligand required to observe half of MBNL1 in the free form, IC50, depends on actual concentrations of MBNL1 and (CUG)4. The curves were generated based on the experimentally determined affinities shown in Table 1.
At micromolar concentrations, many small molecules self-associate into colloidal aggregates that non-specifically inhibit protein activity. Aggregate-based inhibition is sensitive to detergent (34); therefore, we confirmed that the inhibition by 2 was not affected by the presence of Triton X-100, eliminating the possibility of ligand aggregation.
To ensure that the observed inhibition mode does indeed stem from the (CUG)4–MBNL1–ligand complex formation and not an experimental artifact, we analyzed the (CUG)4–MBNL1 binding in the presence of the unlabeled MBNL1 protein, which should act a competitive inhibitor of Cy3-MBNL1 binding to the immobilized (CUG)4. Addition of unlabeled MBNL1 had no effect on k−1 for the (CUG)4–MBNL1 complex; in contrast k1 decreased proportionally to the fraction of Cy3-MBNL1 (Supplementary Figure S9). Not only did this control experiment confirm that we can clearly distinguish the different modes of inhibition but also that Cy3-labeling does not interfere with (CUG)4–MBNL1 interaction.
Binding affinity and MBNL1 inhibition potency of ligand 2 are nearly 100-fold that of 1 in bulk experiments
To measure the binding constants of 1 and 2 to CUG repeat by a more traditional approach, a steady-state fluorescence titration method with TAMRA-(CUG)6 was used. It is known that guanosine quenches the TAMRA fluorescence through photoinduced electron transfer (35,36). Therefore, it is possible that binding of a ligand to the UU mismatch close to 5′-TAMRA makes a structural change that can lead to quenching of the 5′-TAMRA by the 3′-G in 5′-TAMRA-(CUG)6-3′. Ligands 1 and 2 were titrated into TAMRA-(CUG)6 solution.
On increasing the ligand concentration, TAMRA fluorescence intensity gradually decreased as a result of fluorophore quenching by the bound ligand. A plot of normalized fluorescence intensity versus increased concentrations of each ligand yielded a binding isotherm with KD of 66 ± 2 µM and 318 ± 15 nM for 1 and 2, respectively (Figure 6). These KD values are similar to what we derived from the single-molecule study (Table 1). To confirm that the observed change in TAMRA fluorescence is a direct result of the ligand binding and is not caused by RNA aggregation, we compared the absorbance of the RNA–ligand solution before and after it was filtered through 0.02-µm (Whatman Anotop 6809-1002) filter. No loss of RNA because of aggregation or precipitation was observed in the presence of the two assayed ligands (data not shown).
Figure 6.
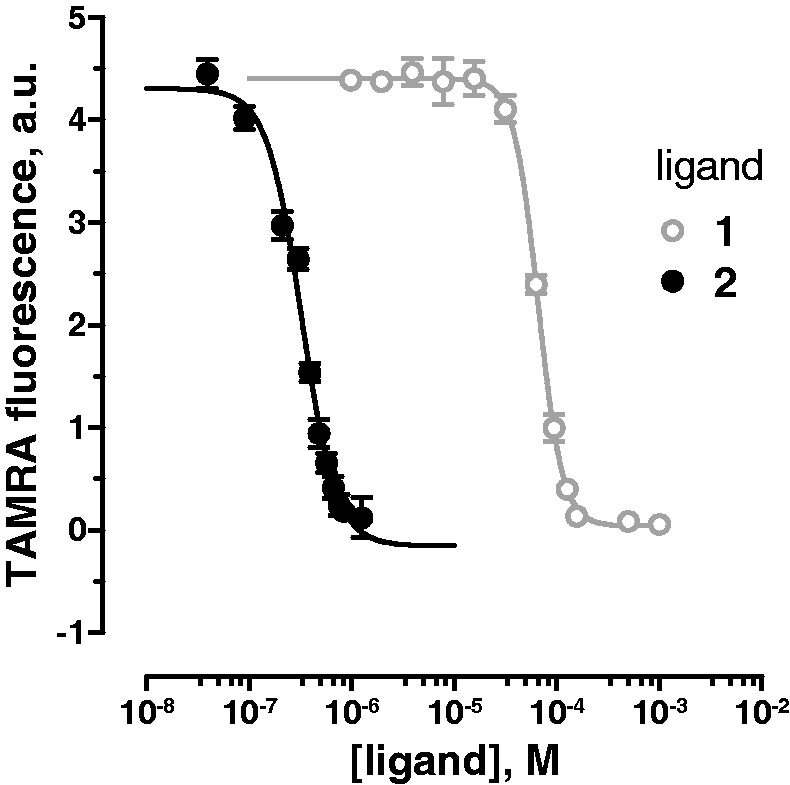
Fluorescence titrations of TAMRA-(CUG)6 with 1 and 2. Fluorescence intensity of TAMRA-(CUG)6 in the presence of increasing concentrations of 1 (gray open circles) and 2 (black filled circles). TAMRA was excited at 560 nm, and its emission was recorded at 590 nm. Error bars represent mean ± standard deviation of three replicates.
To provide a more conventional confirmation of single-molecule results, we used SPR to study the inhibition of MBNL1 binding to (CUG)4 and (CUG)12. To distinguish binding of ligands from MBNL1 to the immobilized RNA constructs, the injection of MBNL1 was preceded by ligand injection. The experiments were done in the presence of a large excess of competitor tRNA (1 µM yeast tRNA) to confirm the specificity of ligands for CUG repeat. The maximum Response Unit (RU) on MBNL1 injection at various concentrations of each inhibitor was recorded. These values were normalized and plotted versus increasing concentration of each ligand to yield a binding isotherm. Apparent IC50 values against (CUG)4 and (CUG)12 for 1 were 174 ± 12 and 293 ± 19 µM, respectively, whereas for 2, the values were 1.3 ± 0.2 and 1.1 ± 0.1 µM, respectively (Supplementary Note S3 and Supplementary Figure S11). Because of the non-competitive inhibition mechanism, IC50 values should depend on the concentration of immobilized (CUG)n on the SPR sensor chip, which is not measurable. Nonetheless, obtaining smaller IC50 value for 2, compared with 1, is in agreement with the single-molecule results. It is noteworthy that 1 and 2 showed no binding to MBNL1 (Supplementary Note S4 and Supplementary Figure S12).
DISCUSSION
RNA is emerging as an attractive drug target because of its central role in transcription, translation and an ever-growing number of key cellular processes (37). Most traditional RNA targeted drug discovery has focused on antibiotics and anti-HIV drug candidates (38). However, non-coding RNA, pre-mRNA and mRNA as a drug target have recently gained significant attention (39). In many diseases, such as DM1, targeting RNA is the most appropriate option, as the RNA is the causative agent of the pathology, whereas the protein function should not be altered (40). Despite their potential health benefits, the development of small molecules that specifically target RNA is still in its infancy. Previous studies, including our own, have identified small molecule inhibitors of the MBNL1–CUGexp interaction. In each case, it was assumed that the identified small molecule acts as a conventional competitive inhibitor. To further develop this approach to drug discovery, it is vital to establish the exact mechanism of the inhibition and ideally to provide a full kinetic and thermodynamic picture of the process (11). Thus, we undertook the first study of its kind to provide a full description of the mechanism by which a protein–RNA interaction is inhibited at the single-molecule level.
We developed and used a TIRFM-based single-molecule analysis to study the interaction of MBNL1, a key regulatory protein in alternative splicing, and (CUG)4, a validated model of CUGexp as well as the inhibition of this interaction by the small molecule RNA-binding ligands. This model system represents a single independent unit of RNA–protein or RNA–ligand–protein interaction. Our analysis revealed the affinity of MBNL1 for (CUG)4 to be 3.1 ± 0.1 nM, whereas using Electrophoretic Mobility Shift Assay (EMSA), we (21) and others (30) reported KD values of 26 ± 4 and 170 ± 20 nM, respectively. This was not surprising because the single-molecule measurements were carried out under true equilibrium conditions, whereas EMSA can underestimate affinity because the short-lived complexes may dissociate while being resolved. Additionally, only productive (native) interactions between (CUG)4 and MBNL1 molecules were detected by the single-molecule measurements, thereby canceling out any possible errors resulting from heterogeneity of MBNL1 or (CUG)4.
Single-molecule analyses of the MBNL1–(CUG)4 interaction in the presence of two inhibitors allowed us to determine the affinities of the two tested ligands for (CUG)4, as well as how they affect MBNL1–(CUG)4 complex formation. Notably, the equilibrium dissociation constants for the ligand–(CUG)n binding determined in bulk and at the single-molecule level were similar. Converging of the binding affinity of the 1–(CUG)n and 2–(CUG)n complexes (n = 4 or 6) measured by the fluorescence and single-molecule methods to the same value validated the single-molecule method. Our single-molecule analysis was model independent and suggested an inhibition mode that has not been considered previously: instead of competing for the same binding site, the ligands and MBNL1 can form a ternary complex on (CUG)4.
In the more physiologically relevant buffer used in this study, ligand 1 was a relatively weak inhibitor, which bound to (CUG)4 with a low affinity (i.e. KD = 22 ± 3 µM). Moreover, it allowed MBNL1 to bind to the 1–(CUG)4 complex with very high affinity (KD = 10.2 ± 0.8 nM). This interesting finding prompted us to design more effective small molecules. We pursued a bivalent ligand with the idea that it would: (i) bind to CUGexp with a higher affinity and ideally weaken the MBNL1 interaction with ligand–CUGexp; (ii) exhibit a longer occupancy time and increase the RNA ds stabilization, if MBNL1 indeed binds preferentially to the ss form of RNA as has been suggested (41,42); and (iii) bind a longer segment of RNA to achieve more effective coverage and steric blocking of CUGexp.
To reach the drug development goals outlined earlier in the text, we designed a library of dimeric ligands and found 2 to be the most potent inhibitor of CUG repeat–MBNL1 interaction. The affinity of 2 for (CUG)4 was measured by the single-molecule method to be KD = 450 ± 20 nM, a value that is almost 50-fold lower than that measured for 1. Despite this large increase in affinity, MBNL1 can still bind to the 2–(CUG)4 complex. This unexpected observation of a non-competitive inhibition mode for both 1 and 2 is the key finding of this study. Its implication for drug design is obvious. The 2–(CUG)4 complex reduces the MBNL1 affinity only 6-fold relative to naked (CUG)4 and 2-fold relative to that measured for the 1–(CUG)4 complex. Thus, tight binding is an insufficient criterion for effective inhibition because it may shift the overall equilibrium into manifold 2 of Table 1a (i.e. low KD2) without necessarily affecting MBNL1 binding. Highly effective inhibition will result from agents that exhibit both tight and selective binding to CUGexp (low KD2) and high KD3 values.
Another significant result is that the apparent IC50 values for the ligands depend markedly on both (CUG)4 and MBNL1 concentrations (Figure 5). Thus, the CUGexp length and cellular copy number and the MBNL1 concentration will determine the drug effectiveness. Because the CUG repeat continues to expand over the patient’s life, an ideal therapeutic agent should be able to inhibit binding of MBNL1 to CUGexp over a broad range of nuclear CUGexp copy number and repeat lengths. By single-molecule and bulk methods (Figure 5, Supplementary Figure S11 and Table 1), we found that, given the same concentration of MBNL1, 2 effectively inhibits (CUG)n–MBNL1 (n = 4 or 12) interaction to a greater extent than 1 and over a broader range of (CUG)n concentrations, a trend that can be further improved in the future ligands.
In summary, this study revealed that inhibition of (CUG)4–MBNL1 interaction by small molecules does not occur by a simple competitive mechanism. These findings aided in the design of a more potent (CUG)4–MBNL1 inhibitor and are guiding our current optimization efforts. More generally, the increasingly frequent discovery of key roles for RNA outside its established role in protein synthesis suggests new opportunities for RNA-targeted therapeutics. Indeed, RNA is now a validated, yet underused drug target, with less off-pathway binding. As the development of inhibitors for protein–RNA interactions becomes more widespread, we believe the inhibition model revealed in this study will need to be considered. It is likely that the dynamic and versatile structure of RNA allows it to form ternary ligand–protein–RNA complexes, although with lower stability (43). Finally, the single-molecule methodology described herein may prove to be a powerful method to unravel the inhibition mechanism for any biomacromolecular interaction.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–12, Supplementary Notes 1–5, Supplementary Methods, Supplementary References [21,43–45] and NMR spectra of compounds.
FUNDING
National Institutes of Health (NIH) [R01AR058361 to S.C.Z.]; Howard Hughes Medical Institute (Early Career Scientist Award to M.S.). Funding for open access charge: NIH and University of Iowa start-up funds.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Yuan Fu and Kali Miller for their assistance with MBNL1 expression, purification and the synthetic procedures and Dr Tom Meek (GSK) for valuable discussions.
REFERENCES
- 1.Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Rourke JR, Swanson MS. Mechanisms of RNA-mediated disease. J. Biol. Chem. 2009;284:7419–7423. doi: 10.1074/jbc.R800025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Echeverria GV, Cooper TA. RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res. 2012;1462:100–111. doi: 10.1016/j.brainres.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488:111–115. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Lopez A, Llamusi B, Orzaez M, Perez-Paya E, Artero RD. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc. Natl Acad. Sci. USA. 2011;108:11866–11871. doi: 10.1073/pnas.1018213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foff EP, Mahadevan MS. Therapeutics development in myotonic dystrophy type 1. Muscle Nerve. 2011;44:160–169. doi: 10.1002/mus.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl Acad. Sci. USA. 2009;106:18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ofori LO, Hoskins J, Nakamori M, Thornton CA, Miller BL. From dynamic combinatorial ‘hit' to lead: in vitro and in vivo activity of compounds targeting the pathogenic RNAs that cause myotonic dystrophy. Nucleic Acids Res. 2012;40:6380–6390. doi: 10.1093/nar/gks298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, Hoskins J, Tran T, Housman D, Thornton CA, et al. Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database and chemical similarity searching. J. Am. Chem. Soc. 2012;134:4731–4742. doi: 10.1021/ja210088v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, Disney MD. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J. Am. Chem. Soc. 2009;131:9767–9779. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holdgate GA, Gill AL. Kinetic efficiency: the missing metric for enhancing compound quality? Drug Discov. Today. 2011;16:910–913. doi: 10.1016/j.drudis.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Blow N. Proteins and proteomics: life on the surface. Nat. Methods. 2009;6:389–393. [Google Scholar]
- 13.Navratilova I, Myszka DG. Investigating biomolecular interactions and binding properties using SPR biosensors. Surf. Plasmon Reson. Based Sens. 2006;4:155–176. [Google Scholar]
- 14.Elenko MP, Szostak JW, van Oijen AM. Single-molecule imaging of an in vitro-evolved RNA aptamer reveals homogeneous ligand binding kinetics. J. Am. Chem. Soc. 2009;131:9866–9867. doi: 10.1021/ja901880v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moffitt JR, Chemla YR, Bustamante C. Methods in statistical kinetics. Methods Enzymol. 2010;475:221–257. doi: 10.1016/S0076-6879(10)75010-2. [DOI] [PubMed] [Google Scholar]
- 16.Karunatilaka KS, Solem A, Pyle AM, Rueda D. Single-molecule analysis of Mss116-mediated group II intron folding. Nature. 2010;467:935–939. doi: 10.1038/nature09422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, Swanson MS. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007;35:5474–5486. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galletto R, Amitani I, Baskin RJ, Kowalczykowski SC. Direct observation of individual RecA filaments assembling on single DNA molecules. Nature. 2006;443:875–878. doi: 10.1038/nature05197. [DOI] [PubMed] [Google Scholar]
- 19.Kim YT, Tabor S, Churchich JE, Richardson CC. Interactions of gene 2.5 protein and DNA polymerase of bacteriophage T7. J. Biol. Chem. 1992;267:15032–15040. [PubMed] [Google Scholar]
- 20.Honda M, Park J, Pugh RA, Ha T, Spies M. Single-molecule analysis reveals differential effect of ssDNA-binding proteins on DNA translocation by XPD helicase. Mol. Cell. 2009;35:694–703. doi: 10.1016/j.molcel.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arambula JF, Ramisetty SR, Baranger AM, Zimmerman SC. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc. Natl Acad. Sci. USA. 2009;106:16068–16073. doi: 10.1073/pnas.0901824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 23.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem. Biol. 2012;7:856–862. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong CH, Richardson SL, Ho YJ, Lucas AM, Tuccinardi T, Baranger AM, Zimmerman SC. Investigating the binding mode of an inhibitor of the MBNL1. RNA complex in myotonic dystrophy Type 1 (DM1) leads to the unexpected discovery of a DNA-selective binder. ChemBioChem. 2012;13:2505–2509. doi: 10.1002/cbic.201200602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmerman SC, Lamberson CR, Cory M, Fairley TA. Topologically Constrained bifunctional intercalators—DNA intercalation by a macrocyclic bisacridine. J. Am. Chem. Soc. 1989;111:6805–6809. [Google Scholar]
- 26.Fechter EJ, Olenyuk B, Dervan PB. Design of a sequence-specific DNA bisintercalator. Angew. Chem. Int. Ed. Engl. 2004;43:3591–3594. doi: 10.1002/anie.200454231. [DOI] [PubMed] [Google Scholar]
- 27.Holman GG, Zewail-Foote M, Smith AR, Johnson KA, Iverson BL. A sequence-specific threading tetra-intercalator with an extremely slow dissociation rate constant. Nat. Chem. 2011;3:875–881. doi: 10.1038/nchem.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van den Wildenberg SM, Prevo B, Peterman EJ. Single molecule analysis. Methods. 2011;783:81–99. doi: 10.1007/978-1-61779-282-3_5. [DOI] [PubMed] [Google Scholar]
- 29.Reck-Peterson SL, Derr ND, Stuurman N. Imaging single molecules using total internal reflection fluorescence microscopy (TIRFM) Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.top73. pdb.top1173. [DOI] [PubMed] [Google Scholar]
- 30.Warf MB, Berglund JA. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA. 2007;13:2238–2251. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cass D, Hotchko R, Barber P, Jones K, Gates DP, Berglund JA. The four Zn fingers of MBNL1 provide a flexible platform for recognition of its RNA binding elements. BMC Mol. Biol. 2011;12:20. doi: 10.1186/1471-2199-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin F, Auerbach A, Sachs F. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys. J. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teplova M, Patel DJ. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat. Struct. Mol. Biol. 2008;15:1343–1351. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vamosi G, Gohlke C, Clegg RM. Fluorescence characteristics of 5-carboxytetramethylrhodamine linked covalently to the 5' end of oligonucleotides: multiple conformers of single-stranded and double-stranded dye-DNA complexes. Biophys. J. 1996;71:972–994. doi: 10.1016/S0006-3495(96)79300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qu P, Chen XD, Zhou XX, Li X, Zhao XS. Fluorescence quenching of TMR by guanosine in oligonucleotides. Sci. China, Ser. B. 2009;52:1653–1659. [Google Scholar]
- 37.Xu Y, Komiyama M. Structure, function and targeting of human telomere RNA. Methods. 2012;57:100–105. doi: 10.1016/j.ymeth.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 38.Vicens Q, Westhof E. RNA as a drug target: the case of aminoglycosides. ChemBioChem. 2003;4:1018–1023. doi: 10.1002/cbic.200300684. [DOI] [PubMed] [Google Scholar]
- 39.Vicens Q. RNA’s coming of age as a drug target. J. Incl. Phenom. Macro. 2009;65:171–188. [Google Scholar]
- 40.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu Y, Ramisetty SR, Hussain N, Baranger AM. MBNL1-RNA recognition: contributions of MBNL1 sequence and RNA conformation. ChemBioChem. 2012;13:112–119. doi: 10.1002/cbic.201100487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laurent FX, Sureau A, Klein AF, Trouslard F, Gasnier E, Furling D, Marie J. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res. 2012;40:3159–3171. doi: 10.1093/nar/gkr1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, Al-Hashimi HM. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011;7:553–559. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.