

Abstract
G•C Hoogsteen base pairs can form transiently in duplex DNA and play important roles in DNA recognition, replication and repair. G•C Hoogsteen base pairs are thought to be stabilized by protonation of cytosine N3, which affords a second key hydrogen bond, but experimental evidence for this is sparse because the proton cannot be directly visualized by X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy. Here, we combine NMR and constant pH molecular dynamics (MD) simulations to directly investigate the pKa of cytosine N3 in a chemically trapped N1-methyl-G•C Hoogsteen base pair within duplex DNA. Analysis of NMR chemical shift perturbations and NOESY data as a function of pH revealed that cytosine deprotonation is coupled to a syn-to-anti transition in N1-methyl-G, which results in a distorted Watson-Crick geometry at pH > 9. A four-state analysis of the pH titration profiles yields a lower bound pKa estimate of 7.2 ± 0.1 for the G•C Hoogsteen base pair, which is in good agreement with the pKa (7.1 ± 0.1) value calculated independently using constant pH MD simulations. Based on these results and pH dependent NMR relaxation dispersion measurements, we estimate that under physiological pH (pH 7 to 8), G•C Hoogsteen base pairs in naked DNA have a population of 0.02 to 0.002% as compared to 0.4% for A•T Hoogsteen base pairs and likely exist primarily as a protonated species.
Keywords: Hoogsteen base pair, cytosine protonation, pKa shift, NMR, MD simulations
We recently showed using NMR relaxation dispersion techniques1,2 that A•T and G•C base pairs in duplex DNA can transiently form Hoogsteen base pairs4 with populations in the range of 0.1 – 0.5 % and lifetimes of 0.3 – 1.1 ms at pH ~ 6.3,5 Transition from Watson-Crick (WC) to Hoogsteen (HG) base pairs requires a 180° rotation of the purine base about the glycosidic bond and, therefore, a change in the base orientation from anti to syn conformation.6 While A•T HG base pairs retain two hydrogen bonds (H-bonds) upon this conformational change, G•C HG base pairs retain only a single H-bond unless cytosine N3 becomes protonated to form a second stabilizing H-bond (Figure 1A).
Figure 1.
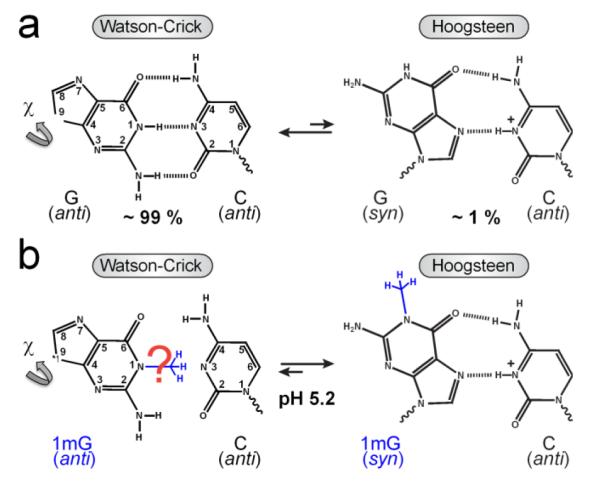
Schematic of the equilibrium between a G•C WC and HG base pair. (a) The transition from a ground state WC to a transient state HG base pair, with relative populations measured by NMR relaxation dispersion,3 requires an anti-to-syn rotation around the glycosidic bond (χ) and creates a stabilizing H-bond upon C N3 protonation. (b) Methylation at G N1 favors formation of a ground state HG base pair at pH 5.2.3
To date, N3-protonated cytosine in a G•C+ HG base pair has only been directly observed by NMR for triplex DNA, where the protonation constant (or pKa) of cytosine N3 was shown to be elevated by more than 5 units for G•C+ HG7 as compared to the value of ~ 4.2 in free nucleotides.8 However, the protonation state of cytosine N3 in G•C HG base pairs within duplex DNA has not been determined. The pKa of free cytosine is far from neutrality (~ 4.2)8 and the cytosine imino H3 proton cannot be directly visualized in crystal structures or by NMR measurements owing to rapid exchange with solvent. Indeed, the initial proposal that replication by human DNA polymerase ι(hPolι) proceeds via HG rather than WC pairing9 was challenged on the grounds that at physiological pH, G•C would not exist as a stable HG base pair due to lack of protonation.10 Although X-ray structures of duplex DNA bound to proteins, including hPolι (at pH~6.5)11 and TATA-binding protein (at pH~6)12 suggest that cytosine N3 and guanine N7 atoms are within H-bonding distance, protonation of cytosine N3 could not be unambiguously established. Determining the protonation state of cytosine N3 and its pKa value becomes significantly more challenging in naked duplex DNA, where the HG base pairs exist only transiently in solution. Here, we combine NMR and computational methods to directly examine the pKa of cytosine N3 in naked duplex DNA and relative stability of HG base pairs under physiological pH.
We previously showed that G•C HG base pairs can be trapped inside naked duplex DNA by installing a methyl group at the G imino nitrogen N1 position.3 This N1-methylguanine (1mG) modification introduces a bulky substituent at the WC interface and precludes formation of the WC (G)N1H1⋯N3(C) H-bond, tipping the equilibrium towards the HG base pair at low pH (Figure 1B).3 Based on chemical shift analysis, we showed that trapped HG base pairs have similar characteristics to their transient unmodified counterparts. We confirmed formation of the 1mG15•C10 HG base pair in A6-DNA1mG10 at pH 5.2 based on observation of nuclear Overhauser effect (NOE) connectivity and proton/carbon chemical shift signatures that indicate a syn conformation for the 1mG10 base (Figure 2A).3
Figure 2.

Estimating the pKa for cytosine N3 inside a trapped 1mG•C HG base pair. (a) 2D 1H, 1H NOESY spectra at pH 5.2 (red) and 9.2 (purple) suggesting a syn conformation at low pH versus an anti conformation at high pH for 1mG10 as well as enhanced conformational exchange and/or distortion for C15 and neighboring sites. (b) pH dependence of 2D 1H,13C HSQC spectra of unlabeled A6-DNA1mG10 showing large conformational changes at the 1mG10•C15 and its two neighboring base pairs. (c) Corresponding chemical shift perturbations (CSP) as a function of pH showing global fitting of the observed pKa ~ 7.2 for the transition from a protonated G•C+ HG to a distorted WC* base pair.
While the protonation state of cytosine could not be deduced directly in either transient or trapped HG base pairs, several indirect lines of evidence suggest that in both cases, the cytosine N3 is protonated to form a G•C+ HG base pair. The 1mG10 modification resulted in significant chemical shift perturbations at the C15 base, which are consistent with N3 protonation. This includes an upfield shift of amino protons (~2 ppm), which is a known characteristic of protonated G•C+ HG base pairs in triplex DNA,7 and a large downfield shift (~2.3 ppm) in C15 C6, which is also expected upon N3 protonation based on density functional theory (DFT) calculations.3 Further evidence that these perturbations reflect cytosine N3 protonation comes from observation of only small chemical shift perturbations (<0.5 ppm) in the thymine residue when trapping an A•T HG base pair through N1-methylation of the adenine.3 Finally, the population of the transient HG base pairs measured by NMR relaxation dispersion decreases more strongly with increasing pH for G•C versus A•T base pairs, and falls outside the limits of detection by NMR relaxation dispersion at higher than neutral pH, as might be expected based on destabilization of the G•C HG base pair due to cytosine N3 deprotonation.3
To further characterize the protonation state of C15 N3 in a G•C HG base pair, we measured natural abundance NMR 1H,13C-HSQC spectra for base and sugar resonances for the unlabeled A6-DNA1mG10 sample as a function of pH and monitored the chemical shift perturbations (CSP) at the modified base pair and adjacent range (5.2 to 9.2) that minimally affects the structural stability of B-DNA and that causes little NMR spectral change in an unmodified A6-DNA (Figure S1). If the chemical shift perturbations observed at C15 upon guanine methylation under acidic conditions arise due to protonation of cytosine N3, increasing the pH should undo these effects and result in C15 chemical shifts that are similar to those observed in WC base pairs.
Increasing the pH from 5.2 to 9.2 resulted in the expected upfield CSPs for cytosine C6 and C5 that are consistent with deprotonation at the N3 position (Figure 2B). However, we also observed CSPs are that not expected based on N3 deprotonation and that suggest a pH-dependent conformational change. In particular, both the sugar C1’ and base C8 resonances of 1mG experience an upfield shift with increasing pH, resulting in carbon chemical shifts (Figure S1) that are strongly indicative of an anti rather than syn nucleobases orientation, as expected for a WC-like geometry. This was supported by large changes in the NOESY cross-peaks at pH 9.2, including a much weaker 1mG10 H8-H1’ cross-peak and a stronger 1mG10 H8-H2’/2” cross-peak than seen for the syn base at pH 5.2, but consistent with an anti base orientation (Figure 2A). We also observed a weak cross-peak between 1mG10 H8 and the 3’ neighboring T9 H1’, confirming that an anti/anti configuration in the sequentially stacked bases, with some structural distortion and/or enhanced dynamics at the 1mG residue (Figure 2A). Increasing the pH resulted in an unusual downfield CSP for C15 C1’ that suggests a change in sugar pucker towards the C3’-endo conformation (Figure S1). A structural and/or dynamic perturbation at C15 could also be inferred from a weaker cross-peak between C15 H1’ and the 3’ adjacent A16 H8 at pH 9.2 than normally observed in B-DNA (Figure 2A). These data suggest that upon deprotonation of cytosine N3 at high pH, an HG base pair stabilized by a single H-bond is no longer energetically favorable as compared to a distorted WC-like geometry (WC*), which could be stabilized by at least one H-bond. Evidently, the 1mG modification does not fully trap the transient HG base pair at pH 5.2 but, rather, inverts the relative populations of the WC and HG species so that the WC* conformation now becomes the transient state. This is further supported by detectable line broadening at the 1mG10•C15 base pair observed at low pH. Such inversion of ground and excited state has previously been observed with targeted mutagenesis in proteins.13
Our findings suggest a complex pH-dependent equilibrium involving at least two pathways between a protonated HG+ and a neutral WC* base pair and four species (HG+, HG, WC*, and WC*+) (Scheme S2). This makes it impossible to determine the pKa of cytosine N3 based on the NMR CSP data without additional assumptions. To a good approximation, the cytosine base CSPs report on the transition from protonated (HG+ and WC*+) to neutral (HG and WC*) species, and can be fit to a 2-state equilibrium (Scheme S1) to extract an observed pKa (pKa,obs). Fitting of the pH dependent cytosine CSPs (C5H5 and C6H6) to a modified 2-state Henderson-Hasselbalch equation describing the change in NMR chemical shift with pH yielded a pKa,obs ~7.2 ± 0.1. Interestingly, similar pKa,obs values in the range of 6.7 – 7.2 were obtained by fitting the CSPs for 1mG and adjacent residues (Table S1) which primarily sense the conformational transition from HG (HG+ and HG) to WC* (WC* and WC*+) states. These data suggest that deprotonation of cytosine N3 is tightly coupled to the HG-to-WC conformational change.
In this expression, fHG+ and fWC+ are the equilibrium fractions of HG+ and WC*+ ([HG+]/([HG+]+[WC*+]) and [WC*+]/([HG+]+[WC*+]), respectively) and KHG+ and KWC+ are the deprotonation equilibrium constants for HG+ and WC*+ (pKHG+ = −log(KHG+ and pKWC+ = −log(KWC+)). This equation shows that the value for pKa,obs is bound between the pKa values for the HG+ and WC*+ base pair, which means that at least one of the protonated species has a pKa value equal to or greater than pKa,obs. We can impose further constraints by assuming that (i) HG+ is the major protonated species based on direct observation of NMR spectra at low pH and (ii) the pKa for WC*+ (pKWC+) is close to that of free cytosine because C15 N3 would be distorted and more solvent exposed relative to HG+ and, thus, not optimally positioned for H-bonding with the potential acceptor, 1mG O6. Thus, without exact knowledge of or pKWC+, or fHG+, we can conclude that pKHG+ is at least as large as pKa,obs or 7.2 ± 0.1, which represents a significant shift of 3 or more units from the intrinsic value for free cytosine.8 These experimental results clearly indicate that protonated HG+ base pairs can exist at physiological pH and reinforce the replication mechanism for the lesion bypass polymerase hPolι proposed by Nair et al.9
To obtain additional insights into the protonation equilibria, we performed constant pH molecular dynamics(CPHMDMSλD) simulations15,16 on the HG G•C+ base pair and its 1mG analog using the same NMR experimental conditions. As shown in Figure 3A, we calculated a pKHG+ value of 7.1 ± 0.1, where the major neutral HG conformation was stabilized by two weaker H-bonds (Figure 3B). Moreover, this pKa prediction was not significantly altered by guanine N1-methylation (Figure 3A). Analysis of the H-bond lengths at pH 7 confirmed that an HG-like conformation was maintained throughout the simulations (Figure S2). These results represent an independent estimate of pKHG+, which is in line with the experimentally bounded pKHG+ value of at least 7.2 ± 0.1, and point to a nearly equal stability of the neutral and protonated species at physiological pH. As in the NMR experiments, the MD simulations may underestimate the value of pKHG+ because polarization effects from the charged G•C+ base pair, which could strengthen these interactions, were not accounted for in the simulation parameters. In contrast, control simulations for a canonical WC base pair (Figure S2), where the protonated species featured a cytosine base shifted towards the major groove to accommodate a wobble configuration with two H-bonds (Figure 3B), yielded a much lower pKa value of 2.4 ± 0.1 that fits the large decrease expected for a helical WC base pair. Due to the lack of accurate structures for the protonated and neutral WC* states, identical simulations could not be carried out for the 1mG-modified WC* base pair.
Figure 3.
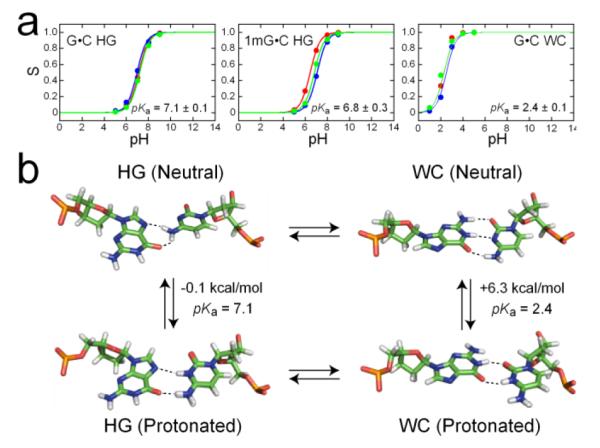
Constant pH MD simulations of WC and HG base pair protonation. (a) Titration curves obtained from 3 independent runs of single-site CPHMDMSλD simulations of a G•C HG base pair, its 1mG analog, and a G•C WC base pair. (b) Corresponding structures for the neutral and protonated WC and HG base pairs and predicted free energy differences at pH 7, depicted in the context of the proposed 4-state equilibrium.
To relate the above observations to transient HG base pairs, we measured relaxation dispersion data over the detectable pH range (4.3 to 6.8) to examine variations in the HG population (pB). Assuming that the neutral G•C HG base pair is significantly destabilized relative to its protonated counterpart, we would predict that at pH > pKa of cytosine N3 (≥7.2), G•C HG base pairs would fall outside the limit of detection by NMR dispersion. This would not be the case for A•T HG base pairs whose populations should remain independent of pH. Indeed, this is what is observed – transient G•C+ HG base pairs are undetectable at pH 7.6, while A•T retains a pB~0.4% (Figure S3). By extrapolating the pH dependence of pB, we estimate a pB~0.02 to 0.002 % for transient G•C+ HG base pairs at physiological pH 7 to 8. This is at least ~20-fold less abundant than for transient A•T HG base pairs, and this difference in abundance increases with metal ion concentration (Figure S4). A comprehensive survey of X-ray structures also reveals a greater abundance of A•T as compared to G•C HG base pairs in duplex DNA (data not shown). Interestingly, we also observed an increase in pB with decreasing pH below 6, which is much more pronounced for G•C+ as compared to A•T base pairs (Figure S3). Fitting of pB as a function of pH yielded pKa(obs) values of 3.2 and 2.7 for G•C and A•T base pairs, respectively (see Supporting Information). This increase in pB with acidic pH arises primarily from an increase in the forward rate constant (Figure S3) and could reflect acid-induced destabilization17 of WC relative to HG states, possibly due to protonation of other groups. For G•C base pairs, this increase in pB could still be explained by cytosine N3 protonation in the context of a 4-state equilibrium (Supporting information).
In conclusion, our data suggests that the pKa of cytosine N3 is ~7.2 and comparable to the pKa of adenine N1 in A•C+ mismatches.18,19 Thus, transient G•C HG base pairs, can significantly populate protonated over neutral species near biological pH with potential implications in DNA recognition and binding by cellular factors. Moreover, we show that, at physiological pH, G•C base pairs containing N1-methyl-G damage exist as a nearly equal mixture of protonated HG+ and disstorted WC-like conformers that could be specifically recognized by DNA repair enzymes in search for damaged DNA.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the Michigan Economic Development Cooperation and the Michigan Technology Tri-Corridor for support in the purchase of a 600 MHz spectrometer and thank Dr. Vivekanandan Subramanian for maintenance of the NMR instrument. This work was supported by NIH grants, GM089846 awarded to H.M.A., and GM057053 and GM037554 awarded to C.L.B.
ABBREVIATIONS
- NMR
Nuclear magnetic resonance
- MD
molecular dynamics
- CSP
chemical shift perturbation
- NOE
nuclear Overhauser effect
- ppm
parts per million
Footnotes
Supporting Information. Details of NMR sample preparation, resonance assignments, CSP analysis, relaxation dispersion experiments, and MD simulations. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Korzhnev DM, Orekhov VY, Kay LE. J. Am. Chem. Soc. 2005;127:713. doi: 10.1021/ja0446855. [DOI] [PubMed] [Google Scholar]
- (2).Palmer AG, III, Massi F. Chem. Rev. 2006;106:1700. doi: 10.1021/cr0404287. [DOI] [PubMed] [Google Scholar]
- (3).Nikolova EN, Kim E, Wise AA, O’Brien PJ, Andricioaei I, Al-Hashimi HM. Nature. 2011;470:498. doi: 10.1038/nature09775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hoogsteen K. Acta Crystallogr., Sect. A: Found. Crystallogr. 1963;16:907. [Google Scholar]
- (5).Nikolova EN, Gottardo FL, Al-Hashimi HM. J. Am. Chem. Soc. 2012;134:3667. doi: 10.1021/ja2117816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang AH, Ughetto G, Quigley GJ, Hakoshima T, van der Marel GA, van Boom JH, Rich A. Science. 1984;225:1115. doi: 10.1126/science.6474168. [DOI] [PubMed] [Google Scholar]
- (7).Asensio JL, Lane AN, Dhesi J, Bergqvist S, Brown T. J. Mol. Biol. 1998;275:811. doi: 10.1006/jmbi.1997.1520. [DOI] [PubMed] [Google Scholar]
- (8).Izatt RM, Christensen JJ, Rytting JH. Chem. Rev. 1971;71:439. doi: 10.1021/cr60273a002. [DOI] [PubMed] [Google Scholar]
- (9).Nair DT, Johnson RE, Prakash S, Prakash L, Aggarwal AK. Nature. 2004;430:377. doi: 10.1038/nature02692. [DOI] [PubMed] [Google Scholar]
- (10).Wang J. Nature. 2005;437:E6. doi: 10.1038/nature04199. [DOI] [PubMed] [Google Scholar]
- (11).Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Structure. 2005;13:1569. doi: 10.1016/j.str.2005.08.010. [DOI] [PubMed] [Google Scholar]
- (12).Patikoglou GA, Kim JL, Sun L, Yang SH, Kodadek T, Burley SK. Genes Dev. 1999;13:3217. doi: 10.1101/gad.13.24.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bouvignies G, Vallurupalli P, Hansen DF, Correia BE, Lange O, Bah A, Vernon RM, Dahlquist FW, Baker D, Kay LE. Nature. 2011;477:111. doi: 10.1038/nature10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shi C, Wallace JA, Shen JK. Biophys. J. 2012;102:1590. doi: 10.1016/j.bpj.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Goh GB, Knight JL, Brooks CL., III J. Chem. Theory Comput. 2012;8:36. doi: 10.1021/ct2006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Goh GB, Knight JL, Brooks CL., III J. Chem. Theory Comput. 2013;9:935. doi: 10.1021/ct300942z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hermann P, Fredericq E. Nucleic Acids Res. 1977;4:2939. doi: 10.1093/nar/4.8.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Moody EM, Brown TS, Bevilacqua PC. J. Am. Chem. Soc. 2004;126:10200. doi: 10.1021/ja047362h. [DOI] [PubMed] [Google Scholar]
- (19).Siegfried NA, O’Hare B, Bevilacqua PC. Biochemistry. 2010;49:3225. doi: 10.1021/bi901920g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.