

Abstract
Shortly after their discovery at the end of the 80s, stable singlet carbenes have been recognized as excellent ligands for transition metal based catalysts, and as organo-catalysts in their own right. At the end of the 2000s, it has been shown that they can coordinate main group elements in their zero oxidation state, and even activate small molecules. This review covers examples in the literature dealing with the most recent application of stable singlet carbenes, namely their use to stabilize radicals and radical ions that are otherwise unstable.
Introduction
Radicals play essential roles in numerous chemical1 and biological processes.2 Many transition metal complexes and a few non-metal compounds such as molecular oxygen are examples of stable radicals under ambient conditions. However, most paramagnetic main group species are unstable as the unpaired electron makes the molecule highly reactive. Since the pioneering work by Gomberg over 100 years ago, demonstrating that the triphenylmethyl radical could be generated in a flask,3 radical species have been exciting synthetic targets. Typical methods for stabilizing such reactive molecules involve delocalizing the unpaired electron and/or utilizing bulky groups to prevent dimerization.4
Singlet carbenes have a lone pair and a vacant orbital, they are perhaps most well known for their great utility as ligands in transition metal catalysis.5 In their infancy,6 these species were viewed as strong σ-donors with negligible π-accepting character.7 Recent reports indicate that the substitution on the carbene centre is highly influential on the donor ability as well as electron accepting properties, giving ligands with diverse properties.8 The empty p-orbital can indeed engage in π-backbonding interactions and delocalize electron density. The recent realization that the π-accepting nature of carbenes can be exploited has led to new opportunities in transition metal catalysis9 and the activation of small molecules with the free carbenes themselves.10,11
In recent years the electrophilicity of carbenes has also been utilized for stabilizing radicals and radical ions. With respect to the term stabilizing, we refer to either generating or observing species that can otherwise not be detected spectroscopically, and in some cases even isolating such compounds. This review focuses on the observation and isolation of main group radicals that are supported by carbene ligands. Although many interesting paramagnetic carbene-metal complexes have been observed or isolated,12,13 this area is beyond the scope of this review.
Carbene-stabilized boron centred radicals
Anionic boron radicals with the formula BR3·− have been studied for a long time,14 but other types of boron radicals are rare.15 The preparation of BR3·− is simply the population of the vacant p-orbital of an electron deficient borane with a single electron. A neutral analog requires a Lewis base (L) coordinate to the boron centre giving a L-BR2· adduct. Carbenes are ideal ligands and can readily coordinate to borenium cations or BH3 which are precursors for generating such species.
Recently, significant research has been performed on the parent species, BH2·, as NHC-stabilized complexes 1· (NHC: N-heterocyclic carbene) (Figure 1).16 The UV irradiation of a mixture of NHC-BH3 1H and di-tert-butylperoxide generates the corresponding radical, which can be characterized by EPR spectroscopy.16c The EPR and corroborating theoretical results indicate that the spin density of the resulting radical is delocalized throughout the carbene ligand and boron centre. There is planarity through the NHC-B unit in contrast to the previously studied amine adducts R3N-BH2·, which feature a pyramidalized boron centre.17 The nitrogen ligated species are σ-type radicals whereas with NHCs they are of π-type. This is a direct result of the carbene being a better π-acceptor than amines.
Fig 1.
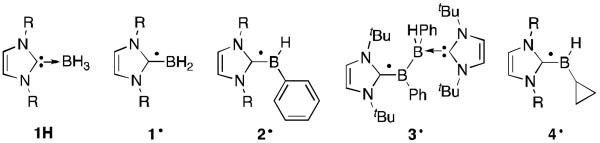
NHC-borane precursors 1H, and radicals 1·-4· observed by EPR spectroscopy.
The synthesis of NHC-BH2· radicals is facile as the boron-hydrogen bond dissociation energy in the precursor is small (80-88 kcal/mol).16b Although the rate of hydrogen atom donation of the NHC-BH3complexes is slower than Bu3GeH, (TMS)3SiH and Bu3SnH, it is faster than Et3SiH, making them useful in certain transformations. These complexes have been used with success as co-initiators of acrylate photopolymerizations as well as radical reductions of halides and xanthates.16
Hydrogen atoms can also be easily abstracted from NHC-BH2Ar complexes to give the corresponding radicals (e.g. 2·).16g The EPR spectra showed delocalization of the spin density throughout the NHC, boron centre and aryl groups. The H,Ar substituted derivatives dimerize two orders of magnitude slower than the corresponding H,H analogues (half life of the former ~10 s). This is rationalized by the extended π-system of the aryl group and by an increase in sterics at boron. Interestingly, a diboranyl radical 3· generated by hydrogen atom abstraction from the NHC-BH2Ph precursor has been observed by EPR.16g The proposed mechanism to form 3· involved H· abstraction from the starting material to give a radical of type 2·, subsequent dimerization to the diborane, then a hydrogen atom is abstracted from the diborane to afford the diboranyl radical 3·. The SOMO of 3· is primarily localized on the NHC-BPh moiety, much like 2·.
Alkyl substituted boron radicals (BHR·) are more challenging to prepare as hydrogen atom abstraction from the NHC-BH2R precursors usually occurs at the β-carbon on the alkyl chain with subsequent elimination of alkene and NHC-BH2· generation.16g Among the exceptions is the cyclopropyl radical 4· that can be observed by EPR and is resistant towards ring opening and alkene elimination.
The aforementioned hydrogen substituted boron radicals have only been observed by spectroscopic methods. Their meta-stable nature may be attributed to the lack of bulk at boron. Indeed, Gabbaï et al. were able to isolate the two carbene-BR2· derivatives 5·18 and 6·19 (Figure 2). In the first example (5·), two bulky mesityl groups and a π-accepting acridinyl carbene are on boron as stabilizing groups.18 The EPR spectrum showed that the spin density is delocalized throughout the acridine framework with minimal density on boron; remarkably, negligible electronic contribution of the mesityl groups was observed. Radical 5· could even be structurally characterized, and the solid-state structure indicates that the B-C bond length lies between a single and double bond (1.56 Å vs. 1.59 Å and 1.48 Å, respectively). Interestingly, a second electron could be added to generate the corresponding borataalkene with a drastically contracted B-C bond (1.46 Å) consistent with the population of a π-orbital.20 The NHC stabilized derivative 6· could also be isolated but not structurally characterized.19 The EPR spectrum indicated that the spin density is delocalized throughout the mesityl groups, boron centre and the entire NHC framework. The larger hyperfine coupling with boron (7.90 G for 6· vs. 2.55 G for 5·) and the involvement of the mesityl groups clearly demonstrate the weaker π-accepting properties of the NHC compared to the acridinyl carbene.
Fig 2.
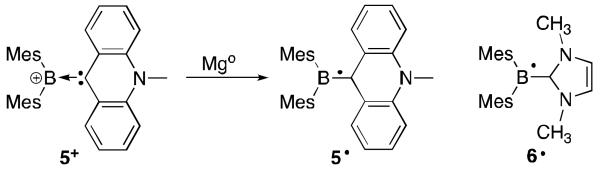
Synthesis of 5· and radical 6·.
Our group has isolated a totally different type of paramagnetic carbene-stabilized boron derivative. The CAAC2BH borylene 7 [CAAC: cyclic (alkyl)(amino)carbene]21 is a compound with a lone pair on boron, thus being isoelectronic with an amine.22 The reaction of borylene 7 with GaCl3 results in the generation of the radical cation 7+·, the same type of oxidation that is observed for an amine (Figure 3). EPR data and theoretical calculations indicate that the SOMO is primarily located on the boron (50 %) and the nitrogen atom of both carbenes (33 % total). The carbon-boron bond lengths elongate upon oxidation (1.58 Å vs. 1.52 Å), and the C-N bonds within the CAAC slightly contract (1.36 Å vs. 1.38 Å). This trend is consistent with decreased electron density at boron and as a consequence decreased π-back donation from the boron to the carbene centres.
Fig 3.
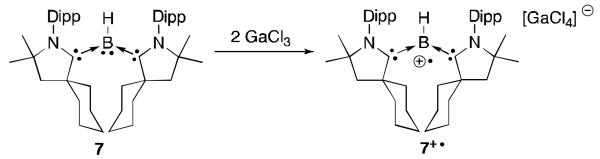
Oxidation of borylene 7 to the radical cation 7+·.
It is noteworthy that the use of carbenes has also allowed for the generation of various boron species, which were hitherto believed to be non-existent as exemplified by derivatives A23 and B24 (Figure 4). These compounds could serve as precursors for new carbene stabilized diboron radicals by one electron reduction or oxidation reactions.
Fig 4.
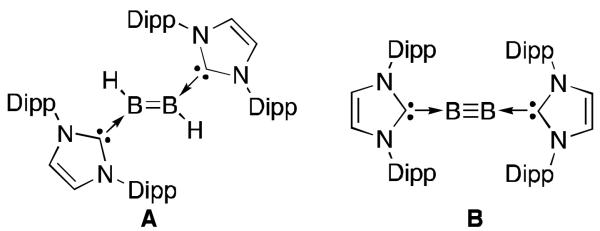
Recently reported unusual boron species stabilized by carbenes.
Carbene-stabilized silicon centred radicals
Silyl radicals of type R3Si· have been widely studied and used in chemical synthesis, as exemplified by the popular (Me3Si)3Si· discovered by Chatigilialoglu et al.25 Not surprisingly, the half-lives of these radicals increase with the steric bulk and (tBu2MeSi)3Si· was found to even be isolable in a crystal form (C, Figure 5).26 Also of note, radical anions D27 and E28 were isolated and structurally characterized; they were prepared by reduction of a disilene.
Fig 5.
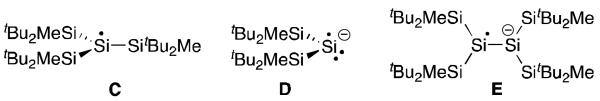
Isolated neutral silyl radical C and radical anions D and E.
Aside from species C-E, stable silicon radicals are rare. It is only in the last two years that carbene-stabilized silicon radicals have been isolated. NHCs can coordinate to the silylene 8, which is based on the β-diketiminate ligand (Figure 6).29 The reaction of muonium (atom made up of an antimuon and an electron, which is chemically similar to hydrogen) with the free silylene 8 resulted in the addition of muonium to two sites; the butadiene moiety in the ligand framework (9a) and the silicon centre (9b).30 In the analogous reaction of muonium with the NHC-complexed silylene 10, butadiene addition was also observed (11a), however the other product showed the unpaired electron on the carbene centre rather than on silicon (11b). This was deduced from the larger μSR hyperfine coupling constants with the nitrogen atoms of the NHC ring compared to those of the silylene and the decreased coupling to silicon in 11b vs. 9b.
Fig 6.

Reaction of the silylene 8 and NHC-silylene adduct 10 with muonium.
NHCs can also coordinate to bis(silyl)silylene as shown by compound 12 reported by Sekiguchi et al. (Figure 7).31 Oxidation with a trityl salt produces the corresponding radical cation 12+·. The EPR spectrum shows strong hyperfine coupling to a single silicon atom and weak coupling to the two NHC nitrogen atoms. The solid state structure of 12+· shows a planar geometry at silicon indicating a π-type radical. A change in hybridization occurs upon removal of an electron from the pyramidal silylene adduct 12. Calculations indicate that the majority of the positive charge (+0.62) resides on the silyl moiety indicating that the dative model drawn is the most accurate. Note that the radical cation 12+· results formally from the replacement of an anionic substituent of C by a neutral NHC ligand.
Fig 7.
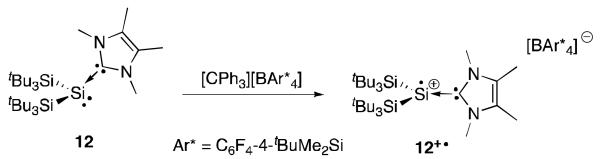
Synthesis of silicon radical cation 12+·.
The most surprising results in the field offered NHC-stabilized silicon radicals have been reported by Roesky, Stalke et al. The colourless NHC stabilized silicon dihalides 13a,b32 are of course diamagnetic with a lone pair on the silicon centre (Figure 8). Addition of a 3-fold excess of CAAC 14 to 13a results in the formation of the (CAAC)2SiCl2 adduct 15a.33 Remarkably, this product is a deep blue biradical with an unpaired electron residing on each carbene centre. Two polymorphs of 15a were isolated, a singlet and a triplet biradical as the unpaired electrons can have the same or opposite spin. The singlet state is calculated to be between 2.2-7.5 kcal/mol higher in energy than the triplet state. The solid-state structures of the two polymorphs were essentially the same. The carbene-silicon bond distances are much shorter in 15a than in the NHC complex 13a (1.85 Å vs. 1.99 Å), indicating that a covalent respresentation is more accurate than the dative model. The N-C bond within the CAAC framework is longer than typical CAAC complexes. This is due to the p-orbital on the carbene carbon being occupied with a single electron, preventing delocalization of the nitrogen lone pair to form an N-C double bond. Remarkably, the singlet polymorph is air stable for up to a week.
Fig 8.
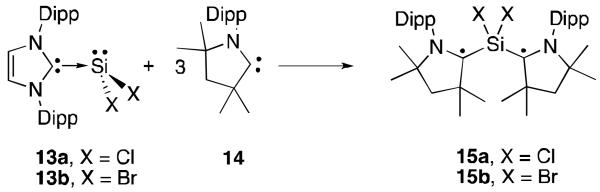
Synthesis of silicon dihalide biradicals 15.
The dibromide derivative 15b could also be prepared from 13b but is much less stable than the chloride analogue 15a;34 it decomposes rapidly in solution. With this knowledge of stability (SiCl2 > SiBr2), more electronegative substituents on silicon should assist in the stabilization of these types of biradicals. However, attempts to generate the SiF2 derivative were unsuccessful,34 although it appears to be a viable synthetic target.
Very interestingly, the silicon dichloride diradical 15a could be reduced with KC8 to generate the bis(carbene)Si compound 16 (Figure 9).35 This species can be viewed as a carbene-stabilized single atom allotrope36 of silicon. Theoretical calculations on 16, indicate that the HOMO is a π-type orbital along the C-Si-C moiety, while the HOMO-1 is the lone pair on the silicon centre. Based on this, the authors describe the bonding between the carbene and silicon as σ-donation from the carbene to the silicon and π-backdonation of one lone pair of electrons on silicon with the two carbon atoms (16”). The bonding model with the electrons localized on the carbon atoms, as a biradical 16′, can also be drawn. No EPR signal was observed, however it is believed that the dark blue color is consistent with a small HOMO-LUMO gap which could introduce biradical character. Crystals of 16 are stable for a day under ambient conditions.
Fig 9.
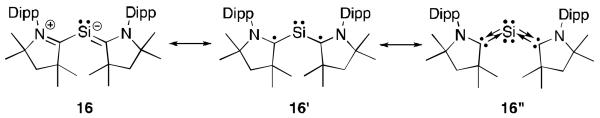
Resonance structures of 16.
In 2008, Robinson et al.37 reported the preparation of the bis(NHC)Si2 complex F (Figure 10) which like 16 bears electron density at silicon.38 It should be interesting if the analogous CAAC complex could be prepared to determine if the electron density is delocalized onto the carbene centres as a biradical. Furthermore, the redox chemistry of the carbene stabilized Si2 allotropes could give rise to interesting radical anions and cations.
Fig 10.
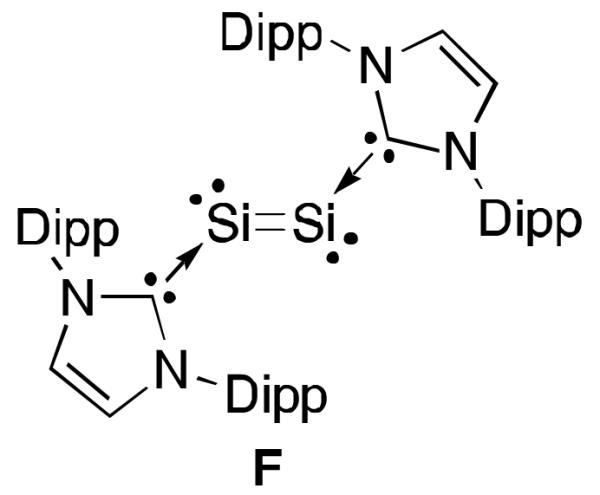
Robinson’s bis(NHC)Si2 complex F.
Carbene-stabilized pnictogen centred radicals
Phosphinyl radicals have been studied for a number of years.39 Although they can be monomeric in solution, they typically dimerize in the solid state.40 Consequently, a few examples of structurally characterized phosphorus radicals G-K have been reported (Figure 11), all of them being stabilized by very bulky subtituents, and/or spin delocalization.41 In addition, two phosphorus centred radical cations L and M have recently been described.42
Fig 11.

Structurally characterized phosphorus radicals.
The first attempt to use a carbene to stabilize a phosphorus radical was reported in 2007 by Apeloig et al.13 The phosphorus radical ·P(O)(OiPr)2 was generated by the irradiation of {(iPrO)2(O)P}2Hg in the presence of free NHC (Figure 12).13 Although the resulting carbene-diisopropylphosphoryl radical complex 17· cannot be isolated, its half life is prolonged to 7.1 seconds. The EPR spectrum and calculations show that considerable electron density resides on the carbon (39%) and the remaining N2C2 portion of the NHC (37%), while only a small portion is on the phosphorus atom (7.5%). Hence, the resonance structure 17a· is a better representation of the molecule rather than the donor-stabilized phosphorus radical 17b·. Accordingly, the optimized geometry indicates that the carbon atom bonded to phosphorus is slightly pyramidalized (Σangles = 352°). The corresponding reaction with the adamantyl benzimadazol-2-ylidene produces radical 18· for which DFT calculations indicate that the carbon atom possesses an even larger portion of spin density (47%) than in 17·.43 a result, the carbene centre of 18· As is more pyramidalized (Σangles = 341°) than in 17·, which hampers the delocalization of the spin density, and destabilizes complex 18· (τ½ < 1s).
Fig 12.

Synthesis and structural representation of persistent NHC-diisopropylphosphoryl radicals 17· and 18·.
In 2010, our group adopted a very different approach to generate a carbene-stabilized phosphorus radical.44 We readily prepared phosphalkene 19 by the addition of a CAAC to an aminodichlorophosphine and subsequent reduction with magnesium. Due to the substitution pattern, 19 is electron-rich and an electron could be removed by reaction with [CPh3][B(C6F5)4] to afford the stable radical cation 19+· (Figure 13). Suprisingly, the spin density is primarily localized on phosphorus (68%) rather than on the π-accepting CAAC ligand. A single crystal X-ray diffraction study showed that the N-C bond in the CAAC is significantly contracted in comparison to the phosphaalkene precursor (1.32 Å vs. 1.38 Å) and is consistent with a double bond. The phospheniumyl cation structure is the most accurate with the positive charge localized on the nitrogen of the carbene. The reaction of 19+· with nBu3SnH produced the phosphine (19H+) confirming the presence of the unpaired electron on phosphorus.
Fig 13.
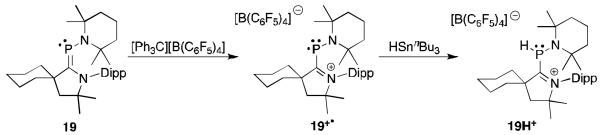
Synthesis and reactivity of the structurally characterized CAAC-phospheniumyl radical 19+·.
In order to isolate a stable neutral radical, we were inspired by the work of Cummins et al. who reported that vanadium–iminato ligands can stabilize phosphinyl radicals (J, Figure 11).41a Since carbenes can behave similarly to transition metals (both have an occupied and vacant orbital),10i we just replaced the metal fragment of J by an NHC. Reduction of the corresponding phosphenium triflate 20 afforded the desired radical 20· (Figure 14).45 This paramagnetic species displayed a significant amount of electron density at the phosphorus centre (64%), greatly exceeding that of the bis(vanadium–iminato) analog J (31%). To directly compare the radical stabilization offered by NHCs versus vanadium centres, the mixed complex 21· was prepared similarly. In 21· only 1% of the spin density resides in the phosphorus 3p orbital, a miniscule amount compared to 67% at the vanadium centre. These results clearly demonstrate that the vanadium fragment is much more efficient than the NHC to delocalize spin density. However, the NHC group still provides sufficient stabilization to allow for the isolation and structural characterization of 20·.
Fig 14.

Phosphinyl radicals featuring vanadium and NHC substituents.
Dinitrogen is the most abundant molecule in the atmosphere but little is known about its heavier congeners, which do not exist under ambient conditions. However, carbene stabilized versions of the heavy group 15 elements36,46 have been isolated for P2,10k,47 As248 as well as the heterodiatomic species PN.49
The structure of the bis(carbene)-P2 species can be drawn as a as a bis-phosphinidene coordinated by two carbenes (22a,b)or as diphosphalkene (22a’,b’) or (Figure 15).47 The latter representation shows the P2 fragment to be electron rich, and therefore prone to oxidation. Cyclic voltammetry on the (NHC)2P2 complex 22a shows two reversible one-electron oxidations whereas the CAAC2P2 complex 22b has only one.50 CAACs are better π-accepting ligands than NHCs and consequently withdraw more electron density from the P2 core. This is a very good demonstration of the different electronic properties of NHCs and CAACs.
Fig 15.

Cyclic voltammograms of 22a,b and their oxidation products.
Both diphosphorus species are easily oxidized by [CPh3][B(C6F5)4] to the radical cations 22a+· and 22b .50 Crystallographically, the metrical parameters only showed slight deviations between the two structures. However, the CAAC derivative 22b+· gave an EPR signal as a triplet of quintets with large hyperfine coupling to the phosphorus centres and small coupling to the two nitrogen atoms of the CAAC ligands. This differed from its NHC counterpart 22a+· which only displayed a triplet indicative of hyperfine coupling only to the two phosphorus atoms. The calculated spin density and the EPR data suggest that CAACs are more efficient than NHCs to delocalize the electron density; this is reflected by the smaller amount of spin density on the P2 moiety in 22b+· (54% vs. 77% in 22a+·). The NHC complex could be oxidized further to the dication 22a++ which was not possible with the CAAC derivative, attributed again to the π-accepting nature of the CAAC.
Arsenic radicals are much less prevalent than phosphorus. In fact the radical cation 23+· is the only structurally characterized arsenic paramagnetic species documented (Figure 16).51 Similarly to the P2 complexes, this compound has been prepared by simple oxidation of the diarsenic precursor 23 with two equivalents of GaCl3. In the EPR spectrum, only hyperfine coupling to the arsenic atoms was observed. The calculated SOMO indicated that considerable electron density is localized on the diarsenic core of 23+· (82%), which is slightly greater than on the diphosphorus core of 22a+· (77%). The As-As bond is contracted upon oxidation (2.32 Å vs. 2.44 Å), indicating partial double bond character. This species could be oxidized a second time like 22a with GaCl3 to form the As2++ dication 23++.
Fig 16.

Oxidation of the NHC stabilized As2 molecule.
The carbene stabilized phosphorus mononitride 24 that features an NHC on nitrogen and a CAAC on phosphorus could also be prepared (Figure 17).49 The cyclic voltammogram of 24 shows a reversible one electron oxidation. In line with the electrochemical results, the PN radical cation 24+· could be generated and isolated by the reaction of 24 with [CPh3][B(C6F5)4]. Calculations on a model compound showed that the spin density is primarily on phosphorus (40%) with less on the central nitrogen (19%) and stikingly almost the same amount on the nitrogen atom of the CAAC (18%). As with the P2 radical cations 22a+· and 22b+·, this is a clear demonstration that CAACs are better π-acceptors in comparison to NHCs.
Fig 17.
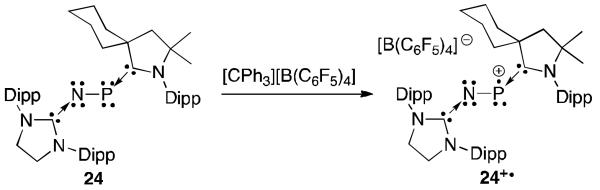
Synthesis of radical cation 24+·.
The activation of P4 with singlet carbenes has produced carbene stabilized P+, P2 , P4, P8 and P12 units.10j-n These derivatives should also serve as good candidates to generate new 60 radical cations by oxidation reactions.
Carbene stabilized organic radicals
So far, applications of stable carbenes for stabilizing organic radicals are very rare. However, already in 1997, Fukuzumi et al. prepared models of the thiamin coenzyme, which feature a thiazolylidene scaffold (Figure 18).52 The electrochemical oxidation of enolate 25− produces the neutral radical 25· identified by EPR spectroscopy that is persistent for several hours. The EPR indicates that almost no spin density resides on the aryl group and calculations show that the latter could be in fact perpendicular to the radical fragment. The oxidation of 25− occurs at a low potential indicating that in vivo thiamine-dependent acetylations by pyruvate could involve one-electron transfers. This electron transfer process was confirmed by the reaction of 25− with [Co(phen)3]2+ (phen = phenanthroline) resulting in the redox reaction producing [Co(phen)3]+ and 25·.
Fig 18.

Oxidation of the thiazolylidene based enolate 25− to 25·.
Reed et al. later conducted studies on the radical intermediate of the thiamin coenzyme and pyruvate.53 EPR data obtained on isotopically labelled variants indicate that a π-type radical is generated within the system and confirmed that the spin density is delocalized throughout the thiazolium ring (25b·). Interestingly, in the enzyme the authors postulated that a protonation can occur giving the corresponding radical cation 25H+· but were not able to localize where the proton binds the thiamin.
We recently investigated the possibility of using carbenes to prepare stable radical cations, with a structure inspired by 25H+·. We reasoned that the use of not only one but two carbenes would allow the isolation of such species. After observing that bulky carbenes react with CO to form amino-ketenes,11b,c species typically unstable,54 we reasoned that a less bulky carbene such as the anti-Bredt NHC 2655 could allow for the attack of two carbenes to CO, giving the oxyallyl derivative 27 (Figure 19). Although the latter could not be isolated, it was characterized by NMR spectroscopy at −40°C. Protonation of 27 gave rise to the corresponding oxyallyl cation 27H+, which by simple oxidation with air afforded the oxyallyl radical cation 27+·.
Fig 19.

Synthesis of oxyallyl radical cation 27H+·.
Interestingly, two isomers of 27+· were isolated (d,l-27+· and meso-27+·). According to X-ray diffraction studies, the former has the two carbenes conjugated with the CO fragment but in the latter only one carbene assists in delocalizing the spin density. Surprisingly, the two derivatives bear similar spin density on the CO core (d,l-27+·: 51.6%; meso-27+·: 48%) indicating that one anti-Bredt NHC is probably sufficient in stabilizing such radical species. Note that, prior to this, oxyallyl radicals were only observed in inert matrices at low temperatures56 or by mass spectrometry.57 These results suggest that a variety of carbene-stabilized organic radicals should be isolable at ambient conditions.
Why carbenes are so effective to stabilize radical species and which carbenes are the best?
Protonated “classical” unsaturated NHCs, as exemplified by the imidazolium cation (28H+), show an irreversible single electron reduction (−2.76 V vs. FeCp2/FeCp2+). 58 The corresponding chemical reduction in a flask with potassium produces the free carbene 28, believed to result from the decomposition of radical 28H·, indicating that this neutral radical is highly reactive (Figure 20).
Fig 20.
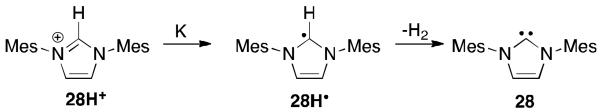
The reduction of an imidazolium cation with potassium.
Experimental results from μSR spectroscopy showed that carbene 28 reacts with muonium exclusively at the carbene centre to generate a radical 28Mu· similar to 28H· with muonium in place of hydrogen (Figure 21).59 Calculations show that the reaction at this site is preferred by 14 kcal/mol over the double bond in the framework giving 29Mu·. Radical 28Mu·, which of course cannot be isolated, is calculated to be pyramidalized at the carbon atom by about 40°, and 74.9% of the spin density resides at this site.
Fig 21.
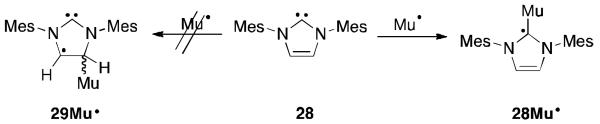
Reaction of muonium with NHC 28.
Although the afforementioned results seem to indicate that carbenes are not prone to stabilize radical species, it is of foremost importance to realize that the experiments have been performed with unsaturated NHCs, which are probably among the weakest π-accepting carbenes. Indeed, an examination of the literature reveals that carbenes, which are only slightly more electrophilic than 28, can accommodate an additional electron. Cyclic voltammetry on the free triazol-5-ylidene 30 indicates that the empty orbital can be populated with a single electron to generate the radical anion 30·- (Figure 22).60 The electrochemically generated radical could even be observed by EPR. Moreover, calculations on annulated versions of NHCs, such as 31, show that in the corresponding radical anion up to 30% of the spin density resides on the carbene carbon.61
Fig 22.
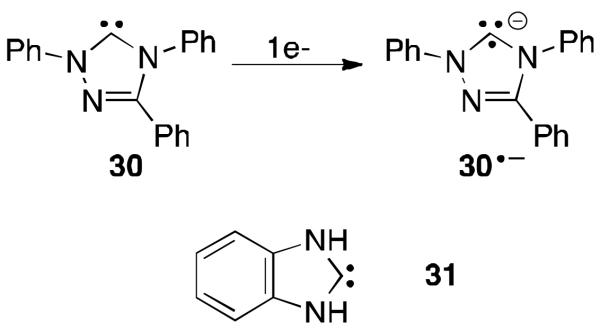
Reduction of triazol-5-ylidene 30 and annulated-NHC 31.
Clearly, all the results presented in this mini-review suggest that π-accepting carbenes are more efficient for stabilizing radicals, because they better delocalize the spin density. This prompted us to develop a method to evaluate the π-accepting properties of carbenes.62 Note that the very popular TEP (Tolman Electronic Parameter)63 measures the overall donor properties of carbenes, which is the σ-donation subtracted by the π-acception. Our method is based on the 31P NMR chemical shift of readily synthesized phenylphosphinidene-carbene adducts 32 (Figure 23). These compounds can be represented by two extreme canonical structures. The resonance form 32a corresponds to a typical phosphaalkene, featuring a formal P=C double bond, and it is well-known that their 31P NMR signals are at low field. In contrast, form 32b corresponds to a carbene-phosphinidene adduct featuring a P-C dative bond with two lone pairs of electrons at phosphorus, and therefore give signals at high field. It is obvious that an increase of the π-accepting property of the carbene favors the contribution of resonance form 32a, and thus more π-accepting carbenes will give a signal further downfield.
Fig 23.
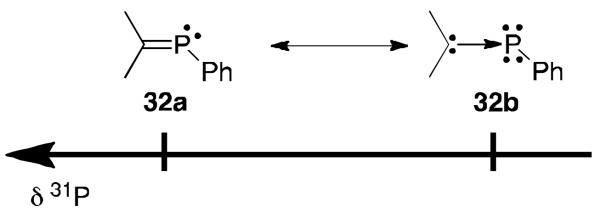
Structures of 32 and their relative 31P NMR chemical shifts.
Conclusions
This report merely documents the infancy of this promising area of research. So far carbene stabilized paramagnetic main group species have only been reported for a few elements, namely boron, silicon, phosphorus, arsenic and carbon. As mentioned in the introduction, radicals play essential roles in numerous chemical processes, and almost nothing is known about the reactivity of carbene stabilized radicals. Importantly, the electronic properties of carbenes can be easily tuned, and therefore the reactivity of the corresponding radicals. The recent work by Roesky, Stalke et al.33,35 shows that carbenes can also stabilize biradicals. Consequently, it is reasonable to believe that they could also allow for the isolation of polyradicals, which can find numerous applications in materials science.
Acknowledgements
We are grateful to the NSF (CHE-1316956), NIH (R01 GM 68825), DOE (DE-SC0009376), for financial support of our work.
Biographies
Caleb Martin was born in 1985 in New Brunswick, Canada. He completed his BSc at Mount Allison University in 2007. He then attended the University of Western Ontario for his PhD under the supervision of Paul Ragogna. Following graduation in 2011, he began working with Guy Bertrand as a Post Doctoral Researcher currently as a member of the UCSD/CNRS Joint Research Laboratory at the University of California, San Diego. In June 2013 he will begin his independent career as an Assistant Professor at Baylor University.
Michèle Soleilhavoup studied chemistry at the University Paul Sabatier in Toulouse and received her PhD in 1993 under the supervision of Guy Bertrand. From 1993 to 1995, she worked for BASF AG at Ludwigshafen. In 1995, she moved to the University Paris VI as a “Chargée de Recherche CNRS”, and from 2000 to 2001, she worked in the Remi Chauvin’s group at the Laboratoire de Chimie de Coordination in Toulouse, before joining the UCR/CNRS Joint Research Laboratory at the University of California, Riverside and in 2012 the UCSD/CNRS. Laboratory at the University of California, San Diego. Her current research interests are focused on carbene chemistry and their application as tunable ligands for transition metal catalysts.
Guy Bertrand was an undergraduate in Montpellier, and obtained his PhD from the University of Toulouse. From 1988 to 1998 he was a “Director of Research” at the Laboratoire de Chimie de Coordination du CNRS, and from 1998 to 2005 the Director of the Laboratoire d’Hétérochimie Fondamentale et Appliquée at the University Paul Sabatier (Toulouse). From 2001 to 2012 he has been Distinguished Professor and Director of the UCR/CNRS Joint Research Chemistry Laboratory at the University of California at Riverside, and now at the University of California, San Diego. His research spans a wide range of topics at the border between organic and inorganic chemistry. He is a member of the French Academy of Sciences.
Notes and references
- 1.(a)" Fossey J, Lefort D, Serba J. Free Radicals in Organic Chemistry. John Wiley and Sons; 1995. [Google Scholar]; (>b) Renaud P, Sibi MP. Radicals in Organic Synthesis. Wiley-VCH; 2008. [Google Scholar]; (c) Chatgilialoglu C, Studer A. Encyclopedia of Radicals in Chemistry, Biology and Materials. Wiley-VCH; 2012. [Google Scholar]
- 2.Halliwell B, Gutteridge JMC. Free Radicals Biology and Medicine. Oxford University Press; 2007. [Google Scholar]
- 3.Gomberg M. J. Am. Chem. Soc. 1900;22:757. [Google Scholar]
- 4.For reviews, see: Power PP. Chem. Rev. 2003;103:789. doi: 10.1021/cr020406p. Lee VY, Nakamoto M, Sekiguchi A. Chem. Lett. 2008;37:128. Lee VY, Sekiguchi A. Eur. J. Inorg. Chem. 2005:1209. Hicks RG. Org. Biomol. Chem. 2007;5:1321. doi: 10.1039/b617142g.
- 5.For recent reviews on carbenes, see: Hahn FE, Jahnke MC. Angew. Chem., Int. Ed. 2008;47:3122. doi: 10.1002/anie.200703883. Schuster O, Yang L, Raubenheimer HG, Albrecht M. Chem. Rev. 2009;109:3445. doi: 10.1021/cr8005087. Melaimi M, Soleilhavoup M, Bertrand G. Angew. Chem., Int. Ed. 2010;49:8810. doi: 10.1002/anie.201000165. Martin D, Melaimi M, Soleilhavoup M, Bertrand G. Organometallics. 2011;30:5304. doi: 10.1021/om200650x. Vignolle J, Cattoën X, Bourissou D. Chem. Rev. 2009;109:3333. doi: 10.1021/cr800549j. Diez-Gonzalez S, Marion N, Nolan SP. Chem. Rev. 2009;109:3612. doi: 10.1021/cr900074m. Vougioukalakis GC, Grubbs RH. Chem. Rev. 2010;110:1746. doi: 10.1021/cr9002424. Grossmann A, Enders D. Angew. Chem., Int. Ed. 2012;51:314. doi: 10.1002/anie.201105415. Díez-González S, editor. N-Heterocyclic carbenes, from laboratory curiosities to efficient synthetic tools. Royal Society of Chemistry Publishing; Cambridge: 2011. Glorius FA, editor. N-Heterocyclic Carbenes in Transition Metal Catalysis. Springer Verlag; 2007. Nolan SP, editor. N-Heterocyclic Carbenes in Synthesis. Wiley-VCH; 2006.
- 6.For the preparation of the first stable carbenes, see: Igau A, Grützmacher H, Baceiredo A, Bertrand G. J. Am. Chem. Soc. 1988;110:6463. Igau A, Baceiredo A, Trinquier G, Bertrand G. Angew. Chem. Int. Ed. Engl. 1989;28:621. Arduengo AJ, III, Harlow RL, Kline M. J. Am. Chem. Soc. 1991;113:361. Arduengo AJ, III, Goerlich JR, Marshall WJ. J. Am. Chem. Soc. 1995;117:11027. Enders D, Breuer K, Raabe G, Runsink J, Teles JH, Melder JP, Ebel K, Brode S. Angew. Chem. Int. Ed. Engl. 1995;34:1021. Hahn FE, Wittenbecher L, Boese R, Bläser D. Chem. Eur. J. 1999;5:1931.
- 7.(a) Arduengo AJ., III Acc. Chem. Res. 1999;32:913. [Google Scholar]; (b) Bourissou D, Guerret O, Gabbaï FP, Bertrand G. Chem. Rev. 2000;100:39. doi: 10.1021/cr940472u. [DOI] [PubMed] [Google Scholar]; (c) Herrmann WA. Angew. Chem., Int. Ed. 2002;41:1290. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]; (d) Peris E, Crabtree RH. Coord. Chem. Rev. 2004;248:2239. [Google Scholar]
- 8.(a) Jacobsen H, Correa A, Costabile C, Cavallo L. J. Organomet. Chem. 2006;691:4350. [Google Scholar]; (b) Jacobsen H, Correa A, Poater A, Costabile C, Cavallo L. Coord. Chem. Rev. 2009;253:687. [Google Scholar]; (c) Hu X, Castro-Rodriguez I, Olsen K, Meyer K. Organometallics. 2004;23:755. [Google Scholar]; (d) Sanderson MD, Kamplain JW, Bielawski CW. J. Am. Chem. Soc. 2006;128:16514. doi: 10.1021/ja067475w. [DOI] [PubMed] [Google Scholar]; (e) Khramov DM, Lynch VM, Bielawski CW. Organometallics. 2007;26:6042. [Google Scholar]; (f) Saravanakumar S, Oprea AI, Kindermann MK, Jones PG, Heinicke J. Chem. Eur. J. 2006;12:3143. doi: 10.1002/chem.200501183. [DOI] [PubMed] [Google Scholar]; (g) Mercs L, Labat G, Neels A, Ehlers A, Albrecht M. Organometallics. 2006;25:5648. [Google Scholar]; (h) Fantasia S, Petersen JL, Jacobsen H, Cavallo L, Nolan SP. Organometallics. 2007;26:5880. [Google Scholar]; (i) Droege T, Glorius F. Angew. Chem. Int. Ed. 2010;49:6940. doi: 10.1002/anie.201001865. [DOI] [PubMed] [Google Scholar]
- 9.Alcarazo M, Stork T, Anoop A, Thiel W, Fürstner A. Angew. Chem., Int. Ed. 2010;49:2542. doi: 10.1002/anie.200907194. [DOI] [PubMed] [Google Scholar]
- 10.(a) Moerdyk JP, Bielawski CW. Nat. Chem. 2012;4:275. doi: 10.1038/nchem.1267. [DOI] [PubMed] [Google Scholar]; (b) Moerdyk JP, Bielawski CW. J. Am. Chem. Soc. 2012;134:6116. doi: 10.1021/ja3014105. [DOI] [PubMed] [Google Scholar]; (c) Hudnall TW, Moerdyk JP, Bielawski CW. Chem. Commun. 2010;46:4288. doi: 10.1039/c0cc00638f. [DOI] [PubMed] [Google Scholar]; (d) Lee Y-G, Moerdyk JP, Bielawski CW. J. Phys. Org. Chem. 2012;25:1027. [Google Scholar]; (e) Chase DT, Moerdyk JP, Bielawski CW. Org. Lett. 2012;14:5510. doi: 10.1021/ol302596r. [DOI] [PubMed] [Google Scholar]; (f) Wiggins KM, Moerdyk JP, Bielawski CW. Chem. Sci. 2012;3:2986. doi: 10.1039/c5sc90019k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Frey GD, Masuda JD, Donnadieu B, Bertrand G. Angew. Chem., Int. Ed. 2010;49:9444. doi: 10.1002/anie.201005698. [DOI] [PubMed] [Google Scholar]; (h) Frey GD, Lavallo V, Donnadieu B, Schoeller WW, Bertrand G. Science. 2007;316:439. doi: 10.1126/science.1141474. [DOI] [PubMed] [Google Scholar]; (i) Martin D, Soleilhavoup M, Bertrand G. Chem. Sci. 2011;2:389. doi: 10.1039/C0SC00388C. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Masuda JD, Schoeller WW, Donnadieu B, Bertrand G. Angew. Chem., Int. Ed. 2007;46:7052. doi: 10.1002/anie.200703055. [DOI] [PubMed] [Google Scholar]; (k) Back O, Kuchenbeiser G, Donnadieu B, Bertrand G. Angew. Chem., Int. Ed. 2009;48:5530. doi: 10.1002/anie.200902344. [DOI] [PubMed] [Google Scholar]; (l) Masuda JD, Schoeller WW, Donnadieu B, Bertrand G. J. Am. Chem. Soc. 2007;129:14180. doi: 10.1021/ja077296u. [DOI] [PubMed] [Google Scholar]; (m) Martin CD, Weinstein CM, Moore CE, Rheingold AL, Bertrand G. Chem. Commun. 2013;49:4486. doi: 10.1039/c3cc42041h. [DOI] [PubMed] [Google Scholar]; (n) Dorsey CL, Squires BM, Hudnall TW. Angew. Chem., Int. Ed. 2013;52:4462. doi: 10.1002/anie.201301137. [DOI] [PubMed] [Google Scholar]
- 11.(a) Siemeling U, Färber C, Bruhn C, Leibold M, Selent D, Baumann W, Hopffgarten M. v., Goedecke C, Frenking G. Chem. Sci. 2010;1:697. [Google Scholar]; (b) Hudnall TW, Bielawski CW. J. Am. Chem. Soc. 2009;131:16039. doi: 10.1021/ja907481w. [DOI] [PubMed] [Google Scholar]; (c) Lavallo V, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. Angew. Chem., Int. Ed. 2006;45:3488. doi: 10.1002/anie.200600987. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Martin D, Moore CE, Rheingold AL, Bertrand G. Angew. Chem., Int. Ed. DOI: 10.1002/anie.201302841. [Google Scholar]
- 12.For examples, see: Roberts JAS, Franz JA, van der Eide EF, Walter Eric D., Petersen JL, DuBois DL, Bullock RM. J. Am. Chem. Soc. 2011;133:14593. doi: 10.1021/ja202754e. Danopoulos AA, Tsoureas N, Wright JA, Light ME. Organometallics. 2003;23:166. Danopoulos AA, Pugh D, Smith H, Sassmannshausen J. Chem. Eur. J. 2009;15:5491. doi: 10.1002/chem.200900027. Danopoulos AA, Wright JA, Motherwell WB, Ellwood S. Organometallics. 2004;23:4807. Pugh D, Wright JA, Freeman S, Danopoulos AA. Dalton Trans. 2006:775. doi: 10.1039/b512133g. Hu X, Castro-Rodriguez I, Meyer K. Chem. Commun. 2004:2164. doi: 10.1039/b409241d. Hu X, Castro-Rodriguez I, Meyer K. J. Am. Chem. Soc. 2004;126:13464. doi: 10.1021/ja046048k. Hu X, Meyer K. J. Am. Chem. Soc. 2004;126:16322. doi: 10.1021/ja044271b. Hu X, Castro-Rodriguez I, Meyer K. J. Am. Chem. Soc. 2003;125:12237. doi: 10.1021/ja036880+. Chen C, Qiu H, Chen W, Wang D. J. Organomet. Chem. 2008;693:3273. Lorber C, Vendier L. Organometallics. 2008;27:2774. Lorber C, Vendier L. Dalton Trans. 2009:6972. doi: 10.1039/b905056f. Abernethy CD, Cowley AH, Jones RA, Macdonald CLB, Shukla P, Thompson LK. Organometallics. 2001;20:3629. Liu T, Darensbourg MY. J. Am. Chem. Soc. 2007;129:7008. doi: 10.1021/ja071851a. Ito M, Matsumoto T, Tatsumi K. Inorg. Chem. 2009;48:2215. doi: 10.1021/ic802276w.
- 13.Tumanskii B, Sheberla D, Molev G, Apeloig Y. Angew. Chem., Int. Ed. 2007;46:7408. doi: 10.1002/anie.200702297. [DOI] [PubMed] [Google Scholar]
- 14.(a) Chu TL, Weismann TJ. J. Am. Chem. Soc. 1956;78:23. [Google Scholar]; (b) Chu TL, Weismann TJ. J. Am. Chem. Soc. 1956;78:3610. [Google Scholar]; (c) Moeller CW, Wilmarth WK. J. Am. Chem. Soc. 1959;81:2638. [Google Scholar]; (d) Leffler JE, Doland E, Tanigaki T. J. Am. Chem. Soc. 1965;87:927. [Google Scholar]; (e) Weissman SI, van Willigen H. J. Am. Chem. Soc. 1965;87:2285. [Google Scholar]; (f) Griffin RG, van Willigen H. Chem. Phys. 1972;57:86. [Google Scholar]; (g) Karplus M, Frankel GK. J. Chem. Phys. 1962;35:1312. [Google Scholar]; (h) Olmstead MM, Power PP. J. Am. Chem. Soc. 1986;108:4235. [Google Scholar]; (i) Kaim W, Schulz A. Angew. Chem., Int. Ed. 1984;23:65. [Google Scholar]; (j) Rajca A, Rajca S, Desai SR. Chem. Commun. 1995:1958. [Google Scholar]; (k) Schulz A, Kaim W. Chem. Ber. 1989;122:1863. [Google Scholar]; (l) Kwan RJ, Harlan J, Norton JR. Organometallics. 2001;20:3818. [Google Scholar]; (m) Berndt A, Klusik H, Schlüter K. J. Organomet. Chem. 1981;222:C25. [Google Scholar]
- 15.For recent examples of stable boron radicals, see: Braunschweig H, Dyakonov V, Jimenez-Halla JOC, Kraft K, Krummenacher I, Radacki K, Sperlich A, Wahler J. Angew. Chem., Int. Ed. 2012;51:2977. doi: 10.1002/anie.201108632. Aramaki Y, Omiya H, Yamashita M, Nakabayashi K, Ohkoshi S, Nozaki K. J. Am. Chem. Soc. 2012;134:19989. doi: 10.1021/ja3094372. Richards VJ, Gower AL, Smith JEHB, Davies ES, Lahaye D, Slater AG, Lewis W, Blake AJ, Champness NR, Kays D. Chem. Commun. 2012;48:1751. doi: 10.1039/c2cc16047a. Wood TK, Piers WE, Keay BA, Parvez M. Chem. Commun. 2009:5147. doi: 10.1039/b911982e. Chiu CW, Gabbai FP. Angew. Chem., Int. Ed. 2007;46:1723. doi: 10.1002/anie.200604119. Tuononen HM, Chivers T, Armstrong A, Fedorchuk C, Boere RT. J. Organomet. Chem. 2007;692:2705. Venkatasubbaiah K, Zakharov LN, Kassel WS, Rheingold AL, Jakle F. Angew. Chem., Int. Ed. 2005;44:5428. doi: 10.1002/anie.200502148. Emslie DJH, Piers WE, Parvez M. Angew. Chem., Int. Ed. 2003;42:1252. doi: 10.1002/anie.200390320. Scheschkewitz D, Amii H, Gornitzka H, Schoeller WW, Bourissou D, Bertrand G. Science. 2002;295:1880. doi: 10.1126/science.1068167. Chivers T, Eisler DJ, Fedorchuk C, Schatte G, Tuononen HM, Boeré RT. Inorg. Chem. 2006;45:2119. doi: 10.1021/ic0520014.
- 16.(a) Lalevée J, Telitel S, Tehfe MA, Fouassier JP, Curran DP, Lacôte E. Angew. Chem., Int. Ed. 2012;51:5958. doi: 10.1002/anie.201200975. [DOI] [PubMed] [Google Scholar]; (b) Solovyev A, Ueng S-H, Monot Julien, Fensterbank L, Malacria M, Lacôte E, Curran DP. Org. Lett. 2010;12:2998. doi: 10.1021/ol101014q. [DOI] [PubMed] [Google Scholar]; (c) Ueng S-H, Solovyev A, Yuan X, Geib SJ, Fensterbank L, Lacôte E, Malacria M, Newcomb M, Walton JC, Curran DP. J. Am. Chem. Soc. 2009;131:11256. doi: 10.1021/ja904103x. [DOI] [PubMed] [Google Scholar]; (d) Walton JC, Brahmi MM, Fensterbank L, Lacôte E, Malacria M, Chu Q, Ueng S-H, Solovyev A, Curran DP. J. Am. Chem. Soc. 2010;132:2350. doi: 10.1021/ja909502q. [DOI] [PubMed] [Google Scholar]; (e) Tehfe M-A, Brahmi MM, Fouassier J-P, Curran DP, Malacria M, Fensterbank L, Lacôte E, Lalevée J. Macromolecules. 2010;43:2261. [Google Scholar]; (f) Curran DP, Solovyev A, Brahmi MM, Fensterbank L, Malacria M, Lacôte E. Angew. Chem., Int. Ed. 2011;50:10294. doi: 10.1002/anie.201102717. [DOI] [PubMed] [Google Scholar]; (g) Walton JC, Brahmi MM, Monot J, Fensterbank L, Malacria M, Curran DP, Lacôte E. J. Am. Chem. Soc. 2011;133:10312. doi: 10.1021/ja2038485. [DOI] [PubMed] [Google Scholar]; (h) Ueng S-H, Fensterbank L, Lacôte E, Malacria M, Curran DP. Org. Biomol. Chem. 2011;9:3415. doi: 10.1039/c0ob01075h. [DOI] [PubMed] [Google Scholar]; (i) Pan X, Lacôte E, Lalevée J, Curran DP. J. Am. Chem. Soc. 2012;134:5669. doi: 10.1021/ja300416f. [DOI] [PubMed] [Google Scholar]; (j) Telitel S, Schweizer S, Morlet-Savary F, Graff B, Tschamber T, Blanchard N, Fouassier JP, Lelli M, Lacôte E, Lalevée J. Macromolecules. 2013;46:43. [Google Scholar]; (k) Tehfe M-A, Monot J, Brahmi MM, Bonin-Dubarle H, Curran DP, Malacria M, Fensterbank L, Lacôte E, Lalevée J, Fouassier J-P. Polym. Chem. 2011;2:625. [Google Scholar]; (l) Tehfe M-A, Monot J, Malacria M, Fensterbank L, Fouassier J-P, Curran DP, Lacôte E, Lalevée J. ACS Macro Lett. 2012;1:92. doi: 10.1021/mz200087g. [DOI] [PubMed] [Google Scholar]
- 17.Kirwan JN, Roberts BP. J. Chem. Soc. Perkin Trans. 2. 1989:539. [Google Scholar]
- 18.Chiu C-W, Gabbaï FP. Angew. Chem., Int. Ed. 2007;46:1723. doi: 10.1002/anie.200604119. [DOI] [PubMed] [Google Scholar]
- 19.Matsumoto T, Gabbaï FP. Organometallics. 2009;28:4252. [Google Scholar]
- 20.Chiu C-W, Gabbaï FP. Angew. Chem., Int. Ed. 2007;46:6878. doi: 10.1002/anie.200702299. [DOI] [PubMed] [Google Scholar]
- 21.(a) Lavallo V, Canac Y, Prasang C, Donnadieu B, Bertrand G. Angew. Chem., Int. Ed. 2005;44:5705. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jazzar R, Dewhurst RD, Bourg JB, Donnadieu B, Canac Y, Bertrand G. Angew. Chem., Int. Ed. 2007;46:2899. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zeng X, Frey GD, Kinjo R, Donnadieu B, Bertrand G. J. Am. Chem. Soc. 2009;131:8690. doi: 10.1021/ja902051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Kinjo R, Donnadieu B, Celik MA, Frenking G, Bertrand G. Science. 2011;333:610. doi: 10.1126/science.1207573. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Robinson GH. Science. 2011;333:530. doi: 10.1126/science.1209588. [DOI] [PubMed] [Google Scholar]; (c) Celik MA, Sure R, Klein S, Kinjo R, Bertrand G, Frenking G. Chem. Eur. J. 2012;18:5676. doi: 10.1002/chem.201103965. [DOI] [PubMed] [Google Scholar]
- 23.(a) Wang Y, Quillian B, Wei P, Wannere CS, Xie Y, King RB, Schaefer HF, III, Schleyer P. v. R., Robinson GH. J. Am. Chem. Soc. 2007;129:12412. doi: 10.1021/ja075932i. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Quillian B, Wei P, Xie Y, Wannere CS, King RB, Schaefer HF, III, Schleyer P. v. R., Robinson GH. J. Am. Chem. Soc. 2008;130:3298. doi: 10.1021/ja800257j. [DOI] [PubMed] [Google Scholar]
- 24.(a) Braunschweig H, Dewhurst RD, Hammond K, Mies J, Radacki K, Vargas A. Science. 2012;336:1420. doi: 10.1126/science.1221138. [DOI] [PubMed] [Google Scholar]; (b) Frenking G, Holzmann N. Science. 2012;336:1394. doi: 10.1126/science.1224003. [DOI] [PubMed] [Google Scholar]
- 25.For reviews, see: Chatigilialoglu C. Adv. Organomet. Chem. 2008;57:117. Chatigilialoglu C. Chem. Rev. 1995;95:12229. Chatigilialoglu C. Chem. Eur. J. 2008;14:2310.
- 26.Sekiguchi A, Fukawa T, Nakamoto M, Lee VY, Ichinoe M. J. Am. Chem. Soc. 2002;124:9865. doi: 10.1021/ja0126780. [DOI] [PubMed] [Google Scholar]
- 27.Inoue S, Ichinohe M, Sekiguchi A. J. Am. Chem. Soc. 2007;129:6096. doi: 10.1021/ja0711314. [DOI] [PubMed] [Google Scholar]
- 28.Sekiguchi A, Inoue S, Ichinohe M, Arai Y. J. Am. Chem. Soc. 2004;126:9626. doi: 10.1021/ja040079y. [DOI] [PubMed] [Google Scholar]
- 29.Xiong Y, Yao SL, Driess M. J. Am. Chem. Soc. 2009;131:7562. doi: 10.1021/ja9031049. [DOI] [PubMed] [Google Scholar]
- 30.Percival PW, McCollum BM, Brodovitch J-C, Driess M, Mitra A, Mozafari M, West R, Xiong Y, Yao S. Organometallics. 2012;31:2709. [Google Scholar]
- 31.Tanaka H, Ichinohoe M, Sekiguchi A. J. Am. Chem. Soc. 2012;134:5540. doi: 10.1021/ja301180v. [DOI] [PubMed] [Google Scholar]
- 32.Ghadwal RS, Roesky HW, Merkel S, Henn J, Stalke D. Angew. Chem., Int. Ed. 2009;48:5683. doi: 10.1002/anie.200901766. [DOI] [PubMed] [Google Scholar]
- 33.Mondal KC, Roesky HW, Schwarzer MC, Frenking G, Tkach I, Wolf H, Kratzert D, Herbst-Irmer R, Niepötter B, Stalke D. Angew. Chem., Int. Ed. 2013;52:1801. doi: 10.1002/anie.201204487. [DOI] [PubMed] [Google Scholar]
- 34.Mondal KC, Samuel PP, Tretiakov M, Singh AP, Roesky HW, Stückl AC, Niepötter B, Carl E, Wolf H, Herbst-Irmer R, Stalke D. Inorg. Chem. 2013;52:4736. doi: 10.1021/ic400357p. [DOI] [PubMed] [Google Scholar]
- 35.Mondal KC, Roesky HW, Schwarzer MC, Frenking G, Niepötter B, Wolf H, Herbst-Irmer R, Stalke D. Angew. Chem., Int. Ed. 2013;52:2963. doi: 10.1002/anie.201208307. [DOI] [PubMed] [Google Scholar]
- 36.Dyker CA, Bertrand G. Science. 2008;321:1050. doi: 10.1126/science.1162926. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Xie Y, Wei P, King RB, Schaefer HF, III, Schleyer P. v. R., Robinson GH. Science. 2008;321:1069. doi: 10.1126/science.1160768. [DOI] [PubMed] [Google Scholar]
- 38.For the germanium and tin analogues, see: Sidiropoulos JA, Jones C, Stasch A, Klein S, Frenking G. Angew. Chem., Int. Ed. 2009;48:9701–9704. doi: 10.1002/anie.200905495. Jones C, Sidiropoulos A, Holzmann N, Frenking G, Stasch A. Chem. Commun. 2012;48:9855–9857. doi: 10.1039/c2cc35228a.
- 39.For reviews, see: Armstrong A, Chivers T, Boere RT. ACS Symposium Series. 2006;917:66. Marque S, Tordo P. Top. Curr. Chem. 2005;250:43. Geoffroy M. Recent Res. Dev. Phys. Chem. 1998;2:311.
- 40.(a) Hinchley SL, Morrison CA, Rankin DWH, Macdonald CLB, Wiacek RJ, Voigt A, Cowley AH, Lappert MF, Gundersen G, Clyburne JAC, Power PP. J. Am. Chem. Soc. 2001;123:9045. doi: 10.1021/ja010615b. [DOI] [PubMed] [Google Scholar]; (b) Giffin NA, Hendsbee AD, Roemmele TL, Lumsden MD, Pye CC, Masuda JD. Inorg. Chem. 2012;51:11837. doi: 10.1021/ic301758k. [DOI] [PubMed] [Google Scholar]; (c) Puntigam O, Förster D, Giffin NA, Burck S, Bender J, Ehret F, Hendsbee AD, Nieger M, Masuda JD, Gudat D. Eur. J. Inorg. Chem. 2013:2041. [Google Scholar]; (d) Förster D, Dilger H, Ehret F, Nieger M, Gudat D. Eur. J. Inorg. Chem. 2012:3989. [Google Scholar]; (e) Hinchley SL, Morrison CA, Rankin DWH, Macdonald CLB, Wiacek RJ, Cowley AH, Lappert MF, Gundersen G, Clyburne JAC, Power PP. Chem. Commun. 2000:2045. doi: 10.1021/ja010615b. [DOI] [PubMed] [Google Scholar]; (f ) Bezombes JP, Borisenko KB, Hitchcock PB, Lappert MF, Nycz JE, Rankin DWH, Robertson HE. Dalton Trans. 2004:1980. doi: 10.1039/b402926g. [DOI] [PubMed] [Google Scholar]
- 41.(a) Agarwal P, Piro NA, Meyer K, Muller P, Cummins CC. Angew. Chem., Int. Ed. 2007;46:3111. doi: 10.1002/anie.200700059. [DOI] [PubMed] [Google Scholar]; (b) Ishida S, Hirakawa F, Iwamoto T. J. Am. Chem. Soc. 2011;133:12968. doi: 10.1021/ja205001m. [DOI] [PubMed] [Google Scholar]; (c) Armstrong A, Chivers T, Parvez M, Boere RT. Angew. Chem., Int. Ed. 2004;43:502. doi: 10.1002/anie.200353108. [DOI] [PubMed] [Google Scholar]; (d) Ito S, Kikuchi M, Yoshifuji M, Arduengo AJ, III, Konovalova TA, Kispert LD. Angew. Chem., Int. Ed. 2006;45:4341. doi: 10.1002/anie.200600950. [DOI] [PubMed] [Google Scholar]; (e) Scheer M, Kuntz C, Stubenhofer M, Linseis M, Winter RF, Sierka M. Angew. Chem., Int. Ed. 2009;48:2600. doi: 10.1002/anie.200805892. [DOI] [PubMed] [Google Scholar]
- 42.(a) Pan X, Su Y, Chen X, Zhao Y, Li Y, Zuo J, Wang X. J. Am. Chem. Soc. 2013;135:5561. doi: 10.1021/ja402492u. [DOI] [PubMed] [Google Scholar]; (b) Pan X, Chen X, Li T, Li Y, Wang X. J. Am. Chem. Soc. 2013;135:3414. doi: 10.1021/ja4012113. [DOI] [PubMed] [Google Scholar]
- 43.Sheberla D, Tumanskii B, Tomasik AC, Mitra A, Hill NJ, West R, Apeloig Y. Chem. Sci. 2010;1:234. [Google Scholar]
- 44.Back O, Ali Celik M, Frenking G, Melaimi M, Donnadieu B, Bertrand G. J. Am. Chem. Soc. 2010;132:10262. doi: 10.1021/ja1046846. [DOI] [PubMed] [Google Scholar]
- 45.Back O, Donnadieu B, Hopffgarten M. v., Klein S, Tonner R, Frenking G, Bertrand G. Chem. Sci. 2011;2:858. [Google Scholar]
- 46.For reviews on carbene stabilized allotropes, see: Wang Y, Robinson GH. Dalton Trans. 2012;41:337. doi: 10.1039/c1dt11165e. Wang Y, Robinson GH. Inorg. Chem. 2011;50:12326. doi: 10.1021/ic200675u.
- 47.Wang Y, Xie Y, Wei P, King RB, Schaefer HF, III, Schleyer P. v. R., Robinson GH. J. Am. Chem. Soc. 2008;130:14970. doi: 10.1021/ja807828t. [DOI] [PubMed] [Google Scholar]
- 48.Abraham MY, Wang Y, Xie Y, Wei P, Schaefer HF, III, Schleyer P. v. R., Robinson GH. Chem. Eur. J. 2010;16:432. doi: 10.1002/chem.200902840. [DOI] [PubMed] [Google Scholar]
- 49.Kinjo R, Donnadieu B, Bertrand G. Angew. Chem., Int. Ed. 2010;49:5930. doi: 10.1002/anie.201002889. [DOI] [PubMed] [Google Scholar]
- 50.Back O, Donnadieu B, Parameswaran P, Frenking G, Bertrand G. Nat. Chem. 2010;2:369. doi: 10.1038/nchem.617. [DOI] [PubMed] [Google Scholar]
- 51.Abraham MY, Wang Y, Xie Y, Gilliard RJ, Jr., Wei P, Vaccaro BJ, Johnson MK, Schaefer HF, III, Schleyer P. v. R., Robinson GH. J. Am. Chem. Soc. 2013;135:2486. doi: 10.1021/ja400219d. [DOI] [PubMed] [Google Scholar]
- 52.(a) Nakanishi I, Otoh S, Suenobu T, Fukuzumi S. Chem. Commun. 1997:1927. [Google Scholar]; (b) Nakanishi I, Otoh S, Fukuzumi S. Chem. Eur. J. 1999;5:2810. [Google Scholar]
- 53.Mansoorabadi SO, Seravalli J, Furdui C, Krymov V, Gerfen GJ, Begley TP, Melnick J, Ragsdal SW, Reed GH. Biochemistry. 2006;45:7122. doi: 10.1021/bi0602516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tidwell TT. Angew. Chem., Int. Ed. 2006;45:5580. doi: 10.1002/anie.200601643. [DOI] [PubMed] [Google Scholar]
- 55.Martin D, Lassauque N, Donnadieu B, Bertrand G. Angew. Chem., Int. Ed. 2012;51:6172. doi: 10.1002/anie.201202137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ikeda H, Tanaka F, Akiyama K, Tero-Kubota S, Miyashi T. J. Am. Chem. Soc. 2004;126:414. doi: 10.1021/ja038068c. [DOI] [PubMed] [Google Scholar]
- 57.Turecek F, Drinkwater DE, McLafferty FW. J. Am. Chem. Soc. 1991;113:5950. [Google Scholar]
- 58.Gorodetsky B, Ramnial T, Branda NR, Clyburne JAC. Chem. Commun. 2004:1972. doi: 10.1039/b407386j. [DOI] [PubMed] [Google Scholar]
- 59.McKenzie I, Brodovitch J-C, Percival PW, Ramnial T, Clyburne JAC. J. Am. Chem. Soc. 2003;125:11565. doi: 10.1021/ja028770t. [DOI] [PubMed] [Google Scholar]
- 60.Enders D, Breuer K, Raabe G, Simonet J, Ghanimi A, Stegmann HB, Teles JH. Tet. Lett. 1997;38:2833. [Google Scholar]
- 61.Pause L, Robert M, Heinicke J, Kühl O. J. Chem. Soc., Perkin Trans. 2. 2001:1383. [Google Scholar]
- 62.Back O, Henry-Ellinger M, Martin CD, Martin D, Bertrand G. Angew. Chem., Int. Ed. 2013;52:2913. doi: 10.1002/anie.201209109. [DOI] [PubMed] [Google Scholar]
- 63.Tolman CA. Chem. Rev. 1977;77:313. [Google Scholar]