

Abstract
Nitrogenase is an enzyme found in many bacteria and archaea that catalyzes biological dinitrogen fixation, the reduction of N2 to NH3, accounting for the major input of fixed nitrogen into the biogeochemical N cycle. In addition to reducing N2 and protons, nitrogenase can reduce a number of small, non-physiological substrates. Among these alternative substrates are included a wide array of carbon containing compounds. These compounds have provided unique insights into aspects of the nitrogenase mechanism. Recently, it was shown that carbon monoxide (CO) and carbon dioxide (CO2) can also be reduced by nitrogenase to yield hydrocarbons, opening new insights into the mechanism of small molecule activation and reduction by this complex enzyme as well as providing clues for the design of novel molecular catalysts.
1. Introduction
Nitrogenase, found in the bacterial and archaeal domains of life, catalyzes all biological N2 fixation (eqn 1), accounting for approximately 60% of the fixed N input from N2 into the global biogeochemical nitrogen cycle [1].
| (eqn 1) |
The industrial Haber-Bosch reaction provides most of the remaining input of fixed N [1]. Three different classes of nitrogenase have been reported, with a key difference among the classes being the identity of the heterometal contained in their active site metal cluster [2,3]. The most widely distributed and best-studied class of nitrogenase contains molybdenum (Mo) as the heterometal; hence, the designation “FeMo-cofactor” for the active site metal cluster, while the other forms apparently contain V or Fe in place of Mo. These latter enzymes are often referred to as “alternative” nitrogenases because they are not as widely distributed and are expressed only when Mo is unavailable in the case of Azotobacter vinelandii, which has the ability to produce all three nitrogenase types. The alternative nitrogenases are also less efficient at N2 reduction when compared to Mo-nitrogenase [2].
Mo-nitrogenase is composed of two component proteins designated the Fe protein and the MoFe protein [4]. The Fe protein is a homodimeric protein having two ATP-binding sites, one on each subunit, and a single Fe4-S4 cluster bridging the two identical subunits [5] (Figure 1). Catalysis is initiated when the reduced, MgATP-bound form of the Fe protein, docks with the MoFe protein, triggering intermolecular electron transfer and hydrolysis of the two ATP molecules [4,6]. The MoFe protein is an α2β2 heterotetramer, having two types of metal clusters [7]. One of these, the P (Fe8-S7) cluster, is located at each αβ interface in the MoFe protein near the Fe protein docking face where it acts as an intermediary for electron transfer from the Fe protein to the active site metal cluster, called the FeMo-cofactor (Fe7-S9-Mo1-C-R-homocitrate) (Figure 1). The FeMo-cofactor is the site of substrate binding and reduction [8] (Figure 2).
Figure 1.

Mo-nitrogenase with cofactors. Shown is one functioning half of the Monitrogenase. The top shows the Fe protein (left) and an αβ-dimer half of the MoFe protein (right). Shown below are the metal clusters and ATP, with Fe in rust, S in yellow, C in gray, O in red, N in blue, and Mo in magenta. Taken from PDB 2AFK.
Figure 2.

FeMo-cofactor and key residues. Shown is the FeMo-cofactor with key MoFe protein amino acid side chains. Colors are Fe in rust, S in yellow, C in gray, O in red, N in blue, and Mo in magenta. PDB 2AFK.
A proposed model for the nitrogenase macroscopic mechanism indicates that Fe protein binding to the MoFe protein induces large protein conformational changes (~800 Å2) within the two proteins that gate the electron transfer (ET) events [9]. The first ET event is proposed to involve intramolecular ET from the resting P cluster (designated PN) to the resting FeMo-cofactor (MN), resulting in an oxidized P cluster (P1+) and a reduced FeMo-cofactor (MR) [10] (Figure 1). In the second ET event, Fe protein transfers an electron to the oxidized P1+ cluster, resulting in reduction of the P cluster back to the PN state and oxidized Fe protein. This model has been designated the “deficit-spending” model to reflect the creation of an electron deficit at the P cluster that is then backfilled. Recent results support ATP hydrolysis following the electron transfer events [11], although this sequence has yet to be definitively established. The cycle is completed by the dissociation of the Fe protein from the MoFe protein [12]. A reduced and MgATP-bound Fe protein then associates again to the partially reduced MoFe protein, and the cycle of electron transfer and ATP hydrolysis is repeated to accomplish the accumulation of sufficient electrons to affect substrate binding and reduction [13].
Important aspects of the nitrogenase mechanism includes defining exactly how and where substrates bind, understanding how electrons are accumulated, and defining the roles of ATP binding and hydrolysis in coordinating electron transfer and substrates reduction. Recent progress in analysis of both trapped N2 intermediates and Ccontaining substrate intermediates are beginning to provide insights into various aspects of this complex mechanism [14]. Here, recent insights from the study of trapped N2 reduction intermediates are presented, together with a summary of results involving examination of the reduction of C-containing compounds, and how study of the reduction of such compounds could provide new mechanistic insights.
2. N2 Reduction Mechanism
2.1. Accumulating electrons on FeMo-cofactor
One of the key challenges in understanding the nitrogenase mechanism is explaining how the FeMo-cofactor accumulates multiple electrons to activate the system for N2 binding and subsequent reduction. Kinetic studies revealed that the FeMo-cofactor must accumulate 3–4 electrons before N2 actually binds to the active site [15]. The number of electrons accumulated within the MoFe protein is designated by the nomenclature En, where n represents the number of electrons received from the Fe protein. A condition of the deficit-spending ET model described above [10] is that all electrons passed to the MoFe protein from the Fe protein must accumulate on the FeMo-cofactor or one of its bound activated intermediate states [14].
Progress in understanding how nitrogenase accumulates electrons on FeMo-cofactor has come from recent studies investigating intermediates trapped on the FeMo-cofactor during substrate reduction using a combination of MoFe proteins having amino acid substitutions and rapidly freezing the samples during turnover, with subsequent characterization of trapped states using advanced spectroscopic methods (e.g., electron paramagnetic resonance (EPR) and electron nuclear double resonance (ENDOR))[14,16–18]. Using these approaches, it has been possible to trap and characterize intermediates during turnover with protons, N2, diazene (HN=NH), hydrazine (H2N-NH2), and several alkynes (-C≡C-) [13,19]. Characterization of one of these trapped states, the proton turnover state, has provided a provisional answer to the question of how the FeMo-cofactor accumulates electrons and provides guidance for a likely mechanism for early steps in N2 reduction [16,20,21].
The proton-trapped state observed in the α-70Ile substituted MoFe protein has been definitively assigned to the 4-electron reduced state of the FeMo-cofactor (E4) [16,22]. Surprisingly, it was found that in this multiply reduced state, the metal core of the FeMocofactor, is in the same oxidation state as the resting state (E0) [16]. This apparent contradiction was explained when it was discovered that the trapped E4 state has two Fe-bound bridging hydrides [16] (Figure 3). Thus, it appears that electrons are located on the bound metal-hydrides, leaving the metal core of the FeMo-cofactor in the resting oxidation state. Such “parking” of electron pairs on metal bound hydrides indicates that FeMo-cofactor might formally access only two oxidation states during the catalytic process [14]. These metal bound hydrides figure prominently in a proposed draft nitrogenase mechanism, as described in the next section.
Figure 3.

Metal hydrides and the FeMo-cofactor. Shown are provisional binding sites for bridging hydrides (H−) and protons (H+) on one 4Fe-4S face of the FeMo-cofactor. All hydrogen species are highlighted in red.
2.2. Provisional N2 reduction mechanism
One of the most important features of the nitrogenase N2 reduction mechanism is the observation that, once bound, N2 is quantitatively converted to 2 NH3, with no observed “leaked” intermediates for Monitrogenase [23]. During N2 reduction by the V-nitrogenase, a trace of hydrazine has been reported [24]. Thus, nitrogenase is designed to bind N2 and conduct multiple rounds of reduction and protonation without the release of semi-reduced states. This feature, the ability to capture a substrate for multiple rounds of reduction, is important towards understanding how nitrogenase can achieve the multiple electron reduction of N2 or C-compounds.
The emerging mechanism indicates that nitrogenase catalyzes N2 reduction by the sequential addition of electrons and protons to a FeMo-cofactor bound N2 [14]. In such a sequential reduction mechanism, it is expected that N2 reduction intermediates bound to the FeMo-cofactor following even number electron additions (2 or 4) respectively correspond to metal-bound diazene and hydrazine, as shown in eqn 2.
| (eqn 2) |
How the electrons and protons are added to the FeMo-cofactor bound N2 is not yet established. However, a “reductive elimination” model has been proposed wherein N2 binding to the E4 state involves the loss of two metal-bound hydrides as H2, with electron and proton addition to the bound N2 resulting in a metal-bound diazene [14]. Subsequent reduction of the bound diazene by addition of electrons/protons would yield a hydrazine bound state and, ultimately, release of two NH3.
3. Early C-Compounds and Nitrogenase
3.1. Alkynes as substrates
Among the earliest studies on isolated nitrogenase was the observation by Dilworth that nitrogenase could reduce acetylene (HC≡CH) by two electrons and two protons to yield ethylene (H2C=CH2) [25,26]. This discovery contributed to the realization that nitrogenase could reduce a number of “alternative” substrates other than just the physiological substrates N2 and protons [4]. Over the years, the list of alternative substrates for nitrogenase has grown long, with common features being small molecules, with multiple bonds. The C-containing alternative substrate family is the largest and represents a focus of the current discussion (Table 1). Kinetic and spectroscopic studies on how nitrogenase reduces acetylene or other alkyne substrates have provided key insights into different aspects of the nitrogenase mechanism [4,27]. For example, it was observed that Mo-dependent nitrogenase has the capacity to reduce acetylene by two electrons to yield ethylene, with very little production of the four electron reduced product ethane (H3C-CH3) [28–31]. In contrast, it has been shown that both the V- and Fe-dependent forms of nitrogenase can catalyze the reduction of acetylene by either two or four electrons, yielding ethylene or ethane, respectively [32–36]. Nevertheless, ethylene is the most abundant reduction product for all of the nitrogenases when acetylene is used as the substrate. Although the Modependent nitrogenase does not have a significant ability to reduce acetylene by four electrons to yield ethane, the enzyme can be remodeled by amino acid substitution such that it gains the capacity for measurable formation of ethane as a reduction product [37]. This observation indicates that the polypeptide environment of the respective active sites of the various nitrogenases predominately controls product distribution when acetylene is used as the substrate, rather than representing a feature of the heterometal.
Table 1.
Carbon-containing substrates for nitrogenases
| Substrate | Reaction | Nitrogenase | References |
|---|---|---|---|
| Carbon-carbon substrate reduction | |||
| Acetylene | HC≡CH + 2H+ + 2e− → CH2=CH2 | Mo, V, and Fe | [25,32,34–36] |
| HC≡CH + 4H+ + 4e− → CH3 –CH3 | Mo, V, and Fe | [28–31,34–36] | |
| Propyne | CH3C≡CH + 2H+ + 2e− → CH3CH=CH2 | Mo | [47,49,52] |
| 1-Butyne | C2H5C≡CH + 2H+ + 2e− → C2H5CH=CH2 | Mo | [47,49,101] |
| 2-Butyne | CH3C≡CCH3 + 2H+ + 2e− → cis-CH3CH=CHCH3 | Mo | [86] |
| Propargyl alcohol | HC≡CCH2OH + 2H+ + 2e− → CH2=CHCH2OH | Mo | [52] |
| Propargyl amine | HC≡CCH2NH2 + 2H+ + 2e− → CH2=CHCH2NH2 | Mo | [50] |
| Allene | CH2=C=CH2 + 2H+ + 2e− → CH3CH=CH2 | Mo | [57] |
| Ethylene | CH2=CH2 + 2H+ + 2e− → CH3–CH3 | Mo and V | [33,56] |
| Cyclopropene | 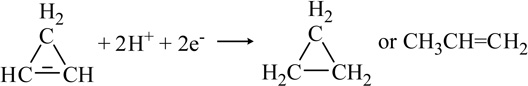 |
Mo | [54] |
| 3,3-Difluorocyclopropene | 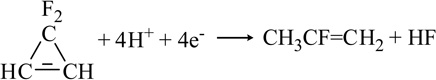 |
Mo | [55] |
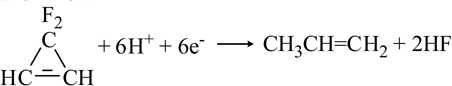 | |||
| Carbon-nitrogen substrate reduction | |||
| Hydrogen cyanide | HC≡N + 2H+ + 2e− → [CH2=NH] + H2O → CH2O + NH3 | Mo and V | [59,61] |
| HC≡N + 4H+ + 4e− → CH3 –NH2 | Mo and V | [49,58,59,61] | |
| HC≡N + 6H+ + 6e− → CH4 + NH3 | Mo and V | [49,58,59,61] | |
| 2HC≡N + 8H+ + 8e− → C2H4 + 2NH3 | Mo | [59,62] | |
| 2HC≡N + 10H+ + 10e− → C2H6 + 2NH3 | Mo | [59,62] | |
| Cyanamide | N≡C–NH2 + 6H+ + 6e− → CH3 –NH2 + NH3 | Mo and V | [65] |
| N≡C–NH2 + 8H+ + 8e− → CH4 + 2NH3 | Mo and V | [65] | |
| Alkyl nitriles | RC≡N + 6H+ + 6e− → RCH3 + NH3 (R = CH3, C2H5 and n-C3H7) | Mo and V | [47,49,64] |
| CH3CH2C≡N + 4H+ + 4e− → CH2=CH–CH3 + NH3 | Mo | [49] | |
| Acrylonitrile | CH2=CH–C≡N + 6H+ + 6e− → CH2=CH–CH3 + NH3 | Mo and V | [49,63,64] |
| CH2=CH–C≡N + 8H+ + 8e− → CH3 –CH2 –CH3 + NH3 | Mo and V | [49,63,64] | |
| cis-But-2-ene-1-nitrile | cis-CH3CH=CH–C≡N + 6H+ + 6e− → cis-or trans-CH3CH=CHCH3 or CH3CH2CH=CH2 + NH3 | Mo | [47] |
| cis-CH3CH=CH–C≡N + 8H+ + 8e−→ CH3CH2CH2CH3 + NH3 | Mo | [47] | |
| trans-But-2-ene-1-nitrile | trans-CH3CH=CH–C≡N + 6H+ + 6e−→ trans-CH3CH=CHCH3 or CH3CH2CH=CH2 + NH3 | Mo | [47] |
| trans-CH3CH=CH–C≡N + 8H+ + 8e−→ CH3CH2CH2CH3 + NH3 | [47] | ||
| But-3-ene-1-nitrile | CH2=CHCH2 –C≡N + 6H+ + 6e−→ CH3CH2CH=CH2 + NH3 | Mo | [47] |
| CH2=CHCH2 –C≡N + 8H+ + 8e−→ CH3CH2CH2CH3 + NH3 | Mo | [47] | |
| Isonitriles | RN≡C + 4H+ + 4e−→ RNH–CH3 (R = CH3) | Mo | [69] |
| RN≡C + 6H+ + 6e−→ RNH2 + CH4 (R = CH3 C2H5, and CH2=CH-) | Mo | [49,62,66] | |
| 2RN≡C + 8H+ + 8e−→ 2RNH2 + CH2 =CH2 (R = CH3 C2H5, and CH2=CH-) | Mo | [49,62,67,69] | |
| 2RN≡C + 10H+ + 10e−→ 2RNH2 + CH3 –CH3 (R = CH3 C2H5, and CH2=CH-) | Mo | [49,62,67,69] | |
| 3CH3N≡C + 12H+ + 12e−→ 3CH3NH2 + CH3CH=CH2 | Mo | [70] | |
| 3CH3N≡C + 14H+ + 14e−→ 3CH3NH2 + CH3 –CH2 –CH3 | Mo | [70] | |
| CH3C≡N + 2H+ + 2e−→ [CH3N=CH2] + H2O → CH2O?? + CH3NH2 (?) | Mo | [69] | |
| CH3N≡C + CO + 8H+ + 8e−→ CH3NH2 + CH2 =CH2 + H2O (???) | Mo | [39,62,69] | |
| CH3N≡C + CO + 10H+ + 10e−→ CH3NH2 + CH3 –CH3 + H2O (???) | Mo | ||
| Diazirine | 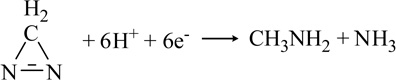 |
Mo | [71] |
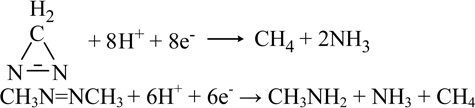 |
Mo | [71] | |
| Dimethyldiazene | CH3N=NCH3 + 6H+ + 6e−→ CH3NH2 + NH3 + CH4 | Mo | [71] |
| Carbon-chalcogen substrate reduction | |||
| Carbon monoxide | C≡O + 6H+ + 6e−→ CH4 + H2O | Mo and V | [93,94] |
| 2C≡O + 8H+ + 8e−→ CH2=CH2 + 2H2O | Mo and V | [91,93,94] | |
| 2C≡O + 10H+ + 10e−→ CH3 –CH3 + 2H2O | Mo and V | [91,93,94] | |
| 3C≡O + 12H+ + 12e−→ CH3CH=CH2 + 3H2O | Mo and V | [92–94] | |
| 3C≡O + 14H+ + 14e−→ CH3CH2CH3 + 3H2O | Mo and V | [91–94] | |
| 4C≡O + 16H+ + 16e−→ CH3CH2CH=CH2 or | Mo and V | [93,94] | |
| (CH3)2C=CH2 + 4H2O | |||
| 4C≡O + 18H+ + 18e−→ CH3CH2CH2CH3 + 4H2O | Mo and V | [93,94] | |
| Carbonyl sulfide | S=C=O + 2H+ + 2e−→ CO + H2S | Mo | [75] |
| Carbon dioxide | O=C=O + 2H+ + 2e−→ CO + H2O | Mo | [75] |
| O=C=O + 2H+ + 2e−→ HCOOH | Mo | [102] | |
| O=C=O + 8H+ + 8e−→ CH4 + 2H2O | Mo | [95] | |
| CO2 + HC≡CH + 8H+ + 8e−→ CH3CH=CH2 + 2H2O | Mo | [95] | |
| CO2 + HC≡CH + 10H+ + 10e−→ CH3 –CH2 –CH3 + 2H2O | Mo | [95] | |
| Carbon disulfide | S=C=S + ?H+ + ?e−→ C?? + ?H2S | Mo | [74] |
| Thiocyanate | S=C=N−+ 3H+ + 2e−→ HC≡N + H2S | Mo | [74] |
| Cyanate | O=C=N−+ 3H+ + 2e−→ HC≡N + H2O | Mo | [74] |
| O=C=N−+ 3H+ + 2e−→ CO + NH3 | Mo | [74] | |
Early investigations of the stereochemistry of H-atom addition to acetylene catalyzed by nitrogenase also provide important mechanistic insights about how substrates are bound and reduced. Analysis of the stereochemistry of the product (cis- or trans-) 1,2-dideuteroethylene (HDC=CDH) formed during reduction of acetylene in the presence of D2O or during the reduction of dideuteroacetylene (DC≡CD) in H2O revealed primarily the cis-product [25,33,38,39]. This observation revealed that reduction of bound acetylene occurs by the addition of both protons to the same side of the bound acetylene [40]. An enzyme-bound η2-vinyl intermediate has been proposed to explain this stereochemistry, with H atom addition to one face of the bound acetylene. This stereospecificity can be perturbed by amino acid substitutions around the active site of Mo-nitrogenase [41]. Further insights into the mechanism of acetylene reduction have come from characterization of the MoFe protein trapped during the course of acetylene reduction using spectroscopic methods such as EPR and ENDOR [42–45]. To summarize a number of such studies, it appears that the trapped state corresponds to the two-electron reduced product ethylene bound to one or more Fe atoms of the FeMo-cofactor [42].
Another important mechanistic insight that has come from kinetic studies of acetylene reduction by nitrogenase is the revelation that there are likely two binding sites for acetylene in both the Mo- and Fe-nitrogenases, whereas it appears that the Vnitrogenase has only one observable acetylene binding site [2,4]. Kinetic studies of acetylene reduction and inhibition using Mo-nitrogenase with an amino acid substitution suggested acetylene binding at a Fe4-S4 face of the FeMo-cofactor [46].
In addition to acetylene, several other alkynes have been investigated as substrates for nitrogenase (Table 1). For example, both propyne (CH3-C≡CH) and 1-butyne (CH3CH2-C≡CH) were found to be reduced by 2 e−/H+ to the corresponding alkenes at very slow rates [47–49]. The triple bonds in propargyl alcohol and propargyl amine are also reduced by nitrogenase to yield the corresponding double-bond compounds (Table 1), which have provided valuable probes for gaining insight into where and how alkyne substrates interact with FeMo-cofactor during catalysis [50–52].
3.2. Alkene substrates
Another important class of C-containing substrates for nitrogenase includes those containing the C=C fragment. For example, cyclopropene (Table 1) can undergo reduction to cleave the single bond forming propene and reduction of the double bond forming cyclopropane [48,53,54]. The production of both 1,3- and 2,3-dideuteropropene when the reaction is run in D2O indicated that there must be an isomerization process of the intermediate derived from the single bond cleavage [48]. Distinguished from cyclopropene reduction, 3,3-difluorocyclopropene can be reduced by 4 e−/H+ to yield 2-fluoropropene or by 6 e−/H+ to yield propene suggesting that the ring-opening reaction occurs through the complete cleavage of the -C=C-double bond [55]. This was the first reported halogen-containing substrate for nitrogenase. Ethylene is also reduced to ethane by both Mo- and V-nitrogenase, but at very slow rates [33,56]. Moreover allene serves as a better substrate than ethylene, even though it is not strictly an alkene due to the sp-hybridized central carbon atom [57]. Allene reduction in D2O confirmed production of 2,3-dideuteropropene (CDH2-CD=CH2) as the exclusive product rather than 1,2-dideuteropropene (CH3-CD=CDH), indicating that isomerization to and reduction of propyne is not involved [57].
3.3. Carbon-nitrogen substrates
Nitrogenase was also found to reduce carbon-nitrogen containing compounds with triple bonds such as hydrogen cyanide, nitriles, and isonitriles (Table 1). Isoelectronic to C2H2 and N2, cyanide (CN−) was found to be both a substrate and inhibitor of nitrogenase [49,58]. It was later shown that hydrogen cyanide (HCN) is the substrate, while CN− is the inhibitor [59]. The 2 e−/H+, 4 e−/H+, and 6 e−/H+ reduction of HCN was observed with the production of methyleneimine (CH2=NH), methylamine (CH3NH2), methane (CH4), and ammonia (NH3) as products for both the Mo- and V-nitrogenases [60,61]. The hydrolysis of unstable methyleneimine led to the production of formaldehyde and ammonia as the final products (Table 1). The further reduction of formaldehyde to methanol was not found [61]. However, very small amounts of C2 products, C2H4 and C2H6, were observed, which might be derived from an insertion mechanism accounting for the isocyanide reduction and coupling [59,62]. Reduction of alkyl nitriles resulted in production of the corresponding alkanes and ammonia as products [47]. However, reduction of acrylonitrile, a better nitrile substrate, can be achieved by 6 e−/H+ or 8 e−/H+, which resulted in the production of propene and ammonia, or propane and ammonia, respectively [63,64]. Propane formed in this reaction is apparently not from the subsequent reduction of propene from the 6 e−/H+ reaction. This is further supported by reduction of the 4C-containing nitrile analogues, for which the reaction patterns might be more complex [47]. However, reduction of up to 4C nitriles demonstrates that substantially larger substrates than N2 and acetylene can be accommodated by the active site of nitrogenase, even though the branched versions were not reduced [47]. The production of 1,1,3-trideuteropropene from reduction of acrylonitrile in D2O indicates a double bound shift reaction. The mechanism of propane production is still not clear [64]. Similar 6 e−/H+ and 8 e−/H+ reduction of cyanamide yielded methylamine and ammonia, and methane and ammonia, respectively. Higher electron flux conditions favor the 8 e− reduction of cyanamide [65]. It is noteworthy that D2O promotes acrylonitrile reduction by increasing the proportion of the total electron flow going to this substrate [64]. This apparent inverse isotope effect likely provides insights into the mechanism of substrate reduction.
As isomers of corresponding nitriles, isonitriles (also called isocyanides) can be reduced by 6 e−/H+ to form methane and corresponding amines [39,62,66–69]. The four electron reduction product dimethylamine has been characterized during methylisocyanide reduction [69]. Careful analysis of the ratio between methylamine and methane from 6 e−/H+ reduction leads to a proposed two electron reduction reaction similar to the reduction of HCN. The 8 e−/H+ and 10 e−/H+ reductions of isonitriles produced the C2 hydrocarbons, C2H4 and C2H6 [62,67,69]. The reduction of isonitriles in D2O resulted in the formation of C2D4 and C2D6. A sequential insertion mechanism has been proposed for this coupling reaction [47]. The production of C3 hydrocarbons was also observed [70].
Cyclic and acyclic diazene derivatives have also been used as substrates for probing the nitrogenase mechanism [71]. Methane, methylamine and ammonia were found as products from the reduction of diazirine and dimethyldiazene. Recently, an intermediate has been trapped during the turnover of the substrate methyldiazene, revealing -NH2 and -NH3 intermediates bound to FeMo-cofactor, supporting an alternating reduction mechanism for N2 reduction [17,72,73].
3.4. Carbon-chalcogen substrates
Nitrogenase can also reduce carbon-chalcogen substrates such as carbonyl sulfide (COS), carbon disulfide (CS2), thiocyanite (CSN−) and cyanite (CON−) [74,75]. It was shown that nitrogenase can reduce both C=S and C=O bonds [75]. The observation that COS interacts with nitrogenase as an inhibitor of acetylene reduction [76] led to the study of the reduction of COS by nitrogenase [75]. These studies demonstrated that nitrogenase could catalyze the 2 e−/H+ reduction of a new class of compounds. The C=S bound in COS is reduced to form the products CO and H2S. In light of these observations it was shown by kinetics analysis and EPR spectroscopy for the COS structural analogues, that the 2 e−/H+ reduction of the C=S bond of CSN− results in H2S and HCN, and the 2 e−/H+ reduction of the C=O and C=N in CON− results in H2O, HCN and CO, NH3, respectively [74]. Finally it was demonstrated that CS2 can be an inhibitor of nitrogenase reduction reaction and total electron flow but also be reduced to H2S and an unknown CS species [74,75].
In addition to these C-containing substrates, carbon monoxide (CO) was early shown to be an inhibitor of all nitrogenase catalyzed reactions except for proton reduction [4,37,77,78]. This inhibition holds true for all three classes of nitrogenase. In the presence of CO, electron flux through nitrogenase is diverted to proton reduction so that only H2 is produced. CO can also inhibit proton reduction, but only at basic pH values [79]. The binding chemistry of CO to FeMo-cofactor is complex and dependent on the partial pressure or concentration of CO present. In the presence of CO and under turnover conditions, EPR and ENDOR studies have identified three different bound states [80–84]. One under low CO conditions (<0.08 atm in the gas phase or <1:1 [CO]:[FeMo-co] in solution) called “lo-CO”, which is proposed to be a single CO bound to the FeMo cofactor, while under high CO concentration or high partial pressure of the gas (> 0.5 atm), two EPR signals are present: hi-CO and hi(5)-CO [84,85]. Each signal is proposed to be at least two CO molecules bound to separate sites on the cofactor. The protein environment around FeMo-cofactor dictates which CO-induced EPR signals are generated; however there is no correlation between either the presence or the absence of any of the three CO-induced EPR signals and CO inhibition of H2 evolution. CO inhibition of H2 evolution may be due to CO binding to a different EPR-silent site [85].
4. Remodeling Nitrogenase
4.1. The role of MoFe protein in defining substrate size range
A key recent advance in understanding nitrogenase reduction of substrates has been the recognition that it is possible to change the size of substrates that can be reduced by Mo-nitrogenase by remodeling the MoFe protein environment surrounding the FeMo-cofactor [86,87]. Progress in this direction was complex because the dynamic nature of nitrogenase catalysis presented a particular challenge to the design of amino acid substitution strategies (remodeling) as a way of gaining mechanistic insight. Contributing to these challenges were the following features: (i) no substrates bind in the resting state, (ii) in the absence of other substrates, the enzyme reduces protons, immediately returning it to the resting state, and (iii) because multiple electrons are required for substrate binding and reduction, any population of the enzyme must exist in multiple states (i.e., the reaction cannot be effectively synchronized). These problems can be partially solved by freeze-quench techniques using the normal nitrogenase prepared under turnover conditions. However, the most significant mechanistic insights were gained by using freeze-quench strategies in combination with advanced spectroscopic techniques together with remodeled forms of the enzymes populated with a particular catalytic state [13]. How this was accomplished in the analysis of alkyne and N2 reduction intermediates is instructive for understanding the recent progress with expanding nitrogenase reduction of C-containing compounds.
Any amino acid substitution approach applied for mechanistic analyses must be grounded in one of three approaches, or some combination of these approaches: (i) serendipity, (ii) rational substitution based on structural information and/or amino acid side chain functionality, or (iii) genetic selection. Recent work with nitrogenase has employed all three approaches with some success, but the genetic selection approach provided the first insight that launched a key mechanistic advance [88]. The genetic selection approach employed depended on the assumption, not firmly established at the time, that all substrates would bind at or near the same place within the enzyme [4]. It was reasoned that by figuring out where the non-physiological substrate acetylene binds by using genetic selection then this would provide a starting place to employ serendipity and rational design for analysis of N2 binding and reduction. Because acetylene and N2 both compete for the same electrons necessary for substrate reduction [78], it was tested, and subsequently demonstrated, that acetylene can be an effective growth inhibitor when cells need to obtain their nitrogen source from N2 [88,89]. It was then reasoned that, because acetylene is slightly larger than N2, it should be possible to select for mutant strains that produce nitrogenase that are resistant to the inhibitor effects of acetylene. In other words, a bacterium that produces a substituted form of nitrogenase was searched for that was slightly changed at the active site such that acetylene, but not N2, is denied access. Perhaps surprisingly, this approach was successful, ultimately leading to identifying how access of substrates to the FeMo-cofactor is gated during catalysis, as well as where and how certain substrates are bound during catalysis [13,19].
In the genetic screen for an altered form of the MoFe protein that has the ability to discriminate between binding of acetylene (excluded) and N2 (unaffected), it was surprising that one substitution yielding this phenotype resulted in replacement of the α-69Gly residue by 69Ser [88]. The reason this result was surprising was because α-69Gly is not in the first shell of non-covalent interactions with the FeMo-cofactor. Rather, it is located adjacent to α-70Val, which closely approaches a particular Fe-S face of the FeMo-cofactor (Figure 4). This led us to suspect that α-69Gly serves as part of a switch mechanism that exquisitely controls the position of the α-70Val side chain, thereby controlling access to the active site. Indeed, when the side chain of α-70Val was progressively shortened by substitution with Ala or Gly, (without substitution of α-69Gly), progressively larger alkynes (propyne and butyne) were able to access the active site and be reduced [50,87,90]. It was also found that as access to the active site for reduction of larger substrates was increased, the ability to reduce smaller substrates was diminished. Thus, it appears there are three possible roles for the amino acid “shrubbery” surrounding the FeMo-cofactor in the reduction process. The first of these is to make the “hot spot(s)” for catalysis available to a particular substrate. The second could be to lock the substrate in place for progressive reduction steps, indicating a dynamic role for the α-69Gly and α-70Val residue positions. A third related aspect could involve intermediate stabilization through amino acid chain functional groups. It would appear that locking down the substrate is particularly important for those substrates that must accept a relatively large number of electrons for complete reduction, such as N2.
Figure 4.

Position of α-69Gly and α-70Val. Shown are the locations of α-69Gly and α-70Val near the FeMo-cofactor, with the van der Waals surface of the surrounding protein shown as mesh.
5. CO and CO2 Reduction to Hydrocarbons
5.1. Nitrogenase reduction of CO2 and CO
In 1995, it was reported that the wild-type Mo-nitrogenase can catalyze the two electron/proton reduction of CO2 to form CO [75] (Table 1). The CO formed in this reaction could be detected by binding to deoxy-hemoglobin, which provides a very sensitive spectrophotometric assay for CO formation. The rates of CO2 reduction to CO were low, but the reaction was confirmed to be catalyzed by nitrogenase. There was no evidence for reduction beyond CO.
In 2010, it was found that one of the alternative nitrogenases (V-nitrogenase) is able to catalyze the reduction and coupling of two or three CO molecules at very low rates forming C2 (ethylene, H2C=CH2, and ethane, CH3-CH3) and C3 (propane, H3C-CH2-CH3) hydrocarbons [91]. Shortly after, propylene (H3C-CH=CH2) was found as an additional product of CO reduction by V-nitrogenase in D2O assays due to an apparent inverse H/D isotope effects for different product [92]. An additional study using large quantities of proteins revealed methane (CH4), α-butene ((CH3)2C=CH2 or CH3CH2C=CH2) and n-butane (n-CH3CH2CH2CH3) as products for V-nitrogenase [93]. Initial studies with wild-type Mo-nitrogenase did not reveal detectable reduction of CO to hydrocarbons. However, it was found that remodeled forms of Mo-nitrogenase could reduce CO to hydrocarbons [94]. The primary products observed were methane, ethylene (H2C=CH2), propylene (H3C-CH=CH2), ethane (H3C-CH3), and propane (H3CCH2-CH3). Trace amounts of iso-butene ((CH3)2C=CH2) and n-butane (CH3CH2CH2CH3) were also detected. In a separate study, it was found that wild type Mo-nitrogenase could reduce CO to traces of C2 (H2C=CH2 and CH3-CH3) and C3 (H3C-CH2-CH3 and H3C-CH=CH2) hydrocarbons [93]. Mechanistically, it was reported that the hydrogen origin for hydrocarbon production is from solvent protons rather than H2 [92]. But the D2 partial pressure applied in this study to trace the hydrogen source of hydrocarbons was much lower than those H2 partial pressures accounting for H2 inhibition of hydrocarbon formation from CO reduction by V-nitrogenase. The finding that Mo-nitrogenase and Vnitrogenase have the ability to reduce and couple multiple CO molecules is consistent with two CO binding sites being located within FeMo-cofactor, as had been suggested from earlier kinetic and spectroscopic studies.
5.2. CO2 reduction
The observation that Mo-nitrogenase can be endowed with the ability to reduce CO by remodeling the protein environment surrounding the active site [94], coupled with the finding that wild-type nitrogenase can reduce CO2 at low rates to yield CO [75], prompted an examination of whether or not remodeled versions of Monitrogenase might also be able to reduce CO2 by multiple electrons to yield methane (Table 1). The α-70Ala substituted MoFe protein, which earlier had shown low CO reduction rates, was found to catalyze essentially no CO2 reduction when the reaction was monitored for methane formation [95]. However, when the doubly substituted MoFe protein (α-70Ala/ α-195Gln) was examined, this variant showed significant rates of formation of CH4 [95]. It was confirmed that CH4 formation is catalyzed by nitrogenase and that the produced methane is derived from CO2 by using 13C labeled CO2 with analysis of products using gas chromatography and mass spectrometry.
Because the nitrogenase active site contains multiple binding sites, at least with respect to carbon-containing substrates and inhibitors, it seemed possible that a remodeled form of the enzyme might reduce and link CO2 to another substrate. This possibility has been tested by preliminary studies in which a doubly substituted (α-70Ala/ α-195Gln) MoFe protein was incubated under turnover conditions in the presence of both CO2 and acetylene [95]. Analysis of the gas phase of this reaction revealed catalytic formation of propylene (H2C=CH-CH3) (Table 1). The source of the C atoms within propylene was established using 13HCO3− as a substrate with product analysis by gas chromatography and mass spectrometry, revealing that one carbon is from CO2 and two are from acetylene. The rate of formation of propylene was found to be strongly dependent on the partial pressure of acetylene at a defined CO2 concentration.
6. Mechanistic Considerations
6.1. Thermodynamic considerations
Two aspects of the recent discoveries regarding nitrogenase reduction of CO and CO2 are particularly noteworthy in providing insights into the nitrogenase mechanism: the ability to catalyze multiple electron reductions of substrates and the ability to link two or more substrates together during reduction. An important feature of nitrogenase is the ability to carry out multi-electron reduction reactions of small molecules without releasing partially reduced intermediates [4]. This point is underscored when the 6 electron reduction of N2 to NH3 or the 8 electron reduction of CO2 to CH4 are considered, a situation under which there is little or no release of partially reduced intermediates. In fact, the ability to carry out such multiple electron reduction reactions of relatively inert molecules is unique to nitrogenase among known enzymes and is rare among any known molecular catalysts. Understanding how nitrogenase binds and sequesters small molecule substrates to achieve their multielectron reduction is not yet understood and is obviously a key frontier research area within the field. The recent discovery of the involvement of metal-hydrides bound to FeMo-cofactor as a key aspect of substrate activation [14] offers an important starting point for exploring the mechanism of the reduction of both N2 and CO2.
It is instructive to consider the thermodynamics of the expected reaction pathway for N2 reduction. In the proposed reductive elimination model, N2 activation results in the “prompt” formation of an initial metal-bound diazene intermediate with subsequent alternating hydrogenation to form a metal-bound hydrazine intermediate with ultimate release of two ammonia [14]. A consideration of the standard Gibbs free energy change (ΔG°) for this reaction pathway (Figure 5) reveals that the first step, the reduction of N2 to diazene, is the most difficult, being largely endergonic. Subsequent reduction steps are downhill, with the overall reaction being exergonic. Nitrogenase must lower the energy of these intermediates by stabilizing them, almost certainly through binding to the metal cluster, and, possibly, by interaction with amino acid side chains. One challenge in defining a molecular mechanism for N2 reduction is to understand the nature of these intermediates at the atomic level. Recent progress in trapping and characterizing intermediates along the N2 reduction pathway has started to provide some insights into this reaction mechanism [13,19]. For example, accumulating evidence indicates that each intermediate trapped so far is bound to one or more Fe atoms. Further, it appears that the N2-trapped state is early in the reaction pathway and different from the diazene and hydrazine trapped states [73]. By analogy to N2 reduction, the reduction of CO2 to CH4 is expected to proceed through a series of partially reduced intermediates including carbon monoxide (CO) or formate (HCOOH), formaldehyde (CH2O), methanol (CH3OH), and methane (CH4) (eqn 3 with waters omitted) with each step being a 2 e− reduction.
| (eqn 3) |
When the standard Gibbs free energy changes for these expected intermediates for CO2 reduction are compared to the energetics of N2 reduction, some interesting parallels are apparent (Figure 5). Both processes are uphill initially, followed by exergonic reactions for subsequent steps. Considering the energetics for both processes, it is tempting to speculate that similar mechanisms for reduction and proton addition are operative for both substrates. Understanding how nitrogenase stabilizes each of the intermediates, especially the initial high-energy states, and how it captures/stabilizes the partially reduced states is an obvious area for future exploration. The possible role of metal hydrides in both reactions is a lead in gaining insights into how these complex reactions are achieved.
Figure 5.

Standard Gibb’s free energy change diagrams for N2 and CO2 reduction. Shown is the standard Gibb’s free energy change (ΔG°) between intermediates for the N2 (panel A) and CO2 (panel B) reduction pathways. Also shown are possible energy states for metal bound intermediates (lower dashed traces in both panels). All reaction standard Gibb’s free energy changes were calculated from known standard Gibb’s free energy of formation values [98–100].
6.2. Multiple binding sites
One of the more intriguing observations from recent work on the reduction of CO and CO2 is the observation that carbon monomers can be linked to make longer chain hydrocarbons (such as C2 and C3 products) [91,94]. These observations fit with the earlier studies indicating that FeMo-cofactor has multiple binding sites for some substrates and inhibitors [4,96]. For example, as described earlier, studies with the inhibitor CO revealed two different CO bound states on the FeMo-cofactor, depending on the concentration of CO (so called hi-CO and lo-CO) [81]. Further analysis of these two states by ENDOR spectroscopy were interpreted as indicating two binding sites for CO [82–84, 97]. Likewise, kinetic studies with the substrate acetylene were interpreted as the FeMo-cofactor presenting two different binding sites [96]. While these, and other studies, suggested multiple binding sites on the FeMo-cofactor, they were not definitive. The recent findings involving CO reduction and coupling [91,94] provides further circumstantial evidence for multiple binding sites for substrates on the FeMo-cofactor. In both the V-nitrogenase and the remodeled Monitrogenase, CO reduction can result in coupling of two or three CO molecules resulting in C2 or C3 hydrocarbon products. One mechanistic explanation for the formation of these products from CO is the activation of two CO molecules on adjacent binding sites, with linkage of the activated M-CHx species with C-C bond formation and polymerization to form longer chain hydrocarbons [94]. It should be noted, however, that an alternative mechanism can also be considered wherein insertion of an incoming CO molecule with the activated M-CHx species might occur, followed by reduction to the final product. This latter mechanism would not necessarily need two unique substrate binding sites. The recent observation of the ability of nitrogenase to link CO2 to acetylene to produce propylene [95] could also be interpreted in terms of the two reaction mechanisms described for CO coupling. Sorting out how two or more substrates are linked during reduction will be a fascinating aspect of the nitrogenase mechanism.
6.3. Possible mechanisms for hydrogenation of substrates
There are several different possible mechanisms for how substrates could be reduced by nitrogenase including through hydride insertion at an electron-deficient center, protonation of an electron-rich center, or reductive elimination reactions between the adjacent metal-hydride (M-H) and metal-ligand (M-X) bond [14]. The recent discovery of bridging metal-hydrides in nitrogenase suggests possible roles for such hydrides directly in substrate reduction [14], although a direct role has yet to be established. Thus, several questions remain to be answered about the roles of such hydrides including: (1) do hydrides attack bound substrates?; (2) is there a stepwise proton coupled electron transfer of the bound substrate or protonation of the reduced substrate-derived anionic ligand bound to the metallic active center?; and (3) is there a reductive elimination reaction of the M-H and M-X (X = C, N, O, or S etc.) to form X-H species? As these different possible mechanisms are considered, it is important to appreciate that nitrogenase catalysis might use different hydrogenation mechanisms at different steps for different substrates. For example, the formation of both cis- and trans-1,2-dideuteroethylene when acetylene is reduced in the presence of D2O can be explained by the stepwise reduction of acetylene first to a bound one electron reduced η2-vinyl intermediate, which could rotate and be further reduced yielding either cis- or trans-product [40]. However, the production of both cis- and trans-isomer products could also be explained by the insertion of an acetylene molecule into an M-H on FeMo-cofactor. This latter mechanism might also explain the observation that acetylene completely inhibits proton reduction [78], by intercepting the M-H that is destined to make H2. Clearly, much work remains to sort out details of how nitrogenase reduces substrates and the utilization of C-containing substrates will be an important tool in these efforts.
Highlights.
Nitrogenase mechanism is reviewed.
Carbon-containing substrates and inhibitors of nitrogenase are reviewed.
Recent findings for CO and CO2 reduction catalyzed by nitrogenases are reviewed.
The mechanistic insights for nitrogenase carbon reduction chemistry are discussed.
Acknowledgments
This work was supported by the grant from the National Institutes of Health (GM59087). The authors acknowledge Dr. Brian M. Hoffman, Northwestern University, for a long standing collaboration to understand nitrogenase mechanism.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smil V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production. Cambridge, MA: The MIT Press; 2004. [Google Scholar]
- 2.Eady RR. Structure-function relationships of alternative nitrogenases. Chem. Rev. 1996;96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]
- 3.Hu Y, Lee CC, Ribbe MW. Vanadium nitrogenase: a two-hit wonder? Dalton Trans. 2012;41:1118–1127. doi: 10.1039/c1dt11535a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burgess BK, Lowe DJ. Mechanism of molybdenum nitrogenase. Chem. Rev. 1996;96:2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 5.Georgiadis MM, Komiya H, Chakrabarti P, Woo D, Kornuc JJ, Rees DC. Crystallographic structure of the nitrogenase iron protein from Azotobacter vinelandii. Science. 1992;257:1653–1659. doi: 10.1126/science.1529353. [DOI] [PubMed] [Google Scholar]
- 6.Howard JB, Rees DC. Nitrogenase: a nucleotide-dependent molecular switch. Annu. Rev. Biochem. 1994;63:235–264. doi: 10.1146/annurev.bi.63.070194.001315. [DOI] [PubMed] [Google Scholar]
- 7.Chan MK, Kim J, Rees DC. The nitrogenase FeMo-cofactor and P-cluster pair: 22 Å resolution structures. Science. 1993;260:792–794. doi: 10.1126/science.8484118. [DOI] [PubMed] [Google Scholar]
- 8.Shah VK, Brill WJ. Isolation of an iron-molybdenum cofactor from nitrogenase. Proc. Natl. Acad. Sci. U.S.A. 1977;74:3249–3253. doi: 10.1073/pnas.74.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seefeldt LC, Hoffman BM, Dean DR. Electron transfer in nitrogenase catalysis. Curr. Opin. Chem. Biol. 2012;16:19–25. doi: 10.1016/j.cbpa.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danyal K, Dean DR, Hoffman BM, Seefeldt LC. Electron transfer within nitrogenase: evidence for a deficit-spending mechanism. Biochemistry. 2011;50:9255–9263. doi: 10.1021/bi201003a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayweather D, Danyal K, Dean DR, Seefeldt LC, Hoffman BM. Temperature invariance of the nitrogenase electron transfer mechanism. Biochemistry. 2012;51:8391–8398. doi: 10.1021/bi301164j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hageman RV, Burris RH. Nitrogenase and nitrogenase reductase associate and dissociate with each catalytic cycle. Proc. Natl. Acad. Sci. U.S.A. 1978;75:2699–2702. doi: 10.1073/pnas.75.6.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seefeldt LC, Hoffman BM, Dean DR. Mechanism of Mo-dependent nitrogenase. Annu. Rev. Biochem. 2009;78:701–722. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman BM, Lukoyanov D, Dean DR, Seefeldt LC. Nitrogenase: a draft mechanism. Acc. Chem. Res. 2013 doi: 10.1021/ar300267m. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thorneley RNF, Lowe DJ. Kinetics and mechanism of the nitrogenase enzyme. In: Spiro TG, editor. Molybdenum Enzymes. New York: Wiley-Interscience Publications; 1985. pp. 221–284. [Google Scholar]
- 16.Doan PE, Telser J, Barney BM, Igarashi RY, Dean DR, Seefeldt LC, Hoffman BM. 57Fe ENDOR spectroscopy and “electron inventory” analysis of the nitrogenase E4 intermediate suggest the metal-ion core of FeMo-cofactor cycles through only one redox couple. J. Am. Chem. Soc. 2011;133:17329–17340. doi: 10.1021/ja205304t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lukoyanov D, Dikanov SA, Yang Z-Y, Barney BM, Samoilova RI, Narasimhulu KV, Dean DR, Seefeldt LC, Hoffman BM. ENDOR/HYSCORE studies of the common intermediate trapped during nitrogenase reduction of N2H2, CH3N2H, and N2H4 support an alternating reaction pathway for N2 reduction. J. Am. Chem. Soc. 2011;133:11655–11664. doi: 10.1021/ja2036018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barney BM, Lukoyanov D, Igarashi RY, Laryukhin M, Yang T-C, Dean DR, Hoffman BM, Seefeldt LC. Trapping an intermediate of dinitrogen (N2) reduction on nitrogenase. Biochemistry. 2009;48:9094–9102. doi: 10.1021/bi901092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman BM, Dean DR, Seefeldt LC. Climbing nitrogenase: toward a mechanism of enzymatic nitrogen fixation. Acc. Chem. Res. 2009;42:609–619. doi: 10.1021/ar8002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Igarashi RY, Laryukhin M, Dos Santos PC, Lee H-I, Dean DR, Seefeldt LC, Hoffman BM. Trapping H− bound to the nitrogenase FeMo-cofactor active site during H2 evolution: characterization by ENDOR spectroscopy. J. Am. Chem. Soc. 2005;127:6231–6241. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]
- 21.Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Connecting nitrogenase intermediates with the kinetic scheme for N2 reduction by a relaxation protocol and identification of the N2 binding state. Proc. Natl. Acad. Sci. U.S.A. 2007;104:1451–1455. doi: 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC, Hoffman BM. Is Mo involved in hydride binding by the four-electron reduced (E4) intermediate of the nitrogenase MoFe protein? J. Am. Chem. Soc. 2010;132:2526–2527. doi: 10.1021/ja910613m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thorneley RNF, Eady RR, Lowe DJ. Biological nitrogen fixation by way of an enzyme-bound dinitrogen-hydride intermediate. Nature. 1978;272:557–558. [Google Scholar]
- 24.Dilworth MJ, Eady RR. Hydrazine is a product of dinitrogen reduction by the vanadiumnitrogenase from Azotobacter chroococcum. Biochem. J. 1991;277(Pt 2):465–468. doi: 10.1042/bj2770465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dilworth MJ. Acetylene reduction by nitrogen-fixing preparations from Clostridium pasteurianum. Biochim. Biophys. Acta. 1966;127:285–294. doi: 10.1016/0304-4165(66)90383-7. [DOI] [PubMed] [Google Scholar]
- 26.Schöllhorn R, Burris RH. Acetylene as a competitive inhibitor of N2 fixation. Proc. Natl. Acad. Sci. U.S.A. 1967;58:213–216. doi: 10.1073/pnas.58.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seefeldt LC, Dance IG, Dean DR. Substrate interactions with nitrogenase: Fe versus Mo. Biochemistry. 2004;43:1401–1409. doi: 10.1021/bi036038g. [DOI] [PubMed] [Google Scholar]
- 28.Dilworth MJ, Eldridge ME, Eady RR. The molybdenum and vanadium nitrogenases of Azotobacter chroococcum: effect of elevated temperature on N2 reduction. Biochem. J. 1993;289:395–400. doi: 10.1042/bj2890395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim CH, Newton WE, Dean DR. Role of the MoFe protein alpha-subunit histidine-195 residue in FeMo-cofactor binding and nitrogenase catalysis. Biochemistry. 1995;34:2798–2808. doi: 10.1021/bi00009a008. [DOI] [PubMed] [Google Scholar]
- 30.Schneider K, Müller A, Krahn E, Hagen WR, Wassink H, Karl-Heinz K. The molybdenum nitrogenase from wild-type Xanthobacter autotrophicus exhibits properties reminiscent of alternative nitrogenases. Eur. J. Biochem. 1995;230:666–675. doi: 10.1111/j.1432-1033.1995.0666h.x. [DOI] [PubMed] [Google Scholar]
- 31.Scott DJ, Dean DR, Newton WE. Nitrogenase-catalyzed ethane production and CO-sensitive hydrogen evolution from MoFe proteins having amino acid substitutions in an α-subunit FeMo cofactor-binding domain. J. Biol. Chem. 1992;267:20002–20010. [PubMed] [Google Scholar]
- 32.Dilworth MJ, Eady RR, Robson Rl, Miller RW. Ethane formation from acetylene as a potential test for vanadium nitrogenase in vivo. Nature. 1987;327:167–168. [Google Scholar]
- 33.Dilworth MJ, Eady RR, Eldridge ME. The vanadium nitrogenase of Azotobacter chroococcum: Reduction of acetylene and ethylene to ethane. Biochem. J. 1988;249:745–751. doi: 10.1042/bj2490745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eady RR, Robson Rl, Richardson TH, Miller RW, Hawkins M. The vanadium nitrogenase of Azotobacter chroococcum: Purification and properties of the VFe protein. Biochem. J. 1987;244:197–207. doi: 10.1042/bj2440197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pau RN, Eldridge ME, Lowe DJ, Mitchenall LA, Eady R. Molybdenum-independent nitrogenases of Azotobacter vinelandii - a functional species of alternative nitrogenase-3 isolated from a molybdenum-tolerant strain contains an iron molybdenum cofactor. Biochem. J. 1993;293:101–107. doi: 10.1042/bj2930101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider K, Gollan U, Selsemeiervoigt S, Plass W, Muller A. Rapid purification of the proteincomponents of a highly-active iron only nitrogenase. Naturwissenschaften. 1994;81:405–408. doi: 10.1007/BF01132694. [DOI] [PubMed] [Google Scholar]
- 37.Fisher K, Dilworth MJ, Kim CH, Newton WE. Azotobacter vinelandii nitrogenases containing altered MoFe proteins with substitutions in the FeMo-cofactor environment: effects on the catalyzed reduction of acetylene and ethylene. Biochemistry. 2000;39:2970–2979. doi: 10.1021/bi992092e. [DOI] [PubMed] [Google Scholar]
- 38.Hardy RW, Holsten RD, Jackson EK, Burns RC. The acetylene-ethylene assay for N2 fixation: laboratory and field evaluation. Plant Physiol. 1968;43:1185–1207. doi: 10.1104/pp.43.8.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kelly M. Some properties of purified nitrogenase of Azotobacter chroococcum. Biochim. Biophys. Acta. 1969;171:9–22. doi: 10.1016/0005-2744(69)90101-6. [DOI] [PubMed] [Google Scholar]
- 40.Benton PM, Christiansen J, Dean DR, Seefeldt LC. Stereospecificity of acetylene reduction catalyzed by nitrogenase. J. Am. Chem. Soc. 2001;123:1822–1827. doi: 10.1021/ja003662x. [DOI] [PubMed] [Google Scholar]
- 41.Fisher K, Dilworth MJ, Newton WE. Differential effects on N2 binding and reduction, HD formation, and azide reduction with alpha-195His- and alpha-191Gln-substituted MoFe proteins of Azotobacter vinelandii nitrogenase. Biochemistry. 2000;39:15570–15577. doi: 10.1021/bi0017834. [DOI] [PubMed] [Google Scholar]
- 42.Lee H-I, Sørlie M, Christiansen J, Yang T-C, Shao J, Dean DR, Hales BJ, Hoffman BM. Electron inventory, inetic assignment (En), structure, and bonding of nitrogenase turnover intermediates with C2H2 and CO. J. Am. Chem. Soc. 2005;127:15880–15890. doi: 10.1021/ja054078x. [DOI] [PubMed] [Google Scholar]
- 43.McLean PA, True A, Nelson MJ, Lee H-I, Hoffman BM, Orme-Johnson WH. Effects of substrates (methyl isocyanide, C2H2) and inhibitor (CO) on resting-state wild-type and NifV− Klebsiella pneumoniae MoFe proteins. J. Inorg. Biochem. 2003;93:18–32. doi: 10.1016/s0162-0134(02)00580-9. [DOI] [PubMed] [Google Scholar]
- 44.Lee HI, Sorlie M, Christiansen J, Song RT, Dean DR, Hales BJ, Hoffman BM. Characterization of an intermediate in the reduction of acetylene by the nitrogenase alpha-Gln(195) MoFe protein by Q-band EPR and C-13,H-1 ENDOR. J. Am. Chem. Soc. 2000;122:5582–5587. [Google Scholar]
- 45.Sørlie M, Christiansen J, Dean DR, Hales BJ. Detection of a new radical and FeMo-cofactor EPR signal during acetylene reduction by the α-H195Q mutant of nitrogenase. J. Am. Chem. Soc. 1999;121:9457–9458. [Google Scholar]
- 46.Christiansen J, Cash VL, Seefeldt LC, Dean DR. Isolation and characterization of an acetylene-resistant nitrogenase. J. Biol. Chem. 2000;275:11459–11464. doi: 10.1074/jbc.275.15.11459. [DOI] [PubMed] [Google Scholar]
- 47.Hardy RW, Burns RC, Parshall GW. The biochemistry of N2 fixation. In: Dessy R, Dillard J, Taylor L, editors. Bioinorganic Chemistry. Washington, D.C.: American Chemical Society; 1971. pp. 219–247. [Google Scholar]
- 48.McKenna CE, McKenna MC, Huang CW. Low stereoselectivity in methylacetylene and cyclopropene reductions by nitrogenase. Proc. Natl. Acad. Sci. U.S.A. 1979;76:4773–4777. doi: 10.1073/pnas.76.10.4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hardy RWF, Jackson EK. Reduction of model substrates - nitriles and acetylenes - by nitrogenase (N2ase) Fed. Proc. 1967;28:725. [Google Scholar]
- 50.Igarashi RY, Dos Santos PC, Niehaus WG, Dance IG, Dean DR, Seefeldt LC. Localization of a catalytic intermediate bound to the FeMo-cofactor of nitrogenase. J. Biol. Chem. 2004;279:34770–34775. doi: 10.1074/jbc.M403194200. [DOI] [PubMed] [Google Scholar]
- 51.Lee H-I, Igarashi RY, Laryukhin M, Doan PE, Dos Santos PC, Dean DR, Seefeldt LC, Hoffman BM. An organometallic intermediate during alkyne reduction by nitrogenase. J. Am. Chem. Soc. 2004;126:9563–9569. doi: 10.1021/ja048714n. [DOI] [PubMed] [Google Scholar]
- 52.Mayer SM, Niehaus WG, Dean DR. Reduction of short chain alkynes by a nitrogenase α-70Ala-substituted MoFe protein. J. Chem. Soc. Dalton Trans. 2002:802–807. [Google Scholar]
- 53.Gemoets JP, Bravo M, McKenna CE, Leigh GJ, Smith BE. Reduction of cyclopropene by NifV− and wild-type nitrogenases from Klebsiella pneumoniae. Biochem. J. 1989;258:487–491. doi: 10.1042/bj2580487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKenna CE, McKenna MC, Higa MT. Chemical probes of nitrogenase 1 Cyclopropene Nitrogenase-catalyzed reduction to propene and cyclopropane. J. Am. Chem. Soc. 1976;98:4657–4659. doi: 10.1021/ja00431a059. [DOI] [PubMed] [Google Scholar]
- 55.McKenna CE, Eran H, Nakajima T, Osumi A. Active site probes for nitrogenase. In: Gibson AH, Newton WE, editors. Current Perspectives in Nitrogen Fixation. Canberra: Australia Academy of Science; 1981. p. 358. [Google Scholar]
- 56.Ashby GA, Dilworth MJ, Thorneley RNF. Klebsiella pneumoniae nitrogenase Inhibition of hydrogen evolution by ethylene and the reduction of ethylene to ethane. Biochem. J. 1987;247:547–554. doi: 10.1042/bj2470547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burns RC, Hardy RW, Phillips WD. Azotobacter nitrogenase: mechanism and kinetics of allene reduction. In: Stewart WDP, editor. Nitrogen Fixation in Free-Living Organisms. Cambridge: Cambridge University Press; 1975. pp. 447–452. [Google Scholar]
- 58.Hardy RW, Knight EJ. ATP-dependent reduction of azide and HCN by N2-fixing enzymes of Azotobacter vinelandii and Clostridium pasteurianum. Biochim. Biophys. Acta. 1967;139:69–90. doi: 10.1016/0005-2744(67)90114-3. [DOI] [PubMed] [Google Scholar]
- 59.Li J, Burgess BK, Corbin JL. Nitrogenase reactivity: cyanide as substrate and inhibitor. Biochemistry. 1982;21:4393–4402. doi: 10.1021/bi00261a031. [DOI] [PubMed] [Google Scholar]
- 60.Fisher K, Dilworth MJ, Kim C-H, Newton WE. Azotobacter vinelandii nitrogenases with substitutions in the FeMo-cofactor environment of the MoFe protein: effects of acetylene or ethylene on interactions with H+, HCN, and CN−. Biochemistry. 2000;39:10855–10865. doi: 10.1021/bi0001628. [DOI] [PubMed] [Google Scholar]
- 61.Fisher K, Dilworth MJ, Newton WE. Azotobacter vinelandii vanadium nitrogenase: formaldehyde is a product of catalyzed HCN reduction, and excess ammonia arises directly from catalyzed azide reduction. Biochemistry. 2006;45:4190–4198. doi: 10.1021/bi0514109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kelly M, Postgate JR, Richards RL. Reduction of cyanide and isocyanide by nitrogenase of Azotobacter chroococcum. Biochem. J. 1967;102:1C–3C. doi: 10.1042/bj1020001c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burns RC, Fuchsman WH, Hardy RW. Nitrogenase from vanadium-grown Azotobacter: isolation, characteristics, and mechanistic implications. Biochem. Biophys. Res. Commun. 1971;42:353–358. doi: 10.1016/0006-291x(71)90377-9. [DOI] [PubMed] [Google Scholar]
- 64.Fuchsman WH, Hardy RWF. Nitrogenase-catalyzed acrylonitrile reductions. Bioinorg. Chem. 1972;1:195–213. [Google Scholar]
- 65.Miller RW, Eady RR. Cyanamide: a new substrate for nitrogenase. Biochim. Biophys. Acta. 1988;952:290–296. doi: 10.1016/0167-4838(88)90129-x. [DOI] [PubMed] [Google Scholar]
- 66.Hwang JC, Burris RH. Nitrogenase-catalyzed reactions. Biochim. Biophys. Acta. 1972;283:339–350. doi: 10.1016/0005-2728(72)90250-2. [DOI] [PubMed] [Google Scholar]
- 67.Kelly M. The kinetics of the reduction of isocyanides, acetylenes and the cyanide ion by nitrogenase preparation from Azotobacter chroococcum and the effects of inhibitors. Biochem. J. 1968;107:1–6. doi: 10.1042/bj1070001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munson TO, Burris RH. Nitrogen fixation by Rhodospirillum rubrum grown in nitrogen-limited continuous culture. J. Bacteriol. 1969;97:1093–1098. doi: 10.1128/jb.97.3.1093-1098.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rubinson JF, Corbin JL, Burgess BK. Nitrogenase reactivity: methyl isocyanide as substrate and inhibitor. Biochemistry. 1983;22:6260–6268. doi: 10.1021/bi00295a034. [DOI] [PubMed] [Google Scholar]
- 70.Biggins DR, Postgate JR. Nitrogen fixation by cultures and cell-free extracts of Mycobacterium flavum 301. J. Gen. Microbiol. 1969;56:191–193. doi: 10.1099/00221287-56-2-181. [DOI] [PubMed] [Google Scholar]
- 71.McKenna CE, Simeonov AM, Eran H, Bravo-Leerabhandh M. Reduction of cyclic and acyclic diazene derivatives by Azotobacter vinelandii nitrogenase: diazirine and trans-dimethyldiazene. Biochemistry. 1996;35:4502–4514. doi: 10.1021/bi950964g. [DOI] [PubMed] [Google Scholar]
- 72.Barney BM, Lukoyanov D, Yang T-C, Dean DR, Hoffman BM, Seefeldt LC. A methyldiazene (HN=N-CH3)-derived species bound to the Nitrogenase active-site FeMo Cofactor: implications for mechanism. Proc. Natl. Acad. Sci. U.S.A. 2006;103:17113–17118. doi: 10.1073/pnas.0602130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lukoyanov D, Yang Z-Y, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Unification of reaction pathway and kinetic scheme for N2 reduction catalyzed by nitrogenase. Proc. Natl. Acad. Sci. U.S.A. 2012;109:5583–5587. doi: 10.1073/pnas.1202197109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rasche ME, Seefeldt LC. Reduction of thiocyanate, cyanate, and carbon disulfide by nitrogenase: kinetic characterization and EPR spectroscopic analysis. Biochemistry. 1997;36:8574–8585. doi: 10.1021/bi970217e. [DOI] [PubMed] [Google Scholar]
- 75.Seefeldt LC, Rasche ME, Ensign SA. Carbonyl sulfide and carbon dioxide as new substrates, and carbon disulfide as a new inhibitor, of nitrogenase. Biochemistry. 1995;34:5382–5389. doi: 10.1021/bi00016a009. [DOI] [PubMed] [Google Scholar]
- 76.Madden MS, Kindon ND, Ludden PW, Shah VK. Diastereomer-dependent substrate reduction properties of a dinitrogenase containing 1-fluorohomocitrate in the iron-molybdenum cofactor. Proc. Natl. Acad. Sci. U.S.A. 1990;87:6517–6521. doi: 10.1073/pnas.87.17.6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hwang JC, Chen CH, Burris RH. Inhibition of nitrogenase-catalyzed reductions. Biochim. Biophys. Acta. 1973;292:256–270. doi: 10.1016/0005-2728(73)90270-3. [DOI] [PubMed] [Google Scholar]
- 78.Rivera-Ortiz JM, Burris RH. Interactions among substrates and inhibitors of nitrogenase. J. Bacteriol. 1975;123:537–545. doi: 10.1128/jb.123.2.537-545.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pham DN, Burgess BK. Nitrogenase reactivity: effects of pH on substrate reduction and carbon monoxide inhibition. Biochemistry. 1993;32:13725–13731. doi: 10.1021/bi00212a043. [DOI] [PubMed] [Google Scholar]
- 80.Yates MG, Lowe DJ. Nitrogenase of Azotobacter chroococcum: A new electronparamagnetic-resonance signal associated with a transient species of the MoFe protein during catalysis. FEBS Letters. 1976;72:127–130. doi: 10.1016/0014-5793(76)80826-5. [DOI] [PubMed] [Google Scholar]
- 81.Davis LC, Henzl MT, Burris RH, Orme-Johnson WH. Iron-sulfur clusters in the molybdenum-iron protein component of nitrogenase Electron paramagnetic resonance of the carbon monoxide inhibited state. Biochemistry. 1979;18:4860–4869. doi: 10.1021/bi00589a014. [DOI] [PubMed] [Google Scholar]
- 82.Pollock RC, Lee H-I, Cameron LM, DeRose VJ, Hales BJ, Orme-Johnson WH, Hoffman BM. Investigation of CO bound to inhibited forms of nitrogenase MoFe protein by 13C ENDOR. J. Am. Chem. Soc. 1995;117:8686–8687. [Google Scholar]
- 83.Christie PD, Lee H-I, Cameron LM, Hales BJ, Orme-Johnson WH, Hoffman BM. Identification of the CO-binding cluster in nitrogenase MoFe protein by ENDOR of 57Fe isotopomers. J. Am. Chem. Soc. 1996;118:8707–8709. [Google Scholar]
- 84.Lee H-I, Cameron LM, Hales BJ, Hoffman BM. CO binding to the FeMo cofactor of CO-inhibited nitrogenase: 13CO and 1H Q-band ENDOR investigation. J. Am. Chem. Soc. 1997;119:10121–10126. [Google Scholar]
- 85.Maskos Z, Fisher K, Sørlie M, Newton WE, Hales BJ. Variant MoFe proteins of Azotobacter vinelandii: effects of carbon monoxide on electron paramagnetic resonance spectra generated during enzyme turnover. J. Biol. Inorg. Chem. 2005;10:394–406. doi: 10.1007/s00775-005-0648-2. [DOI] [PubMed] [Google Scholar]
- 86.Dos Santos PC, Mayer SM, Barney BM, Seefeldt LC, Dean DR. Alkyne substrate interaction within the nitrogenase MoFe protein. J. Inorg. Biochem. 2007;101:1642–1648. doi: 10.1016/j.jinorgbio.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dos Santos PC, Igarashi RY, Lee H-I, Hoffman BM, Seefeldt LC, Dean DR. Substrate interactions with the nitrogenase active site. Acc. Chem. Res. 2005;38:208–214. doi: 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]
- 88.Christiansen J, Cash VL, Seefeldt LC, Dean DR. Isolation and characterization of an acetyleneresistant nitrogenase. J. Biol. Chem. 2000;275:11459–11464. doi: 10.1074/jbc.275.15.11459. [DOI] [PubMed] [Google Scholar]
- 89.Christiansen J, Seefeldt LC, Dean DR. Competitive substrate and inhibitor interactions at the physiologically relevant active site of nitrogenase. J. Biol. Chem. 2000;275:36104–36107. doi: 10.1074/jbc.M004889200. [DOI] [PubMed] [Google Scholar]
- 90.Barney BM, Igarashi RY, Dos Santos PC, Dean DR, Seefeldt LC. Substrate interaction at an iron-sulfur face of the FeMo-cofactor during nitrogenase catalysis. J. Biol. Chem. 2004;279:53621–53624. doi: 10.1074/jbc.M410247200. [DOI] [PubMed] [Google Scholar]
- 91.Lee CC, Hu Y, Ribbe MW. Vanadium nitrogenase reduces CO. Science. 2010;329:642. doi: 10.1126/science.1191455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee CC, Hu Y, Ribbe MW. Tracing the hydrogen source of hydrocarbons formed by vanadium nitrogenase. Angew. Chem. Int. Ed. 2011;50:5545–5547. doi: 10.1002/anie.201100869. [DOI] [PubMed] [Google Scholar]
- 93.Hu Y, Lee CC, Ribbe MW. Extending the carbon chain: hydrocarbon formation catalyzed by vanadium/molybdenum nitrogenases. Science. 2011;333:753–755. doi: 10.1126/science.1206883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang Z-Y, Dean DR, Seefeldt LC. Molybdenum nitrogenase catalyzes the reduction and coupling of CO to form hydrocarbons. J. Biol. Chem. 2011;286:19417–19421. doi: 10.1074/jbc.M111.229344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang Z-Y, Moure VR, Dean DR, Seefeldt LC. Carbon dioxide reduction to methane and coupling with acetylene to form propylene catalyzed by remodeled nitrogenase. Proc. Natl. Acad. Sci. U.S.A. 2012;109:19644–19648. doi: 10.1073/pnas.1213159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shen J, Dean DR, Newton WE. Evidence for multiple substrate-reduction sites and distinct inhibitor-binding sites from an altered Azotobacter vinelandii nitrogenase MoFe protein. Biochemistry. 1997;36:4884–4894. doi: 10.1021/bi9628578. [DOI] [PubMed] [Google Scholar]
- 97.Lee H-I, Hales BJ, Hoffman BM. Metal-ion valencies of the FeMo cofactor in CO-inhibited and resting state nitrogenase by 57Fe Q-band ENDOR. J. Am. Chem. Soc. 1997;119:11395–11400. [Google Scholar]
- 98.Perry RH, Green DW, editors. Perry’s Chemical Engineers’ Handbook. 8th ed. New York, NY: McGraw-Hill, Inc.; 2008. [Google Scholar]
- 99.Linstrom PJ, Mallard WG. NIST Chemistry Webbook. Gathersburg, MD: National Institute of Standards and Technology; 2005. [Google Scholar]
- 100.Dean JA. Lange’s Handbook of Chemistry. 15th ed. New York, NY: McGraw-Hill, Inc.; 1999. [Google Scholar]
- 101.Burns RC, Hardy RW. Nitrogen Fixation in Bacteria and Higher Plants. Berlin: Springer; 1975. [DOI] [PubMed] [Google Scholar]
- 102.Newton WE, Dilworth MJ. Assays of nitrogenase reaction products. Methods Mol. Biol. 2011;766:105–127. doi: 10.1007/978-1-61779-194-9_8. [DOI] [PubMed] [Google Scholar]