

Abstract
OBJECTIVE
Lipodystrophies are categorized by the extent of fat loss (generalized vs. partial) and by inheritance (congenital vs. acquired). We examined whether a group of patients with partial lipodystrophy of the limbs (PLL), type 2 diabetes mellitus (T2DM), and an absence of a family history of lipodystrophy constitute a new clinical subtype.
RESEARCH DESIGN AND METHODS
Ten women with T2DM and PLL were identified in academic diabetes clinics and were matched by age, sex, BMI, ethnicity, and diabetes status with 10 women with control T2DM without lipodystrophy. All patients were characterized by clinical evaluation and hyperinsulinemic clamp.
RESULTS
Patients with T2DM and PLL exhibited symmetrical loss of subcutaneous fat in forearms, or forearms plus calves, and acanthosis nigricans. Maximally stimulated glucose disposal rates were markedly reduced by 56% in the T2DM with PLL group compared with the control T2DM patients, whether normalized by body weight or surface area. Most PLL patients exhibited little or no insulin-mediated glucose uptake after subtraction of non-insulin–mediated glucose uptake. The T2DM with PLL group also had greater elevations in hepatic transaminases and triglycerides and earlier onset of diabetes compared with control T2DM.
CONCLUSIONS
T2DM with PLL represents a previously unrecognized phenotype of lipodystrophy and of T2DM. These T2DM patients exhibit symmetrical lipodystrophy of the distal limbs, acanthosis nigricans, marked insulin resistance with little insulin-mediated glucose uptake, hypertriglyceridemia, and hepatic transaminase elevations, which are greater in severity than observed in patients with common T2DM.
Lipodystrophies are a rare, heterogeneous group of disorders characterized by loss of subcutaneous adipose tissue together with metabolic abnormalities associated with insulin resistance. The conventional diagnostic scheme for lipodystrophy involves the extent of fat loss (general or partial) and inheritance (acquired or congenital) resulting in four categories of disease: congenital generalized lipodystrophy, acquired generalized lipodystrophy, familial partial lipodystrophy (FPL), and acquired partial lipodystrophy (APL) (1–5). In addition to selective loss of body fat, these diseases are also frequently, but not consistently, characterized by hypertriglyceridemia, steatohepatitis, type 2 diabetes mellitus (T2DM), and acanthosis nigricans. Congenital generalized lipodystrophy or Berardinelli-Seip syndrome is an autosomal recessive disorder, which can result from mutations in any one of several genes (AGPAT2, CAV1, BSCL2, PTRF), featuring generalized lack of adipose tissue exhibited at birth or within the first year of life.
Patients with acquired generalized lipodystrophy or Lawrence syndrome develop progressive fat loss beginning in childhood or adolescence involving first the face and upper extremities and eventually the torso. FPL is an autosomal-dominant disorder with identifiable patterns of fat loss, including the Dunnigan variety, Kobberling variety, and mandibuloacral dysplasia. Some patients with the Dunnigan and mandibular dyscrasia phenotypes of FPL have been found to harbor gene mutations involving LMNA, PPARγ, PLIN-1, AKT2, or CIDEC. Patients with APL or the Barraquer-Simons syndrome present with cephalocaudal loss of subcutaneous fat in the face, neck, arms, and thorax, together with membranoproliferative glomerulonephritis, hypocomplementemia, and autoimmune disorders. Other forms of lipodystrophy can accompany distinct disease processes, such as progeria and the acquired partial lipodystrophy in patients with HIV/AIDS, but these are generally considered separately from the forms of congenital and acquired forms described above due to unique differences in presentation and pathophysiology.
This conventional categorization of the lipodystrophies encompasses a heterogeneous group of rare disorders. Nevertheless, metabolic abnormalities observed in many of these patients are also observed in patients with metabolic syndrome and T2DM, including insulin resistance, increased circulating levels of free fatty acids (6,7) and triglycerides (8), ectopic deposition of lipid in skeletal myocytes (9,10) and hepatocytes (11), and dysregulated secretion of adipocytokines (7,12). Indeed, the loss of fat may constitute the primary cause of these metabolic abnormalities, as illustrated by adipose tissue ablation in genetically manipulated mouse models, including A-ZIP/F-1 fatless mice (13) and Agpat2-null lipodystrophic mice (14). By way of illustration, after transplantation of normal subcutaneous adipose tissue into A-ZIP/F-1 fatless mice, redistribution of ectopic lipid in muscle and liver to the transplanted fat occurred, with a significant increase in insulin-mediated glucose uptake (13). These findings suggest that the lipodystrophies and T2DM share a state of defective triglyceride storage in adipose tissue, accompanied by ectopic accumulation of lipid in muscle and liver, contributing to insulin resistance and other abnormalities associated with cardiometabolic disease (15). Also, adipose tissue in patients with metabolic syndrome and T2DM is characterized by infiltration of macrophages, fibrosis, and production of proinflammatory cytokines (16,17). Lipodystrophy can also be accompanied by inflammation, as demonstrated in aP2-nSREBP-1c lipodystrophic mice, which exhibit proinflammatory cytokine tumor necrosis factor-α and interleukin-6 elevations of 2-fold and 10-fold, respectively (18). These observations led to the question whether more subtle forms or patterns of lipodystrophy could exist in insulin-resistant patients with metabolic syndrome or T2DM and contribute to pathophysiology.
In the current study, we describe patients with partial lipodystrophy of the limbs (PLL), which we believe represents a previously unrecognized phenotype of lipodystrophy occurring in patients with T2DM. The physical examination of these patients is characterized by symmetrical loss of subcutaneous fat in forearms and/or calves (or entire limbs) and acanthosis nigricans. They also manifest a profound degree of insulin resistance and marked elevations in hepatic transaminases (indicative of hepatic steatosis) and triglycerides, which are greater in severity than in patients with commonly occurring T2DM lacking PLL.
RESEARCH DESIGN AND METHODS
Subject selection
Patients with T2DM and PLL were identified by one of the authors (W.T.G.) in clinics sponsored by Divisions of Endocrinology or Nutrition Sciences at academic institutions during a 25-year period. Ten of these patients (∼half of the total) agreed to participate in metabolic studies on a research ward and constitute the current study population. Clinical characteristics are reported in Table 1. All patients with PLL in the study were females of various ethnic and racial identities. There are no reasons to suspect men cannot also be affected by T2DM and PLL, even though men may be more difficult to identify than women because of differences in subcutaneous fat. In fact, we encountered one such Hispanic man with symmetrical lipoatrophy of the forearms who did not agree to participate in this study. Nine of the patients with PLL in the study had overt T2DM, whereas one met the criteria for prediabetes. Five of the nine PLL patients with T2DM were treated with insulin plus metformin or a sulfonylurea and the remainder with oral hypoglycemic drugs. A single laboratory conducted the metabolic studies, assuring continuity of methods and comparability of data during this interval.
Table 1.
Characteristics of patients with T2DM with PLL compared with patients with common T2DM
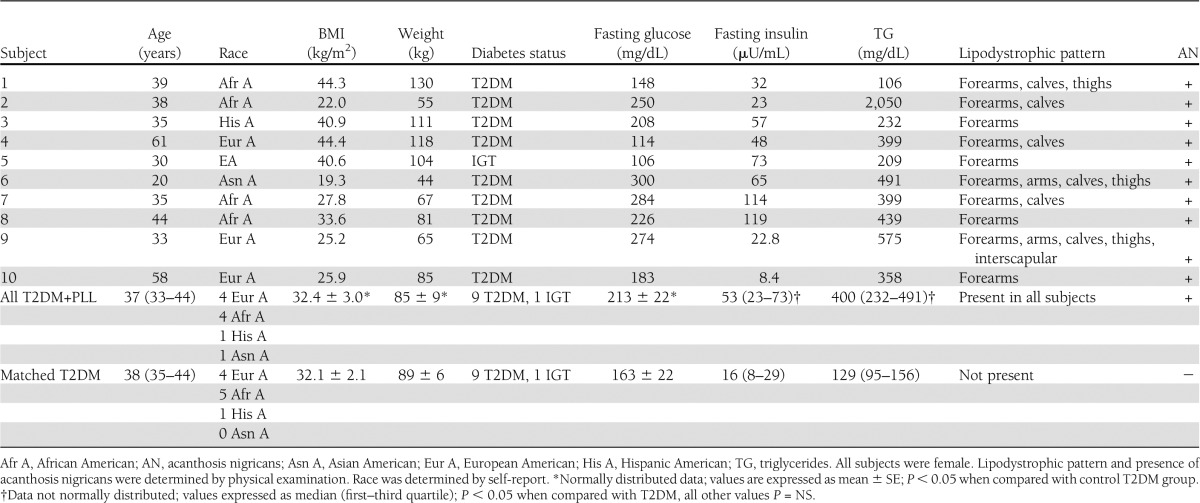
From a larger database of T2DM patients metabolically studied in the same laboratory, the patients with PLL were matched by age, BMI, fasting glucose, ethnicity, and diabetes status to 10 patients with commonly occurring T2DM who did not have clinically evident lipodystrophy. Comparative data in the study groups with and without PLL are also delineated in Table 1. All patients were withdrawn from any medicines used to treat diabetes or dyslipidemia for 1 to 3 weeks so that they could be studied in the untreated state. All patients denied positivity for HIV and explicitly denied that any other family members had the appearance or diagnosis of lipodystrophy to their knowledge. None of the patients engaged in heavy alcohol use (more than 2 drinks/day or 10 drinks/week), and orthostatic hypotension was excluded by sitting and standing blood pressure checks. Race was determined by self-report. This study was approved by the institutional review boards of the institutions, and written informed consent was obtained in all patients.
Protocol and glucose clamp studies
Patients were equilibrated on a metabolic ward on a diet consisting of 20% protein, 30% fat, and 50% carbohydrate for 3 days. Standard 75-g oral glucose tolerance tests were performed after an overnight fast. To assess glucose disposal rates (GDRs), hyperinsulinemic-euglycemic clamps were performed at a maximally effective insulin concentration, as previously described (19). A catheter was inserted into the brachial vein, and a calibrated syringe pump was used to administer regular insulin in a square wave at a rate of 200 mU/m2/min . This infusion provided steady-state serum insulin levels that were maximally effective for promoting glucose uptake in skeletal muscle in patients with T2DM and fully suppressing hepatic glucose production (19). To prevent hypokalemia, a solution of potassium phosphate was simultaneously infused. A variable-rate infusion of a 20% dextrose solution was used to maintain the plasma glucose level at 90 mg/dL for at least 3 h, with plasma glucose levels assessed every 5 min throughout the clamp. During the final three 20-min intervals, the mean glucose infusion rate was used to calculate maximal glucose uptake for each individual. Whole-body glucose uptake was calculated based on the glucose infusion rate corrected for changes in the glucose pool size, assuming a distribution volume of 19% body weight and a pool fraction of 0.65. Under these conditions, the glucose infusion rate in milligrams per minute is equal to the GDR, and the data were normalized to total body weight (kg) or body surface area (m2).
Statistical analysis
The Kolmogorov-Smirnov test was performed to test normality of the data. If normally distributed, the data were expressed as mean ± SE, and the Student t test was used to analyze whether differences between the patients with T2DM with and without PLL were statistically significant. If data were not normally distributed, values were presented as median (first–third quartile), and statistical significance was determined using the nonparametric Wilcoxon rank test. Results were controlled for any differences in age, race, and BMI. P values < 0.05 were considered significant. Statistical analysis was performed using SAS 9.2 software (SAS Institute, Cary, NC) and Microsoft Excel. Serum alanine aminotransaminase (ALT), aspartate aminotransaminase (AST), and alkaline phosphatase (ALP) values were expressed as a percentage of the clinical laboratory’s established upper limit of the normal reference range.
RESULTS
Characteristics of patients
The characteristics of patients with T2DM and PLL are summarized in Table 1. All patients were females, and the race/ethnicity distribution was four European Americans, four African Americans, one Asian American, and one Hispanic American. Nine patients had T2DM with variable severity of hyperglycemia, and one patient had prediabetes with impaired glucose tolerance (IGT). The age range was 20–61 years, with most patients in their 30s. As assessed by BMI (kg/m2), patients ranged from lean (<25) to overweight (25–29.9) or obese (>30). The lipodystrophy pattern was symmetrical in all patients and involved only the limbs. Loss of subcutaneous fat in the forearms only (4 patients) and forearms and calves (3 patients) were the most common presentations, with lipodystrophy involving forearms, calves, and thighs in one patient (Fig. 1.) and forearms, calves, thighs, and arms in two patients, with interscapular region involvement in one of these latter patients. With the loss of subcutaneous fat in the forearms and calves, we cannot rule out the possibility that there was a gain in fat above the level of lipoatrophy, for example, in the arms and thighs (i.e., when the arms and thighs were not affected by lipoatrophy). All patients had acanthosis nigricans. One patient aged 20 years was also diagnosed to have polycystic ovarian syndrome.
Figure 1.

Patient 1 is shown with arrows indicating lipodystrophy of the forearms, thighs, and legs.
The patients with PLL were matched with 10 patients without PLL based on T2DM status, race, age, and BMI. Accordingly, as reported in Table 1, subgroups with and without PLL both consisted of nine T2DM patients and one IGT patient, and there were no statistically significant differences in mean BMI (P = 0.94), age (P = 0.78), and fasting glucose levels (P = 0.19). In addition, the mean waist-to-hip ratio was equally elevated in both subgroups at 0.9, and mean blood pressures were comparable (data not shown; P = NS). However, the mean HbA1c was somewhat higher in the PLL patients (10.6 ± 0.6%) than in the control T2DM subgroup (8.0 ± 1.0%; P < 0.05). Importantly, the reported age of onset for T2DM was earlier in the patients with PLL than in the controls by more than a full decade (28.9 ± 4.8 vs. 39.9 ± 2.6 years, respectively; P < 0.05). Four patients were not cognizant of the lipodystrophy or thought it was a function of aging and did not have a clear notion regarding the onset of the PLL. Six patients related that they believed they began to notice signs referable to the PLL several years before the diagnosis of diabetes. None of the patients without PLL happened to have acanthosis nigricans, and the subgroups differed in other key clinical and metabolic parameters, as described below.
Insulin resistance: GDRs and fasting insulin levels
Patients with T2DM and PLL were markedly more insulin-resistant than their diabetic counterparts without PLL. First, the mean fasting insulin level was increased by more than threefold in diabetic patients with PPL than in patients without PLL (56 ± 12 vs. 18 ± 3 µU/mL, P < 0.05; Table 1). More importantly, maximal GDRs during hyperinsulinemic clamps were significantly lower in the PLL group (179.6 ± 29.14 vs. 404.4 ± 57.7 mg/min, P < 0.05). When normalized by body weight, GDR was reduced by 50% in T2DM with PLL compared with the control T2DM patients (2.3 ± 0.4 vs. 4.7 ± 0.7 mg/kg/min, P < 0.05; Fig. 2A) and by 53% when normalized by body surface area (96.7 ± 15.05 vs. 206.3 mg/m2/min, P < 0.05; data not shown). These GDR values represent the combination of insulin-mediated and non-insulin–mediated glucose uptake rates. To derive values for insulin-mediated glucose uptake, we subtracted 1.6 mg/kg/min, the previously determined rate of non-insulin–mediated glucose uptake at euglycemia in patients with diabetes and those without diabetes (20,21), from the measured GDR values normalized by weight (mg/kg/min) in each subject. The mean values for insulin-mediated glucose uptake are shown in Fig. 2B. Patients with T2DM and PLL had little or no insulin-mediated glucose uptake during the clamps (0.6 ± 0.4 mg/kg/min), and the mean value was reduced by 80% compared with the diabetes subgroup without PLL (3.1 ± 0.8 mg/kg/min, P < 0.05).
Figure 2.

Comparison of GDR and total triglycerides and cholesterol. A: GDRs were assessed using the hyperinsulinemic-euglycemic clamp technique and normalized per kilogram of body weight. Mean ± SE values were significantly lower in 10 patients with T2DM and PLL (T2DM+PLL) compared with 10 patients with common T2DM (2.26 ± 0.42 vs. 4.74 ± 0.77 mg/min/kg; P < 0.05). B: In each subject, the value for non-insulin–mediated glucose disposal (1.6 mg/min/kg; see Insulin resistance: GDRs and fasting insulin levels) was subtracted from the total GDR during the clamp to obtain the insulin-mediated GDR. The mean ± SE value for insulin mediated glucose uptake in T2DM+PLL was minimal and markedly reduced compared with common T2DM (0.64 ± 0.42 vs. 3.12 ± 0.77 mg/min/kg; P < 0.05). C: The mean ± SE value for triglycerides was increased in 10 patients with T2DM and PLL (T2DM+PLL) compared with 10 patients with common T2DM (543 ± 175 vs. 134 ± 28 mg/dL; P < 0.05). D: Mean ± SE levels of total cholesterol were statistically similar in comparing the T2DM groups with and without PLL (223 ± 14 vs. 184 ± 17 mg/dL; P = 0.10). Data in all panels were controlled for BMI, age, and race.
Serum triglycerides and cholesterol
Fasting triglyceride levels were elevated (i.e., >150 mg/dL) in 9 of the 10 patients with PLL, with values ranging from 106 to 2,050 mg/dL. As shown in Fig. 2C, the median triglyceride level was markedly elevated by more than threefold in the PLL patients compared with patients without PLL, with values of 400 (232–491) vs. 129 (95–156) mg/dL, respectively (P < 0.05). Levels of total cholesterol were statistically similar (P = 0.10) between the two subgroups (Fig. 2D).
Liver function tests
Values for ALT, AST, and ALP were expressed as a percentage of the laboratory’s upper limit of the normal reference range. Although liver function tests were not available in all patients, ALT and AST levels were elevated above the normal range in most of the patients with T2DM and PLL (Fig. 3), and mean values were significantly increased by more than twofold when compared with the T2DM patients without PLL (AST: 1.31 ± 0.20- vs. 0.56 ± 0.03-times the upper limit of normal, P < 0.05; and ALT: 1.83 ± 0.28- vs. 0.55 ± 0.05-times the upper limit of normal, P < 0.05; respectfully). In contrast, mean values for ALP were similar in the two subgroups (0.64 ± 0.11- vs. 0.62 ± 0.07-times the upper limit of normal, P = 0.87). The mean total bilirubin value was increased in the PLL group (0.98 ± 0.10 vs. 0.57 ± 0.05 IU/L, P < 0.05; Fig. 3); however, the mean value in the PLL subgroup remained within the normal range (0.1–1.1 IU/L).
Figure 3.

Measurement of liver function. Values for AST (A), ALT (B), and ALP (C) were expressed as percentage of upper limit of the normal reference range and for total bilirubin (D) as IU/L. All panels compare mean ± SE values in patients with T2DM and PLL (T2DM+PLL) and in patients with common T2DM. The mean was statistically increased in T2DM+PLL for AST, ALT, and total bilirubin (all P < 0.05) but not for ALP (P = NS). Data in all panels were controlled for BMI, age, and race.
CONCLUSIONS
We have described both a new form of lipodystrophy and a new phenotype of T2DM. Patients with T2DM and PLL are distinguished by 1) the presence of T2DM or prediabetes, 2) symmetrical lipodystrophy of the forearms, or forearms plus calves, or less commonly, whole limbs, and 3) acanthosis nigricans. Metabolically, these patients have profound insulin resistance, with little or no measurable insulin-mediated glucose disposal during hyperinsulinemic clamps. When compared with the control T2DM patients, the PLL patients will often have greater elevations of hepatic transaminases, suggesting more pronounced hepatic steatosis and more marked hypertriglyceridemia, and report the onset of T2DM a full decade earlier (at ∼29 years). T2DM with PLL can affect patients identifying with multiple ethnic and racial groups and appears to most commonly occur in women.
The pattern of fat loss is dissimilar from that reported for known forms of lipodystrophy with any consistency, and T2DM with PLL does not conform with entities that are conventionally categorized into the two-by-two, congenital versus acquired, generalized versus partial, diagnostic scheme. The lipodystrophy is certainly partial and appears to be acquired, based on the lack of family history and its onset in adulthood. In the conventional scheme, the only form of APL is Barraquer-Simons syndrome, with patients who present with cephalocaudal loss of subcutaneous fat in the face, neck, arms, and thorax, accompanied by expansion, not loss, of adipose tissue in the lower extremities. In addition, although these patients can develop a membranoproliferative glomerulonephritis, they seldom have T2DM and the other metabolic abnormalities associated with severe insulin resistance (1–5). FPL represents a heterogeneous group of autosomal-dominant disorders with onset of lipodystrophy around puberty. The pattern of lipodystrophy in the most common form, the Dunnigan variety, involves the limbs and the trunk, often accompanied by increased adipose in the neck and face. The Kobberling variety of FPL involves women, often with T2DM; however, lipodystrophy consistently involves the limbs and gluteal region, and patients report onset in childhood (22). These features of APL and FPL stand in contrast to patients with T2DM and PLL in whom lipodystrophy primarily involves the distal extremities (forearms or forearms plus calves), with no gluteal or anterior thorax involvement, in whom there is onset of lipodystrophy in adulthood without a family history, and in whom acanthosis nigricans and severe metabolic manifestations of insulin resistance are consistently observed. Although specific gene mutations have been reported in patients with FPL (LMNA, PPARγ, Caveolin-1) (2–5), the characteristics of T2DM and PLL do not conform to a monogenic or familial disease.
T2DM and PLL represent a distinct lipodystrophy phenotype based on current knowledge, but it is possible that with greater recognition of more subtle forms of APL and FPL and a more comprehensive understanding of the relationship between gene mutations and clinical manifestations, the conventional diagnostic scheme may no longer suffice. In this scenario, certain patients with APL, FPL, and T2DM with PLL may be found to share a common pathophysiology necessitating a reconstruction of the diagnostic framework. To illustrate this point, several studies have indicated that FPL may have a greater prevalence than previously thought due to lack of recognition of subtle forms. Patients in one study were screened for lipodystrophy and/or android body habitus, insulin resistance, or altered glucose tolerance. The investigators then tested all patients with one or more of these characteristics for known gene mutations causing FPL (23) and demonstrated that several patients with mutations and associated metabolic features lacked lipodystrophy on physical examination (23). A more recent study showed that screening all nonobese patients with T2DM for known FPL mutations increased its prevalence >400-fold from their baseline assessment (24). Both of these studies suggest strongly that there is an underestimation of prevalence and variability in clinical presentation for the partial lipodystrophies.
From another perspective, T2DM and PLL represent a new phenotype that further exemplifies the heterogeneous nature of T2DM. It is tempting to hypothesize that fat loss in T2DM and PLL is an extreme manifestation of adipose tissue dysfunction generally observed in patients with metabolic syndrome and T2DM, which features macrophage infiltration, fibrosis, inflammation, and dysregulated secretion of adipocytokines (eg, leptin, adiponectin, and resistin). In T2DM with and without PLL, adipose tissue defects lead to ectopic lipid accumulation in other tissues such as liver (25). These considerations suggest that patients with insulin resistance and T2DM may exist along a continuum, with some patients having more subtle or subclinical diminution of subcutaneous fat in the distal extremities. Given the history in several patients that the lipodystrophy preceded the diagnosis of diabetes and that one patient had prediabetes, any normoglycemic patient presenting with PLL should be monitored closely for the eventual development of diabetes. In any event, more detailed studies of patients with T2DM and PLL compared with patients without PLL are needed to elucidate pathophysiology, and additional reports are needed to more precisely define T2DM with PLL as a clinical entity.
Regardless, PLL in patients with T2DM has implications regarding clinical management. The profound degree of insulin resistance and lack of insulin-mediated glucose uptake should be considered in the choices and doses of antidiabetic medications, and careful evaluation and aggressive therapy for marked hypertriglyceridemia and steatohepatitis should be undertaken. Along these same lines, promising results have been observed with leptin replacement therapy in patients with several types of lipodystrophy (26–28). For example, a reduction in serum triglycerides and intrahepatic lipid content was demonstrated in patients with FPL after 6 months of metreleptin treatment (28). It remains to be seen whether pharmacological administration of leptin can ameliorate glycemic control, insulin resistance, hypertriglyceridemia, and hepatic steatosis in T2DM with PLL.
The current study cannot ascertain the prevalence of T2DM with PLL. Even so, partial PLL and the accompanying severity of metabolic abnormalities may not be rare. For the most part, patients in this study presented with T2DM and were initially suspected to have PLL by instinctively inspecting the forearms of each new patient during the introductory shaking of hands. Certainly, all T2DM patients with overt hypertriglyceridemia and hepatic transaminase elevations or with high insulin requirements should be examined closely for PLL. Thus, T2DM with PLL represents a previously unrecognized phenotype of lipodystrophy and of diabetes. These patients exhibit symmetrical lipodystrophy of the distal limbs, acanthosis nigricans, earlier onset T2DM, and marked insulin resistance with little insulin-mediated glucose uptake, as well as hypertriglyceridemia and hepatic transaminase elevations that are greater in severity than observed in patients with commonly occurring T2DM.
Acknowledgments
This work was supported by grants from the National Institutes of Health (DK-083562, DK-038764) to W.T.G, and the Merit Review program of the Department of Veterans Affairs to W.T.G. L.R.S. was supported by a Summer Research Fellowship for Medical Students provided by National Institute of Diabetes and Digestive and Kidney Diseases to the University of Alabama (UAB) Diabetes Research and Training Center (P60-DK-079626) and administered by the UAB Obesity Training Program (T32 DK-062710).
W.T.G. has received speaker honoraria from Merck & Co.; has served on advisory boards for Vivus, Daiichi-Sankyo, LipoScience, Janssen, and Alkermes; and has received research support from Amylin Pharmaceuticals, Inc. and Merck & Co. No other potential conflicts of interest relevant to this article were reported.
L.R.S. researched data and wrote the manuscript. F.G. performed the statistical analysis and reviewed and edited the manuscript. K.L. collected data for the control subjects and reviewed and edited the manuscript. W.T.G. identified the patients, performed metabolic studies, analyzed data, and wrote the manuscript. W.T.G. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank, for their support, the research core facilities of the UAB Diabetes Research and Training Center, the expert contribution of the metabolic study nurses Dana Golson and Penny Wallace, and the research volunteers.
Footnotes
See accompanying commentary, p. 2142.
References
- 1.Garg A. Lipodystrophies. Am J Med 2000;108:143–152 [DOI] [PubMed] [Google Scholar]
- 2.Garg A. Acquired and inherited lipodystrophies. N Engl J Med 2004;350:1220–1234 [DOI] [PubMed] [Google Scholar]
- 3.Garg A, Agarwal AK. Lipodystrophies: disorders of adipose tissue biology. Biochim Biophys Acta 2009;1791:507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab 2011;96:3313–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan JL, Oral EA. Clinical classification and treatment of congenital and acquired lipodystrophy. Endocr Pract 2010;16:310–323 [DOI] [PubMed]
- 6.DeFronzo RA. Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int J Clin Pract Suppl 2004;Oct:9–21 [DOI] [PubMed] [Google Scholar]
- 7.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes 2011;18:139–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adiels M, Olofsson SO, Taskinen MR, Borén J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol 2008;28:1225–1236 [DOI] [PubMed] [Google Scholar]
- 9.Forouhi NG, Jenkinson G, Thomas EL, et al. Relation of triglyceride stores in skeletal muscle cells to central obesity and insulin sensitivity in European and South Asian men. Diabetologia 1999;42:932–935 [DOI] [PubMed] [Google Scholar]
- 10.McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002;51:7–18 [DOI] [PubMed] [Google Scholar]
- 11.Yki-Järvinen H. Liver fat in the pathogenesis of insulin resistance and type 2 diabetes. Dig Dis 2010;28:203–209 [DOI] [PubMed] [Google Scholar]
- 12.Dyck DJ, Heigenhauser GJ, Bruce CR. The role of adipokines as regulators of skeletal muscle fatty acid metabolism and insulin sensitivity. Acta Physiol (Oxf) 2006;186:5–16 [DOI] [PubMed] [Google Scholar]
- 13.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem 2000;275:8456–8460 [DOI] [PubMed] [Google Scholar]
- 14.Agarwal AK, Sukumaran S, Cortés VA, et al. Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2(-/-) gene lipodystrophic mice. J Biol Chem 2011;286:37676–37691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frayn KN. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002;45:1201–1210 [DOI] [PubMed] [Google Scholar]
- 16.Varma V, Yao-Borengasser A, Rasouli N, et al. Muscle inflammatory response and insulin resistance: synergistic interaction between macrophages and fatty acids leads to impaired insulin action. Am J Physiol Endocrinol Metab 2009;296:E1300–E1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhargava P, Lee CH. Role and function of macrophages in the metabolic syndrome. Biochem J 2012;442:253–262 [DOI] [PubMed] [Google Scholar]
- 18.Herrero L, Shapiro H, Nayer A, Lee J, Shoelson SE. Inflammation and adipose tissue macrophages in lipodystrophic mice. Proc Natl Acad Sci U S A 2010;107:240–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garvey WT, Olefsky JM, Griffin J, Hamman RF, Kolterman OG. The effect of insulin treatment on insulin secretion and insulin action in type II diabetes mellitus. Diabetes 1985;34:222–234 [DOI] [PubMed] [Google Scholar]
- 20.Gottesman I, Mandarino L, Gerich J. Estimation and kinetic analysis of insulin-independent glucose uptake in human subjects. Am J Physiol 1983;244:E632–E635 [DOI] [PubMed] [Google Scholar]
- 21.Jumpertz R, Thearle MS, Bunt JC, Krakoff J. Assessment of non-insulin-mediated glucose uptake: association with body fat and glycemic status. Metabolism 2010;59:1396–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herbst KL, Tannock LR, Deeb SS, Purnell JQ, Brunzell JD, Chait A. Köbberling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care 2003;26:1819–1824 [DOI] [PubMed] [Google Scholar]
- 23.Decaudain A, Vantyghem MC, Guerci B, et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J Clin Endocrinol Metab 2007;92:4835–4844 [DOI] [PubMed] [Google Scholar]
- 24.Visser ME, Kropman E, Kranendonk ME, et al. Characterisation of non-obese diabetic patients with marked insulin resistance identifies a novel familial partial lipodystrophy-associated PPARγ mutation (Y151C). Diabetologia 2011;54:1639–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simha V, Garg A. Inherited lipodystrophies and hypertriglyceridemia. Curr Opin Lipidol 2009;20:300–308 [DOI] [PubMed] [Google Scholar]
- 26.Oral EA, Chan JL. Rationale for leptin-replacement therapy for severe lipodystrophy. Endocr Pract 2010;16:324–333 [DOI] [PubMed]
- 27.Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 2010;53:27–35 [DOI] [PubMed] [Google Scholar]
- 28.Simha V, Subramanyam L, Szczepaniak L, et al. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab 2012;97:785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]