

Abstract

A highly stereocontrolled total synthesis of the cytotoxic marine macrolide aplyronine C is described. The route exploits aldol methodology to install the requisite stereochemistry and features a crucial boron-mediated aldol coupling of an N-vinylformamide-bearing methyl ketone with a macrocyclic aldehyde to introduce the full side chain. The synthesis of two novel C21–C34 side chain analogs is also reported.
Actin, the most common protein in eukaryotic cells, is involved in numerous vital cellular functions including cell shape maintenance, division, locomotion, and adhesion.1 Actin dynamics are normally tightly controlled by a number of actin-binding proteins; dysregulation has been implicated in diseases including stroke, cystic fibrosis, and cancer.1a,2
Aplyronines A–H3 (Figure 1) comprise a family of actin-binding marine macrolides that can serve as small molecule mimics of actin-binding proteins. They were isolated in low yield (10–5–10–7% based on wet weight) from the Japanese sea hare Aplysia kurodai by Yamada and co-workers based on their potent cytotoxicity against HeLa-S3 cells.4 Notably, the antiproliferative efficacy of aplyronine A (1) has been demonstrated in vivo against P388 leukemia (T/C 545%, 0.08 mg/kg) and Lewis lung carcinoma (T/C 556%, 0.04 mg/kg), leading to its identification as a promising anticancer drug candidate.5 Aplyronine A forms a 1:1 complex with globular actin (G-actin, Kd = 100 nM), inhibiting polymerization, and also depolymerizes fibrous actin (F-actin).6 X-ray analysis of the actin–aplyronine A crystal structure,7 structure–activity relationship studies,8 and photoaffinity studies9 have highlighted the importance of the C24–C34 tail region in the strong actin depolymerizing activity. The mechanism of cytotoxicity for these macrolides remains unelucidated. Recent work has shown that aplyronine A causes caspase-dependent apoptosis with rapid disassembly of the actin cytoskeleton and dephosphorylation of focal adhesion kinase10 and has suggested an interaction between the aplyronine–actin complex and a secondary biomolecule such as Arp2/3.11
Figure 1.

Structures of selected aplyronines.
The aplyronines have elicited significant interest for their potent antitumor activities and novel actin-binding properties, as well as their unique structures.11−16 Moreover, they hold great potential for the development of biomolecular probes and novel actin-targeting therapeutic agents. It is a testament to the severe challenge presented by these complex polyketides that, despite considerable effort from several groups,12−16 only one prior total synthesis of aplyronines A–C has been achieved, as reported by Yamada and Kigoshi.16a−16c
In our previous work toward the aplyronines (Scheme 1), we reported the synthesis of an advanced macrocyclic intermediate 4(12a,12b) and β-ketophosphonate 5(12c) in pursuit of an aborted HWE fragment coupling strategy. Here, we describe the synthesis of a more suitable aldol coupling partner 6, enabling completion of the total synthesis of aplyronine C (2) and providing access to some novel C21–C34 tail analogs.
Scheme 1. Previous Work and Revised Strategy.
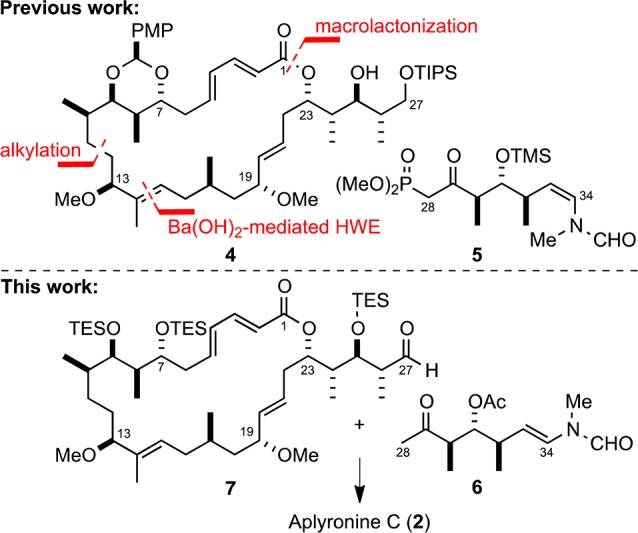
Our revised strategy hinges on a key aldol fragment coupling between the C1–C27 aldehyde 7, derived from previously synthesized macrocycle 4, with the (E)-N- methyl-N-vinylformamide-bearing methyl ketone 6. The strategic decision to incorporate the terminal (E)-vinylformamide into the coupling fragment was influenced by our recent total syntheses of reidispongiolide A17 and rhizopodin,18 and contrasts with more established approaches in which this sensitive moiety is introduced by a testing, late-stage condensation reaction with a highly functionalized C34 aldehyde.
The synthesis of the C28–C34 methyl ketone 6 is outlined in Scheme 2. It commenced with a Sn(II)-mediated aldol reacton19 between known (R)-Roche ester-derived ketone 8(12a) and acetaldehyde to generate the all syn aldol adduct 9 with high yield and selectivity (97%, 15:1 dr).20 A directed 1,3-anti reduction under Evans–Tishchenko conditions21 set the C31 stereocenter and concomitantly capped the C29 alcohol as the ester (10).22 Following silyl protection, the C29 and C33 alcohols were revealed to produce diol 11.
Scheme 2. Synthesis of C28–C34 Ketone 6.

Oxidation of diol 11 to the corresponding keto-aldehyde required that the primary and secondary alcohols be oxidized concurrently, lest intramolecular cyclization onto the nascent aldehyde form an undesired hemiacetal. This was achieved using a double Swern oxidation.23 Following our Wittig protocol for the synthesis of N-methyl-N-vinylformamides,24 the ylide of phosphonium salt 12 (LiHMDS) reacted selectively with the aldehyde to introduce the N-vinylformamide terminus in 13, predominantly in the (Z)-configuration (75% over two steps, 8:1 Z/E). Two further steps introduced the C31 acetate moiety; isomerization to (E)-vinylformamide 6 then proceeded smoothly in the presence of stoichiometric iodine under light-free conditions.24 While all β-acetoxy ketone intermediates were prone to elimination and required careful handling, introduction of the C31 acetate moiety at this early stage proved crucial. Attempted isomerization of fragments bearing a C31-OTES (13) or -OPMB group failed to provide the desired (E)-vinylformamide under a variety of conditions. The C28–C34 fragment 6 was thus accessed by an efficient 10 step sequence in 60% overall yield.
Aldehyde 7 was readily accessible in four steps from our previously reported macrocyclic intermediate 4 (Scheme 3). Accordingly, global deprotection with aqueous HF was followed by TES protection of the resultant tetraol. Selective unveiling of the C27 primary alcohol under mild conditions (THF/H2O/AcOH) and subsequent oxidation provided the desired aldehyde 7.
Scheme 3. Synthesis of C1–C27 Aldehyde 7.

With C1–C27 aldehyde 7 and C28–C34 ketone 6 in hand, attention turned to assembly of the full aplyronine backbone.25 Owing to the complexity of the aldehyde 7 and the acid and base sensitivity of methyl ketone 6, especially mild conditions were required. Initially, we rehearsed this crucial fragment coupling and endgame using 14 as a truncated model for aldehyde 7.26 Boron-mediated aldol coupling conditions proved uniquely effective, leading to two novel C21–C34 tail analogs for the aplyronines (15 and 16; Scheme 4).27
Scheme 4. C21–C34 Side Chain Analogs.

Application of the boron aldol coupling conditions to real aldehyde 7 successfully formed the desired C27–C28 bond (Scheme 5).17 After careful optimization, we were able to perform this delicate aldol coupling using c-Hex2BCl/Et3N for enolization of 6 at −10 °C followed by slow addition of the enolate (3 equiv) to aldehyde 7 at −78 °C. Following a mild, nonoxidative workup, β-hydroxy ketone 17 was obtained in 61% yield with good recovery of the excess ketone 6.
Scheme 5. Fragment Coupling.

Deoxygenation at C27 was achieved using a two-step procedure:17 the β-hydroxy ketone 17 was dehydrated with the Burgess reagent (Et3NSO2NCO2Me)28 to the corresponding enone, which was subsequently reduced in a 1,4-sense to ketone 18 using Stryker’s reagent.29 Under these conditions, elimination of the potentially labile β-acetoxy ketone was minimized, and complete selectivity for reduction of the enone over the sensitive α,β,γ,δ-unsaturated dienoate was obtained.
From this point, all that remained to obtain aplyronine C (2) was stereoselective reduction of the C29 ketone, installation of the appropriate C29 amino ester, and global deprotection. Screening reduction conditions on the model system26 revealed that ketone 19 exhibits moderate inherent diastereoselectivity for the desired C29 epimer (NaBH4, 2:1 dr; entry 1, Table 1). Attempts to enhance this selectivity using bulky reducing agents (entries 2–3) led primarily to formation of elimination-related byproducts. Luche conditions30 (entry 4) appeared more promising, giving an improved 7:1 dr but a disappointingly low yield. Ultimately, Zn(BH4)231 was found to provide the desired alcohol 20 in 10:1 dr and 77% yield (entry 5).
Table 1. Model Studies of C29 Reduction.

| entry | conditions | yield (%) | dr |
|---|---|---|---|
| 1 | NaBH4, MeOH, rt | 55 | 2:1 |
| 2 | l-selectride, THF, –78 °C | –a | – |
| 3 | LiAlH(Ot-Bu)3, THF, −10 °C → rt | –a | – |
| 4 | NaBH4, CeCl3·7H2O, MeOH, 0 °C | 34 | 7:1 |
| 5 | Zn(BH4)2, Et2O, 0 °C | 77 | 10:1 |
Slow reaction rate, forming predominantly elimination and decomposition products.
Pleasingly, this result transferred well to the real system 18 (Scheme 6). The desired diastereomer at C29 (21) was obtained with good selectivity by reduction with Zn(BH4)2 (90%, 10:1 dr). Esterification with (S)-N,N-dimethylalanine under Keck conditions32 (DCC, DMAP, CSA), as precedented by Yamada,16a was followed by global deprotection using HF•py and pyridine to provide (+)-aplyronine C (2). To our satisfaction all 1H and 13C NMR spectroscopic data for this synthetic material correlated with those reported for natural aplyronine C.3a
Scheme 6. Completion of Aplyronine C (2).

In conclusion, this highly stereocontrolled total synthesis of aplyronine C was completed in 28 steps (LLS) and 3.6% overall yield, by a route that is significantly shorter than the previous synthesis (45 steps LLS).16b Alcohol 21 represents an advanced common intermediate from which we can access other members of the aplyronine family. Studies toward these congeners and other novel analogs will be reported in due course.
Acknowledgments
This work was supported by the Dr. Herchel Smith Fellowship from Williams College (S.J.F.), the Croucher Foundation (L.Y.W.L.), the EPSRC (S.J.A.), and the Commonwealth Trust (S.B.B.). Mass spectrometry data were acquired at the EPSRC UK National Mass Spectrometry Facility at Swansea University.
Supporting Information Available
Experimental procedures, details of the model system, and full spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Reviews: ; a Allingham J. S.; Klenchin V. A.; Rayment I. Cell Mol. Life Sci. 2006, 63, 2119. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yeung K.–S.; Paterson I. Angew. Chem., Int. Ed. 2002, 41, 4632. [DOI] [PubMed] [Google Scholar]
- a Pantaloni D.; le Clainche C.; Carlier M. F. Science 2001, 292, 1502. [DOI] [PubMed] [Google Scholar]; b Pollard T. D.; Borisy G. G. Cell 2003, 112, 453. [DOI] [PubMed] [Google Scholar]
- a Yamada K.; Ojika M.; Ishigaki T.; Yoshida Y.; Ekimoto H.; Arakawa M. J. Am. Chem. Soc. 1993, 115, 11020. [Google Scholar]; b Yamada K.; Ojika M.; Kigoshi H.; Suenaga K. In Drugs from the Sea; Fusetani N., Ed.; Karger: Basel, Switzerland, 2000; pp 59–73. [Google Scholar]; c Ojika M.; Kigoshi H.; Suenaga K.; Imamura Y.; Yoshikawa K.; Ishigaki T.; Sakakura A.; Mutou T.; Yamada K. Tetrahedron 2012, 68, 982. [Google Scholar]
- Aplyronine A: IC50 = 0.45 nM; aplyronine C: IC50 = 22.4 nM; aplyronine D: IC50 = 0.071 nM against HeLa-S3 cells (ref (3)).
- Crews P.; Gerwick W. H.; Schmitz F. J.; France D.; Bair K. W.; Wright A. E.; Hallock Y. Pharm. Biol. 2003, 41, 39. [Google Scholar]
- Saito S.; Watabe S.; Ozaki H.; Kigoshi H.; Yamada K.; Fusetani N.; Karaki H. J. Biochem. (Tokyo) 1996, 120, 552. [DOI] [PubMed] [Google Scholar]
- Hirata K.; Muraoka S.; Suenaga K.; Kuroda T.; Kato K.; Tanaka H.; Yamamoto M.; Takata M.; Yamada K.; Kigoshi H. J. Mol. Biol. 2006, 356, 945. [DOI] [PubMed] [Google Scholar]
- Kigoshi H.; Suenaga K.; Takagi M.; Akao A.; Kanematsu K.; Kamei N.; Okugawa Y.; Yamada K. Tetrahedron 2002, 58, 1075. [Google Scholar]
- Kuroda T.; Suenaga K.; Sakakura A.; Handa T.; Okamoto K.; Kigoshi H. Bioconjugate Chem. 2006, 17, 524. [DOI] [PubMed] [Google Scholar]
- a Kita M.; Yoneda K.; Hirayama Y.; Yamagishi K.; Saito Y.; Sugiyama Y.; Miwa Y.; Ohno O.; Morita M.; Suenaga K.; Kigoshi H. ChemBioChem 2012, 13, 1754. [DOI] [PubMed] [Google Scholar]; b Ohno O.; Morita M.; Kitamura K.; Teruya T.; Yoneda K.; Kita M.; Kigoshi H.; Suenaga K. Bioorg. Med. Chem. Lett. 2013, 23, 1467. [DOI] [PubMed] [Google Scholar]
- a Kita M.; Hirayama Y.; Sugiyama M.; Kigoshi H. Angew. Chem., Int. Ed. 2011, 50, 9871. [DOI] [PubMed] [Google Scholar]; b Kita M.; Hirayama Y.; Yamagishi K.; Yoneda K.; Fujisawa R.; Kigoshi H. J. Am. Chem. Soc. 2012, 134, 20314. [DOI] [PubMed] [Google Scholar]
- a Paterson I.; Cowden C. J.; Woodrow M. D. Tetrahedron Lett. 1998, 39, 6037. [Google Scholar]; b Paterson I.; Woodrow M. D.; Cowden C. J. Tetrahedron Lett. 1998, 39, 6041. [Google Scholar]; c Paterson I.; Blakey S. B.; Cowden C. J. Tetrahedron Lett. 2002, 43, 6005. [Google Scholar]
- a Calter M. A.; Guo X. Tetrahedron 2002, 58, 7093. [Google Scholar]; b Calter M. A.; Zhou J. Tetrahedron Lett. 2004, 45, 4847. [Google Scholar]
- Marshall J. A.; Johns B. A. J. Org. Chem. 2000, 65, 1501. [DOI] [PubMed] [Google Scholar]
- a El-Awa A.; Fuchs P. Org. Lett. 2006, 8, 2905. [DOI] [PubMed] [Google Scholar]; b Noshi M. N.; El-Awa A.; Torres E.; Fuchs P. L. J. Am. Chem. Soc. 2007, 129, 11242. [DOI] [PubMed] [Google Scholar]; c Hong W. P.; Noshi M. N.; El-Awa A.; Fuchs P. L. Org. Lett. 2011, 13, 6342. [DOI] [PubMed] [Google Scholar]
- a Kigoshi H.; Ojika M.; Ishigaki T.; Suenaga K.; Mutou T.; Sakakura A.; Ogawa T.; Yamada K. J. Am. Chem. Soc. 1994, 116, 7443. [Google Scholar]; b Suenaga K.; Ishigaki T.; Sakakura A.; Kigoshi H.; Yamada K. Tetrahedron Lett. 1995, 36, 5053. [Google Scholar]; c Kigoshi H.; Suenaga K.; Mutou T.; Ishigaki T.; Atsumi T.; Ishiwata H.; Sakakura A.; Ogawa T.; Ojika M.; Yamada K. J. Org. Chem. 1996, 61, 5326. [Google Scholar]; d Kobayashi K.; Fujii Y.; Hayakawa I.; Kigoshi H. Org. Lett. 2011, 13, 900. [DOI] [PubMed] [Google Scholar]
- Paterson I.; Ashton K.; Britton R.; Cecere G.; Chouraqui G.; Florence G. J.; Knust H.; Stafford J. Chem.—Asian J. 2008, 3, 367. [DOI] [PubMed] [Google Scholar]
- Dalby S. M.; Goodwin-Tindall J.; Paterson I.. Angew. Chem., Int. Ed. 2013, in press [DOI] [PubMed]
- Paterson I.; Tillyer R. D. Tetrahedron Lett. 1992, 33, 4233. [Google Scholar]
- The C29 configuration was confirmed by Mosher ester analysis; see the Supporting Information.
- Evans D. A.; Hoveyda A. H. J. Am. Chem. Soc. 1990, 112, 6447. [Google Scholar]
- The relative configuration was confirmed by NMR analysis of the corresponding acetonide; see the Supporting Information.
- Omura K.; Swern D. Tetrahedron 1978, 34, 1651. [Google Scholar]
- Paterson I.; Cowden C. J.; Rahn V. S.; Woodrow M. D. Synlett 1998, 915. [Google Scholar]
- As outlined in Scheme 1, our synthetic plan had initially anticipated forging the C27–C28 bond via an HWE reaction between aldehyde 7 and β-ketophosphonate 5. While Masamune–Roush conditions (LiCl, DBU) were successful on a model system (ref (12c)), attempts with real aldehyde 7 led only to β-elimination of the C25 silyl ether to give the corresponding enal.
- Attempted formation of lithium enolates of such N-vinylformamide-bearing ketones resulted only in decomposition. For full details of the model fragment coupling and endgame, see the Supporting Information.
- Perrins R. D.; Cecere G.; Paterson I.; Marriott G. Chem. Biol. 2008, 15, 287. [DOI] [PubMed] [Google Scholar]
- Burgess E. M.; Penton H. R.; Taylor E. A. J. Org. Chem. 1973, 38, 26. [Google Scholar]
- Mahoney W. S.; Brestensky D. M.; Stryker J. M. J. Am. Chem. Soc. 1988, 110, 291. [Google Scholar]
- a Luche J. L. J. Am. Chem. Soc. 1978, 100, 2226. [Google Scholar]; b Gemal A. L.; Luche J. L. J. Am. Chem. Soc. 1981, 103, 5454. [Google Scholar]
- Oishi T.; Nakata T. Acc. Chem. Res. 1984, 17, 338. [Google Scholar]
- Boden E. P.; Keck G. E. J. Org. Chem. 1985, 50, 2394. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.