

Abstract
AAA+ proteases, such as Escherichia coli Lon, recognize protein substrates by binding to specific peptide degrons and then unfold and translocate the protein into an internal degradation chamber for proteolysis. For some AAA+ proteases, attaching specific degrons to the N- or C-terminus of green fluorescent protein (GFP) generates useful substrates, whose unfolding and degradation can be monitored by loss of fluorescence, but Lon fails to degrade appropriately tagged GFP variants at a significant rate. Here, we demonstrate that Lon catalyzes robust unfolding and degradation of circularly permuted variants of GFP with a β20 degron appended to the N terminus or a sul20 degron appended to the C terminus. Lon degradation of non-permuted GFP-sul20 is very slow, in part because the enzyme cannot efficiently extract the degron-proximal C-terminal β-strand to initiate denaturation. The circularly permuted GFP substrates described here allow convenient high-throughput assays of the kinetics of Lon degradation in vitro and also permit assays of Lon proteolysis in vivo.
Keywords: ATP-dependent protease, degradation tag, enzyme-mediated protein unfolding
Introduction
AAA+ proteases are molecular machines that convert the chemical energy of ATP binding and hydrolysis into mechanical work that is used to unfold and translocate a protein substrate through the axial pore of a hexameric ring and into a sequestered chamber for degradation (for review see Sauer et al., 2004; Baker and Sauer, 2006; Striebel et al., 2009; Sauer and Baker, 2011). Architecturally, the Lon hexamer is one of the simplest AAA+ proteases, as its family-specific N-terminal domain, its AAA+ ATPase module and its peptidase domain are all connected in a single polypeptide chain. In bacteria, archaea and endosymbiotic organelles, Lon plays important roles in protein quality control by degrading misfolded or damaged proteins and also degrades native proteins that are no longer needed or must be removed for regulatory purposes (Torres-Cabassa and Gottesman, 1987; Gonzalez et al., 1998; Striebel et al., 2009). Lon is also a promising therapeutic target, because it is required for the virulence of several pathogenic bacteria, and inhibition of the human mitochondrial enzyme causes apoptosis of lymphoma cells (Robertson et al., 2000; Ingmer and Brøndsted, 2009; Yang et al., 2011; Bernstein et al., 2012).
Like all AAA+ proteases, Lon recognizes protein substrates via degrons or degradation tags, which can be as simple as specific sequences at the N or C terminus (Sauer and Baker, 2011). Several degrons for Escherichia coli Lon have been identified, including a 20 amino acid sequence within β-galactosidase (called β20), which becomes accessible to Lon only in the unfolded protein, and a sequence from the C-terminus of the cell-division inhibitor SulA (called sul20) (Higashitani et al., 1997; Gonzalez et al., 1998; Gur and Sauer, 2008). Attachment of the sul20 and β20 sequences to model substrates leads to their degradation by E.coli Lon. Interestingly, proteins bearing a C-terminal sul20 degron are degraded with a higher maximal velocity than otherwise identical substrates tagged with a C-terminal β20 degron, suggesting that degron identity regulates Lon activity in some manner (Gur and Sauer, 2009). The β20 sequence also functions as an N-terminal or internal degradation tag for Lon. Although, it is not known how Lon binds the sul20 or β20 degrons, studies with other AAA+ proteases have shown that some degrons bind in the axial pore of the hexameric ring, where they are engaged by the translocation machinery of the enzyme. Subsequent translocation of the degradation tag creates a pulling force when the attached native protein cannot enter the narrow axial channel, eventually leading to unfolding of the protein substrate. Because unfolding is an inherently mechanical process, its rate depends both on the pulling force that the enzyme can exert and on the stability of the local protein structure adjacent to the degradation tag (Lee et al., 2001; Kenniston et al., 2003, 2004).
Green fluorescent protein (GFP) has been an extremely valuable substrate for studies of many AAA+ proteases, as the fluorescent chromophore formed by cyclization and oxidation of residues 65–67 is protected from solvent quenching by the β-barrel structure of the native protein (Heim et al., 1994; Ormö et al., 1996). Enzymatic unfolding and degradation exposes the chromophore to solvent, quenching its fluorescence. Unfortunately, Lon degrades C- and N-terminally tagged variants of GFP extremely slowly (Choy et al., 2007; Gur and Sauer, 2008; Gur et al., 2012), which has hindered mechanistic studies in vitro and in vivo. Here, we demonstrate that Lon can unfold and degrade circularly permuted variants of GFP with appropriate degrons at either the N or C terminus. These substrates allow rapid, high-throughput kinetic analysis of Lon degradation in vitro and can also be used to monitor Lon degradation in vivo. We also show that Lon cannot efficiently extract the degron-tagged C-terminal strand from the β-barrel of non permuted GFP, a substrate that resists Lon degradation.
Materials and methods
Protein cloning, expression and purification
Variants of superfolder GFP (Pédelacq et al., 2006) were cloned into a pCOLADuet-1 plasmid vector with an N-terminal MGSH6SLEVLFQGPGS tag that included a PreScission protease site (Nager et al., 2011). For circular permutations, the normal N and C-termini of GFP were connected by a GGTGGS linker (Baird et al., 1999; Reeder et al., 2010; Nager et al., 2011). The sul20 degron (ASSHATRQLSGLKIHSNLYH) was added to the C-terminus of superfolder GFP variants and the β20 degron (QLRSLNGEWRFAWFPAPEAV) was inserted after residues MG in the N-terminal tag by polymerase chain reaction cloning. The variant with the thrombin cleavage site contained the sequence GGTEGSLVPRGSGESGGS (thrombin site in bold; flanking sequences introduced to limit steric hindrance) inserted between NEK235 and 236RDH, where the residue numbers refer to the sequence of superfolder GFP-sul20. The underlined residues in the GNILGHKLEYNLEASSHAT sequence of cp6-sul20 were deleted in the cp6-Δ8-sul20 variant.
Escherichia coli Lon was expressed from a pBAD33 over-expression vector as described (Gur and Sauer, 2009). Briefly, cells were grown at 37°C until OD600 ∼1, induced with 0.2% arabinose at 37°C for 3.5 h, harvested and resuspended in buffer A [100 mM potassium phosphate (pH 6.5), 1 mM DTT, 1 mM EDTA and 10% glycerol]. After lysis by sonication, insoluble material was removed by high-speed centrifugation, and 2 µl of benzonase (250 U/µl, Sigma) was added to the supernatant, which was incubated on ice for 20 min. The lysate was mixed with P11 phosphocellulose resin (Whatman) equilibrated in buffer A, and the resin was washed twice with buffer A and twice with an otherwise identical buffer containing 200 mM potassium phosphate (pH 6.5). Lon was eluted from the P11 resin with buffer B [400 mM potassium phosphate (pH 6.5), 1 mM DTT, 1 mM EDTA and 10% glycerol], and then chromatographed on a S300 size-exclusion column equilibrated in 50 mM HEPES (pH 7.5), 2 M NaCl and 1 mM DTT. The peak fractions of Lon from this column were >95% pure as assayed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and were buffer exchanged into storage buffer (50 mM HEPES (pH 7.5), 150 mM NaCl, 10 µM EDTA, 1 mM DTT and 10% glycerol) and frozen at –80°C.
Superfolder GFP variants were expressed and purified largely as described (Nager et al., 2011). Cells harboring over-expression plasmids were grown at 37°C to OD600 ∼1, induced with 0.5 mM isopropyl β-d-thiogalactoside (IPTG) at room temperature for 3.5 h, harvested by centrifugation and lysed by sonication. After an initial step of Ni++-NTA (Qiagen) affinity chromatography, material was bound to a MonoQ column (GE Healthcare) equilibrated in 25 mM Tris (pH 8.0), 25 mM NaCl, 1 mM DTT and 10 µM EDTA, and eluted using a linear gradient in the same buffer to 500 mM NaCl over 20 column volumes. The peak fractions of degron-tagged superfolder GFP variants were >90% pure as assayed by SDS-PAGE and were buffer exchanged into 50 mM HEPES (pH 7.5), 150 mM NaCl, 10 µM EDTA, 1 mM DTT and 10% glycerol, and frozen at –80°C.
Biochemical assays
Unless noted, biochemical assays were performed at 37°C in 25 mM Tris (pH 8.0), 100 mM KCl, 10 mM MgCl2. ATPase assays, monitored by absorbance at 340 nm, also contained 5 mM DTT, 2 mM ATP, 1 mM NADH, lactate dehydrogenase (10 U/ml), an ATP-regeneration system—20 mM phosphoenolpyruvate (Sigma) and 10 U/ml of rabbit muscle pyruvate kinase (Sigma)—and MgCl2, which had been warmed to 37°C, was added last to initiate the reaction (Nørby, 1988). Degradation assays contained supplemental 1 mM DTT, 2 mM ATP and an ATP-regeneration system, in addition to E.coli Lon and protein substrates. For degradation reactions assayed by fluorescence, reaction mixtures without ATP were incubated in 96-well flat bottom, 1/2 area plates (Corning) until there was no change in GFP fluorescence caused by thermal equilibration (∼15 min), and degradation was initiated by addition of ATP. For degradation assays monitored by SDS PAGE, 10 µl aliquots were taken at the specified time points and quenched by addition of 3.3 µl of 8% SDS, 250 mM Tris pH 6.8, 40% glycerol, 160 mM DTT, and 0.05% bromophenol blue.
To monitor Lon-mediated tail clipping, cp6-sul20 (50 µM) was incubated with Lon (1 µM hexamer) for 2 h at 37°C, the sample was loaded onto a nickel spin column (Qiagen), and the column was washed with 50 mM HEPES (pH 7.5), 150 mM NaCl, 20 mM imidazole, 10 µM EDTA, 1 mM DTT and 10% glycerol and then eluted with a similar buffer containing 250 mM imidazole. The eluted material from this experiment and purified substrate proteins were submitted for LC-MS analysis using a QSTAR Elite quadrupole-time-of-flight mass spectrometer. Approximately 3 picomoles of sample were loaded onto a reversed phase protein trap, and the sample was desalted on-line and eluted isocratically. Deconvolution of the electrospray data to generate molecular weight spectra was performed with the BioAnalyst software included with the QSTAR Elite data system.
To prepare the split GFP-sul20 substrate, treatment of the sample with 10 U/ml of thrombin (GE Healthcare) for 2 h at 37°C in storage buffer resulted in ∼95% cleavage as assayed by SDS-PAGE, and the split substrate was immediately used for Lon extraction/degradation assays. For the SDS-PAGE assay of degradation, quenched time points were electrophoresed on a 4–20% gradient gel, which was subsequently stained with Coomassie blue. For strand-extraction assays monitored by fluorescence, reactions were performed in a fluorescent plate reader as described above.
Degradation in vivo
The lon– E.coli strain ER2566 (New England Biolabs) was transformed with a pCOLAduet vector encoding cp6-sul20 under transcriptional control of a T7 promoter and/or with pBAD33 encoding wild-type Lon under transcriptional control of the araC promoter. Cells containing one or both plasmids were grown in M9 minimal media with appropriate antibiotics (10 µg/ml chloramphenicol and/or 50 µg/ml kanamycin) at 37°C until OD600 ∼0.3, 1 mM IPTG was added to induce cp6-sul20, and the temperature was dropped to 30°C to allow for more efficient chromophore maturation. After 2 h of CP6-sul20 expression, Lon expression was induced by adding arabinose to a final concentration of 0.2%. At this time, 100 µl of culture was transferred to a corning 96-well 1/2 area flat bottom plate in a SpectraMax M5 plate reader and fluorescence (467 nm excitation; 511 nm emission) was measured at 37°C with shaking of the plate between measurements. Assays were performed in triplicate.
For fluorescence-activated cell sorting (FACS) analysis, E.coli strains W3110 (lon+) and W3110 lon::kan (lon–) were transformed with pCOLAduet encoding cp6-sul20, and grown at 37°C. At OD600 ∼0.3, 1 mM IPTG was added to induce cp6-sul20 expression and the temperature was lowered to 30°C to facilitate more efficient chromophore maturation. After 90 min, a 100 µl aliquot of cells was taken, pelleted in a microcentrifuge and then resuspended in 70% cold ethanol to fix the cells. Samples were stored at –20°C until FACS analysis, and then pelleted, resuspended in phosphate-buffered saline (Boston Bioproducts) at a final concentration of 107 cells/ml, and subjected to FACS on a BD FACScan instrument. Data were collected as phycoerythrin fluorescence versus GFP fluorescence to eliminate auto-fluorescence. ER2566 cells without GFP were used as a negative control to determine the lower limit for the GFP-positive gate.
Results
Lon degrades circularly permuted GFP substrates
For all of the studies presented here, substrates were variants of superfolder GFP, some of which were circularly permuted to place β-strand 6 (cp6) or β-strand 7 (cp7) at the C-terminal end of the 11-stranded β-barrel (Fig. 1A). Each variant had an N-terminal His6 tag and either a β20 degron preceding the His6 sequence, a β20 degron at the C terminus or a sul20 degron at the C terminus of the protein. Because prior studies showed that circular permutation of superfolder GFP enhanced degradation by ClpXP (Nager et al., 2011), we sought to determine if circular permutation would also facilitate degradation of GFP by E.coli Lon.
Fig. 1.

(A) Cartoon representation of the order of secondary structure elements in superfolder GFP and the circularly permuted cp6 and cp7 variants. The N terminus (N) and C terminus (C) of each protein is marked. (B) Proteolysis of degron-tagged variants of the cp6 and cp7 substrates (10 µM) by Lon6 (0.3 µM) was monitored by changes in fluorescence (excitation 467 nm; emission 511 nm). Fluorescence values were normalized by dividing by the time-zero fluorescence. (C) SDS-PAGE assay of degradation of cp6-sul20 (20 µM) by Lon6 (0.6 µM). Following electrophoresis, the gel was stained with Coomassie Blue. (D) Substrate dependence of Lon6 (0.15 µM) degradation of the cp6-sul20, cp7-sul20, β20-cp6 and β20-cp7 proteins. The solid lines are fits to the Hill form of the Michaelis–Menten equation (rate = Vmax/(1 + (KM/[S])n). Values are shown as means ± SEM (N = 3). (E) Dependence on the concentration of substrate proteins of the rate of hydrolysis of ATP (2 mM) by Lon6 (0.15 µM). The solid lines are fits to a Hill equation, rate = basal + amp/(1 + (KM/[S])n), where Vmax = basal + amp. Values are shown as means ± SEM (N ≥ 3).
We constructed and purified nine substrates: β20-GFP, β20-cp6, β20-cp7, GFP-β20, cp6-β20, cp7-β20, GFP-sul20, cp6-sul20 and cp7-sul20. As assayed by loss of native fluorescence, 0.3 µM Lon catalyzed robust degradation of 10 µM concentrations of β20-cp6, β20-cp7, cp6-sul20, cp7-sul20, and slow degradation of the same concentrations of cp6-β20 and cp7-β20 (Fig. 1B). Under the same assay conditions, we observed no change in the fluorescence of β20-GFP, GFP-β20 or GFP-sul20 (not shown), indicating that these non-permuted proteins are extremely poor substrates for Lon degradation. For the four best substrates, Lon degradation of β20-cp6 and β20-cp7 proceeded to a greater extent than degradation of cp6-sul20 and cp7-sul20, which effectively ceased with ∼30% of the initial fluorescence remaining. An SDS-PAGE assay also showed incomplete degradation of cp6-sul20 (Fig. 1C), which was not improved by adding additional ATP, additional Lon or overnight incubation (not shown). We return to the issue of incomplete degradation below.
To determine steady-state kinetic parameters for Lon degradation of the four substrates that were degraded most rapidly, we determined initial rates as a function of the concentration of the β20-cp6, β20-cp7, cp6-sul20 or cp7-sul20 substrates and fit them to the Hill form of the Michaelis–Menten equation (Fig. 1D). KM, Vmax and n (the Hill constant) values for each substrate are listed in Table I. In terms of Vmax values, the substrate rank from highest to lowest was cp6-sul20 > β20-cp6 > β20-cp7 > cp7-sul20 with an ∼5-fold difference between the fastest (∼3.6 min−1 Lon6−1) and slowest (∼0.70 min−1 Lon6−1) rates. For the substrates with C-terminal degrons, the GuHCl concentrations required for 50% unfolding were ∼1.6 M for cp6-sul20, ∼1.8 M for cp7-sul20 and ∼3.8 M for GFP-sul20 (not shown). Thus, the least stable substrates are most susceptible to Lon degradation. The KM's for cp6 or cp7 substrates with the same degron varied, in some cases by as much as 4.5-fold (Table I), indicating that factors in addition to simple degron binding by Lon determine this kinetic parameter.
Table I.
Steady-state kinetic parameters for protein degradation and ATP hydrolysis by E.coli Lon. The error is that of non-linear-least-squares fitting. ATP per substrate was calculated by dividing Vmax for ATP hydrolysis by Vmax for protein degradation
| Substrate | Proteolysis Vmax (min−1 Lon6−1) | Proteolysis apparent KM (µM) | Proteolysis Hill constant | ATPase Vmax (min−1 Lon6−1) | ATPase apparent KM (µM) | ATPase Hill constant | ATP per substrate |
|---|---|---|---|---|---|---|---|
| cp6-sul20 | 3.6 ± 0.2 | 13 ± 2 | 1.2 ± 0.1 | 217 ± 16 | 9.3 ± 1.3 | 1.2 ± 0.1 | 60 |
| β20-cp6 | 2.3 ± 0.3 | 21 ± 6 | 1.2 ± 0.2 | 143 ± 5 | 2.6 ± 0.2 | 1.7 ± 0.1 | 62 |
| cp7-sul20 | 0.70 ± 0.03 | 2.8 ± 0.3 | 1.6 ± 0.3 | 130 ± 22 | 5.2 ± 2.4 | 0.9 ± 0.2 | 186 |
| β20-cp7 | 1.2 ± 0.1 | 8 ± 1 | 1.6 ± 0.3 | 130 ± 14 | 4.8 ± 1.0 | 1.3 ± 0.2 | 108 |
| cp6-Δ8-sul20 | 3.3 ± 0.1 | 13 ± 1 | 1.4 ± 0.1 | 215 ± 50 | 6.0 ± 3.0 | 1.1 ± 0.3 | 65 |
We also varied the concentration of each GFP variant and determined the effects on the steady-state rate of ATP hydrolysis by Lon (Fig. 1E, Table I). For the β20-cp6 substrate, the concentration at which half-maximal ATPase stimulation was observed was substantially lower than the KM for degradation, as previously observed for some other Lon substrates (Gur and Sauer, 2009). The energetic efficiency of Lon degradation of the circularly permuted substrates at saturating concentrations varied, with degradation of cp6-sul20 and β20-cp6 requiring hydrolysis of an average of ∼60 ATPs, β20-cp7 requiring ∼110 ATPs and cp7-sul20 requiring ∼190 ATPs (Table I). Because the energetic cost of translocation of the sul20-tagged substrates should be similar to each other, the ∼3-fold higher energetic cost of degrading cp7-sul20 compared with cp6-sul20 is likely to reflect an increased energetic cost of Lon unfolding. There was no clear correlation between maximum ATPase stimulation and degron identity, unlike the situation observed previously for unfolded substrates (Gur and Sauer, 2009), but this result could simply reflect a more complicated dependence on a combination of degron identity and other features of our substrates.
Tail clipping during Lon proteolysis prevents processive degradation of some molecules
The C-terminal residues of the sul20 degron are known to be important determinants of Lon recognition and degradation (Ishii and Amano, 2001; Gur and Sauer, 2009). It is possible, therefore, that proteolytic clipping of the sul20 degron accounts for the ∼30% of cp6-sul20 and cp7-sul20 proteins that are not degraded. In principle, tail clipping could occur during purification or during the degradation assay in a Lon-dependent manner. Electrospray mass spectrometry of the purified substrates showed 10–15% quantities of tail-clipped species for the cp7-sul20 substrate but effectively no tail-clipped species for the cp6-sul20 substrate (Fig. 2A). To test if tail clipping of cp6-sul20 occurred during Lon degradation, we allowed degradation of this substrate to proceed for 2 h and purified the undegraded His6-tagged substrate by Ni++-NTA chromatography (Fig. 2B). Electrospray mass spectrometry revealed relatively little remaining full-length substrate but a mixture of species with masses expected for removal of 2, 3 or 11 C-terminal amino acids (Fig. 2C). Thus, it appears that Lon-dependent clipping of these residues in some substrates precludes further efficient degradation.
Fig. 2.

(A) Deconvoluted electrospray ionization mass spectra of the purified cp6-sul20 (top) and cp7-sul20 (bottom) proteins. Full-length cp6-sul20 with a mature chromophore but lacking the N-terminal methionine (residue 2–287m) has an expected mass of 31 800.8 Da. Prior to chromophore maturation (2–287i), the expected mass is 20 Da higher. Full-length cp7-sul20 with a mature chromophore but lacking the N-terminal methionine (residues 2–291m) has an expected mass of 32 274.3 Da. Small amounts of proteins missing two (ΔYH) or three residues (ΔLYH) from the C-terminal end of the sul20 degron were observed in the cp7-sul20 but not the cp6-sul20 preparation. (B) The cp6-sul20 protein (50 µM), which contains an N-terminal His6 tag, was incubated with Lon6 (1 µM) for 2 h at 37°C and then purified by Ni++-affinity chromatography. (C) Electrospray ionization mass spectrometry of cp6-sul20 after 2 h of Lon degradation and Ni++-affinity purification revealed substantial tail clipping that removes 2 (ΔYH), 3 (ΔLYH) or 11 (ΔSGLKIHSNLYH) C-terminal residues from the sul20 degron. (D) Degradation of cp6-sul20 or cp6-Δ8-sul 20 (0.5 µM each) by Lon6 (2 µM) at 37°C. Degradation was monitored by loss of 511 nm fluorescence after excitation at 467 nm. Fluorescence was normalized to an initial value of 1.
Tail clipping of the cp6-sul20 substrate by Lon supports a model in which the sul20 degron is the first part of this substrate that is translocated into the degradation chamber, proteolytic removal of 2–11 residues then occurs, and the clipped substrate is subsequently released rather than processively degraded. Release of partially degraded substrates has been observed when AAA+ proteases, including Lon, encounter a domain that is difficult to unfold in multi-domain substrates (Lee et al., 2001; Kenniston et al., 2005; Gur et al., 2012). For the cp6-sul20 substrate, the intact β-barrel of GFP could resist unfolding and result in release of some partially degraded substrates. In an attempt to minimize clipping and partial degradation, we deleted eight residues preceding the sul20 degron to generate cp6-Δ8-sul20, reasoning that degradation might be more processive if a shorter linker between the degron and the β-barrel core necessitated core denaturation before the degron reached the peptidase active sites. Indeed, Lon degraded cp6-Δ8-sul20 to ∼90% completion and cp6-sul20 to ∼70% completion under single-turnover conditions of enzyme excess (Fig. 2D). In this experiment, cp6-Δ8-sul20 degradation was also ∼2-fold faster than cp6-sul20 degradation (Fig. 2D), but the steady-state Vmax and KM kinetic parameters for Lon degradation of cp6-Δ8-sul20 and cp6-sul20 were similar (Table I). These results suggest that unfolding of cp6-Δ8-sul20 by Lon may not be the rate-limiting step under steady-state conditions.
Inefficient Lon extraction of the degron-tagged C-terminal β-strand of GFP-sul20
Why is GFP-sul20 degraded so slowly by Lon? This protein stimulated ATP hydrolysis by Lon (Kapp ∼7 µM; not shown), whereas untagged GFP did not, indicating that Lon recognizes the sul20 degron in GFP-sul20. Previous studies demonstrated that ClpXP degrades superfolder GFP-ssrA in a two-step reaction in which the degron-tagged C-terminal β-strand is initially extracted and the resulting 10-stranded native intermediate (GFP1–10) is subsequently unfolded and degraded (Martin et al., 2008; Nager et al., 2011). In principle, Lon degradation of GFP-sul20 might be very slow because the extraction and/or global unfolding step is inefficient. To test for potential effects on the extraction step, we inserted a thrombin site in the loop between strands 10 and 11 of GFP-sul20 and produced a split substrate by thrombin cleavage (Fig. 3A). We then incubated the split substrate (5 µM) with Lon (0.3 µM) and observed slow degradation of the sul20-tagged C-terminal fragment but no degradation of the untagged N-terminal fragment over the course of 30 min as monitored by SDS-PAGE (Fig. 3B). This result suggests that Lon extraction of the sul20-tagged fragment from the split substrate is inefficient. However, the C-terminal fragment did not run as a sharp band precluding quantification. As a more rigorous test of Lon's ability to extract the sul20-tagged C-terminal fragment, we exploited the fact that native GFP1–10 has the same fluorescence as GFP when excited at 467 nm but no fluorescence when excited at 400 nm, because excitation at the lower wavelength requires excited state transfer of a proton from the chromophore to Glu222 in the 11th β-strand (Stoner-Ma et al., 2006; Kent et al., 2008). When thrombin-split GFP-sul20 (0.5 µM) was incubated with excess Lon (2 µM), an ∼20% reduction in 400-nm fluorescence with little change in 467-nm fluorescence was detected over 30 min (Fig. 3C). Thus, Lon extracts only a small fraction of the C-terminal fragments in the population of substrates. However, the slow rate at which Lon extracted the degron-tagged C-terminal strand was substantially faster then the rate of degradation of the GFP-sul20 substrate, suggesting that inefficient strand extraction and slow global unfolding of the GFP1–10 intermediate combine to account for the resistance of GFP-sul20 to Lon degradation.
Fig. 3.

(A) Cartoon showing the thrombin-cleavage site inserted between β-strands 10 and 11 of GFP-sul20 and the generation of the split substrate by thrombin cleavage. (B) The thrombin-split GFP-sul20 protein (5 µM) was incubated with Lon6 (0.3 µM) and the reaction was monitored by SDS-PAGE, followed by staining with Coomassie Blue. (C) The thrombin-split GFP-sul20 protein (0.5 µM) was incubated with Lon6 (2 µM) and the reaction was monitored by changes in fluorescence at 511 nm after excitation at 400 nm (extraction/degradation of β-strand 11) or 467 nm (global unfolding/degradation).
Fluorescence detection of Lon degradation in vivo
Changes in cellular fluorescence linked directly to Lon degradation would provide a potentially powerful way to monitor proteolysis in vivo. To test for intracellular degradation, we expressed plasmid-borne cp6-sul20 from an IPTG-inducible promoter in an E.coli strain that lacks a functional chromosomal lon gene (ER2566), and after ∼2 h induced expression of plasmid-borne Lon from an arabinose-inducible promoter. Cellular fluorescence began to decrease ∼15 min after Lon induction and, after 60 min, approached the level in an experiment in which cp6-sul20 expression was not induced (Fig. 4A). By contrast, cellular fluorescence was ∼3-fold higher when cp6-sul20 expression was induced but Lon expression was not induced (Fig. 4A). These results demonstrate that cp6-sul20 fluorescence can be used as a reporter of intracellular Lon degradation.
Fig. 4.
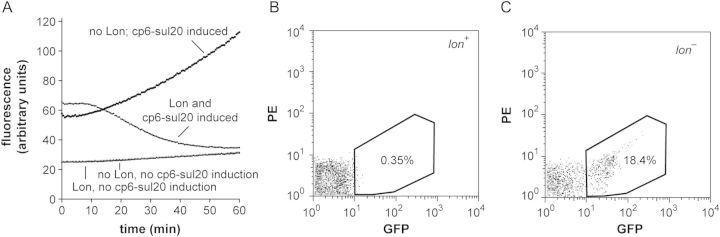
(A) Fluorescence of cells expressing different combinations of the cp6-sul20 GFP variant and Lon protease. At time = 0, arabinose was added to induce expression of Lon from a pBAD33 vector or to lon– cells containing an empty pBAD33 plasmid. Two hours before, IPTG was added to induce expression of plasmid-borne cp6-sul20 or water was added to the un-induced control. (B) FACS analysis of lon+ W3110 cells expressing plasmid-borne cp6-sul20. The x-axis shows GFP fluorescence (excitation 488 nm; emission 530 ± 15 nm); the y-axis shows phycoerythrin (PE) fluorescence (excitation 488 nm; emission 585 ± 21 nm) as an auto-fluorescence control. Approximately 0.4% of the cells fall within the GFP-positive area. (C) FACS analysis of lon::kan W3110 cells expressing plasmid-borne cp6-sul20. Approximately 18% of the cells fall within the GFP-positive area.
To determine if degradation of cp6-sul20 by normal cellular concentrations of Lon could be detected, we transformed lon+ and lon– cells with an over-expressing plasmid, induced cp6-sul20 expression for 90 min and then performed FACS. Only ∼0.4% of the lon+ cells were present in the high-fluorescence gating region (Fig. 4B), whereas ∼18% of the lon– cells were recovered in this region (Fig. 4C). Thus, normal cellular Lon levels are sufficient for reliable detection of cp6-sul20 degradation in vivo by FACS analysis.
Discussion
We have shown that E.coli Lon efficiently degrades circularly permuted variants of superfolder GFP with specific N-terminal or C-terminal degrons. These and related substrates should be useful for characterizing how Lon structure/function mutations alter the steady-state kinetics of protein degradation, for assessing the relationship between degron sequences and their activities in promoting Lon degradation, and for performing high-throughput screens for small-molecule inhibitors of degradation. Importantly, Lon degradation of these circularly permuted GFP substrates is also easily monitored in vivo, facilitating screens for cellular mutations that influence Lon degradation or GFP mutations that enhance or suppress degradation. Fluorescent assays for Lon degradation of peptides or non-native proteins have been reported (Rudyak and Shrader, 2000; Lee and Berdis, 2001; Gur and Sauer, 2008, 2009), but these assays do not require substrate unfolding by Lon, are often independent of the addition of a defined degron, and cannot be used in vivo.
Escherichia coli Lon degrades GFP-ssrA and GFP-β20 but at extremely slow rates (≤0.06 min−1 Lon6−1; Choy et al., 2007; Gur and Sauer, 2008), precluding the use of these substrates for many assays. Consistently, we found that Lon degrades superfolder variants of GFP-β20, GFP-sul20 and β20-GFP very slowly. In combination, these results suggest that Lon has a very difficult time unfolding the β-barrel of non-permuted GFP, either when pulling on the protein from the N-terminus or the C-terminus. Indeed, we find that Lon inefficiently dislodges even the C-terminal β-strand of a split variant of superfolder GFP-sul20. By contrast, Lon degrades the circularly permuted cp6-sul20 and β20-cp6 variants of superfolder GFP at rates ∼50-fold faster than the best non-permuted variants. From the perspective of protein stability, the β-barrels of cp6 and cp7 are less thermodynamically stable than the barrel of non-permuted superfolder GFP (Nager et al., 2011). The detailed rates at which Lon degrades the circularly permuted GFP substrates probably depend on the mechanical stabilities of degron-proximal elements of secondary structure in these proteins (Lee et al., 2001).
The relative unfolding capabilities of Lon and other AAA+ proteases are highly substrate specific. For example, Lon is a very weak unfoldase for some degron-tagged substrates (Koodathingal et al., 2009) but a robust unfoldase for others (Gur et al., 2012). Our current and recent results (Nager et al., 2011) show that circular permutation of GFP can dramatically alter its susceptibility to degradation by different AAA+ proteases. For example, Lon can barely degrade non-permuted GFP but robustly degrades the cp6 variant, whereas ClpXP degrades non-permuted GFP at a much faster rate than Lon but degrades the cp6 variant at a slower rate. We anticipate that circularly permuted variants of GFP will also be useful substrates for different classes of AAA+ proteases, protein-remodeling enzymes and protein-secretion machines.
Lon clipping of the terminal residues of the degron in some cp6-sul20 substrates prevents complete degradation and probably accounts for incomplete proteolysis of cp7-sul20 as well. For such tail clipping to occur, the sul20 degron must enter the degradation chamber but the substrate must not be committed to processive degradation. The build-up of clipped cp6-sul20 substrates to levels higher than the Lon hexamer concentration indicates that clipped substrates can be released from the enzyme, probably as a consequence of an unsuccessful unfolding attempt. Indeed, it seems likely that engaged but unclipped substrates are also released when unfolding fails, accounting for the higher energetic cost of degrading cp7-sul20 compared with cp6-sul20. Previous experiments showed that the sul20 degron can also act as a tethering sequence, facilitating degradation of a substrate by Lon, without itself being degraded (Gur et al., 2012). It will be important to determine the location of the tethering site in the structure and to determine how degron binding to different sites in Lon is coordinated.
Acknowledgements
We thank I. Papayannopoulos and Eyal Gur for help and advice. This work was supported by National Institutes of Health grant AI-16982, National Science Foundation Graduate Research Fellowship to M.L.W., and NCI grant P30CCA14051, supporting the FACS Core of the Koch Institute for Cancer Research.
References
- Baird G.S., Zacharias D.A., Tsien R.Y. Proc. Natl Acad. Sci. USA. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker T.A., Sauer R.T. Trends Biochem. Sci. 2006;31:647–653. doi: 10.1016/j.tibs.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein S.H., Venkatesh S., Li M., et al. Blood. 2012;119:3321–3329. doi: 10.1182/blood-2011-02-340075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy J.S., Aung L.L., Karzai A.W. J. Bacteriol. 2007;189:6564–6571. doi: 10.1128/JB.00860-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M., Frank E.G., Levine A.S., Woodgate R. Genes Dev. 1998;12:3889–3899. doi: 10.1101/gad.12.24.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur E., Sauer R.T. Genes Dev. 2008;22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur E., Sauer R.T. Proc. Natl Acad. Sci. USA. 2009;106:18503–18508. doi: 10.1073/pnas.0910392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur E., Vishkautzan M., Sauer R.T. Protein Sci. 2012;21:268–278. doi: 10.1002/pro.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R., Prasher D.C., Tsien R.Y. Proc. Natl Acad. Sci. USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashitani A., Ishii Y., Kato Y., Koriuchi K. Mol. Gen. Genet. 1997;254:351–357. doi: 10.1007/s004380050426. [DOI] [PubMed] [Google Scholar]
- Ingmer H., Brøndsted L. Res. Microbiol. 2009;160:704–710. doi: 10.1016/j.resmic.2009.08.017. [DOI] [PubMed] [Google Scholar]
- Ishii Y., Amano F. Biochem. J. 2001;358:473–480. doi: 10.1042/0264-6021:3580473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston J.A., Baker T.A., Fernandez J.M., Sauer R.T. Cell. 2003;114:511–520. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- Kenniston J.A., Baker T.A., Sauer R.T. Proc. Natl Acad. Sci. USA. 2005;102:1390–1395. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston J.A., Burton R.E., Siddiqui S.M., Baker T.A., Sauer R.T. J. Struct. Biol. 2004;146:130–140. doi: 10.1016/j.jsb.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Kent K.P., Childs W., Boxer S.G. J. Am. Chem. Soc. 2008;130:9664–9665. doi: 10.1021/ja803782x. [DOI] [PubMed] [Google Scholar]
- Koodathingal P., Jaffe N.E., Kraut D.A., Prakash S., Fishbain S., Herman C., Matouschek A. J. Biol. Chem. 2009;284:18674–18684. doi: 10.1074/jbc.M900783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I., Berdis A.J. Anal. Biochem. 2001;291:74–83. doi: 10.1006/abio.2001.4988. [DOI] [PubMed] [Google Scholar]
- Lee C., Schwartz M.P., Prakash S., Iwakura M., Matouschek A. Mol. Cell. 2001;7:627–637. doi: 10.1016/s1097-2765(01)00209-x. [DOI] [PubMed] [Google Scholar]
- Martin A., Baker T.A., Sauer R.T. Nat. Struct. Mol. Biol. 2008;15:139–145. doi: 10.1038/nsmb.1380. [DOI] [PubMed] [Google Scholar]
- Nager A.R., Baker T.A., Sauer R.T. J. Mol. Biol. 2011;413:4–16. doi: 10.1016/j.jmb.2011.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørby J.G. Methods Enzymol. 1988;156:116–119. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]
- Ormö M., Cubitt A.B., Kallio K., Gross L.A., Tsien R.Y., Remington S.J. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- Pédelacq J.-D., Cabantous S., Tran T., Terwilliger T.C., Waldo G.S. Nat. Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Reeder P.J., Huang Y.-m., Dordick J.S., Bystroff C. Biochemistry. 2010;49:10773–10779. doi: 10.1021/bi100975z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G.T., Kovach M.E., Allen C.A., Ficht T.A., Roop R.M. Mol. Microbiol. 2000;35:577–588. doi: 10.1046/j.1365-2958.2000.01726.x. [DOI] [PubMed] [Google Scholar]
- Rudyak S.G., Shrader T.E. Protein Sci. 2000;9:1810–1817. doi: 10.1110/ps.9.9.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer R.T., Baker T.A. Annu. Rev. Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- Sauer R.T., Bolon D.N., Burton B.M., et al. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner-Ma D., Melief E.H., Nappa J., Ronayne K.L., Tonge P.J., Meech S.R. J. Phys. Chem. B. 2006;110:22009–22018. doi: 10.1021/jp065326u. [DOI] [PubMed] [Google Scholar]
- Striebel F., Kress W., Weber-Ban E. Curr. Opin. Struct. Biol. 2009;19:209–217. doi: 10.1016/j.sbi.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Torres-Cabassa A.S., Gottesman S. J. Bacteriol. 1987;169:981–989. doi: 10.1128/jb.169.3.981-989.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.J., Lee J.S., Cha J.Y., Baik H.S. Mol. Cells. 2011;32:317–323. doi: 10.1007/s10059-011-1017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]