

Abstract
Giant nerve terminals offer a unique opportunity to learn about dynamic changes in intracellular global Ca2+ concentration ([Ca2+]i) because this quantity can be measured precisely with indicator dyes and the composition of the intra-terminal ionic milieu can be controlled. We review here recent literature on [Ca2+]i signalling in the calyx of Held and discuss what these measurements can tell us about endogenous Ca2+ buffers and Ca2+ extrusion mechanisms. We conclude that in spite of the favourable experimental conditions, some unresolved questions still remain regarding absolute values for the Ca2+-binding ratio, the affinity of the basic fixed buffer and the Ca2+ affinities of the major endogenous Ca2+ binding proteins. Uncertainties about some of these presynaptic properties, including the roles of Mg2+ and ATP (as a Mg2+ buffer), however, extend to the point that mechanisms controlling the decay of [Ca2+]i signals in unperturbed terminals may have to be reconsidered.
 |
Erwin Neher earned a MSc in Biophysics from the Univ. of Wisconsin–Madison after studying physics at the Technical Univ. of Munich. He received his PhD in Physics from the Technical Univ. of Munich, working with Dr. Hans Dieter Lux at the Max Planck Institute Munich, and pursued postdoctoral studies with Dr. Charles F. Stevens at Yale Univ. From 1983 to 2011 he has been director of the Membrane Biophysics Department at the Max Planck Institute in Göttingen. Holger Taschenberger received his PhD in neurobiology from the Technical Univ. of Munich, working with Dr. Rosemarie Grantyn at the Max Planck Institute Munich. He pursued postdoctoral studies with Dr. Henrique von Gersdorff at the Vollum Institute in Portland, Oregon and with Dr. Erwin Neher at the Max Planck Institute in Göttingen. He is now a group leader at the Max Planck Institute in Göttingen.
Neurotransmitter release at nerve terminals is triggered by localized domains of elevated calcium ion concentration ([Ca2+]i) in the vicinity of Ca2+ channels (see Eggermann et al., 2012 for a recent review). Numerous simulations have addressed the problem of buffered calcium diffusion, which governs the build-up and collapse of these domains during short episodes of Ca2+ influx (Roberts, 1994; Naraghi & Neher, 1997; Bucurenciu et al., 2008; Pan & Zucker, 2009; Eggermann & Jonas, 2012). Once Ca2+ channels have closed following an action potential (AP), Ca2+ equilibrates with fast Ca2+ buffers and gradients dissipate diffusionally. For small nerve terminals this happens on a time scale of 2–6 ms (see below). Such equilibrated, ‘residual’[Ca2+]i, typically below micromolar, is much lower than the peak [Ca2+]i within the local domains (10–100 μm; for review see Eggermann et al., 2012) and does not trigger rapid release of neurotransmitter. Nevertheless, it is very important to understand its properties, since it may cause asynchronous transmitter release and it plays dominant roles in short-term plasticity (Neher & Sakaba, 2008). ‘Residual calcium’ has already been postulated by Katz & Miledi (1968) to underlie short-term facilitation. More recently, evidence has accumulated that elevated global [Ca2+]i can strongly enhance vesicle recruitment during ongoing activity, thereby counteracting rapid depletion of the pool of readily releasable vesicles, which would otherwise lead to short-term depression of neurotransmitter release (Dittman & Regehr, 1998; Wang & Kaczmarek, 1998). In fact, a recent study at the calyx of Held, a giant nerve terminal of the auditory brainstem, provided evidence that during stimulation with high-frequency AP trains the rate of vesicle recruitment is about 10 times faster than at rest (Hosoi et al., 2007). This study also showed that the globally measured [Ca2+]i in the terminal rises to about 1.5–2 μm during such trains (see also Sabatini & Regehr, 1998; DiGregorio et al., 1999; Korogod et al., 2005; Müller et al., 2007; Lee et al., 2008).
Although temporally and spatially equilibrated Ca2+ signals should be much easier to understand than buffered diffusion within nanodomains, it turns out that no satisfactory description of global [Ca2+]i, which would allow one to precisely predict its changes during and after single APs or AP trains, has yet been presented. The major problem lies in the fact that Ca2+ indicator dyes, which are used to monitor [Ca2+]i transients, themselves act like buffers and thereby distort Ca2+ signals. Several studies have been performed which partially circumvent this problem by using low concentrations of low affinity indicator dyes (Helmchen et al. 1997; Sabatini & Regehr, 1998; Habets & Borst, 2006). A study on the calyx of Held (Müller et al. 2007) presented convincing data showing that the global [Ca2+]i transient following a single AP has a biphasic decay composed of a fast component with a time constant of 20–30 ms that is followed by a slow one with a time constant of about 500 ms (Fig. 1). This work also demonstrated that the fast component of decay was due to Ca2+ binding to a relatively ‘slow’ Ca2+ buffer, most likely parvalbumin. The fast component disappeared upon ‘washout’ during prolonged episodes of tight-seal whole-cell recording and was not present in calyces of parvalbumin-deficient mice. Furthermore, it could be preserved during whole-cell recordings by including a low concentration of either EGTA or parvalbumin in the patch pipette. The time course of paired-pulse facilitation of EPSCs mirrored the decay of the [Ca2+]i transients. The authors concluded that paired-pulse facilitation is, in part, controlled by global [Ca2+]i and that this, in turn, owes its rapid decay to the binding of residual Ca2+ to a mobile Ca2+ buffer with slow binding kinetics, primarily represented by parvalbumin (Müller et al. 2007).
Figure 1. [Ca2+]i transients evoked by single APs (Aa, Ba) or by bursts of 10 APs at 100 Hz (Ab, Bb).
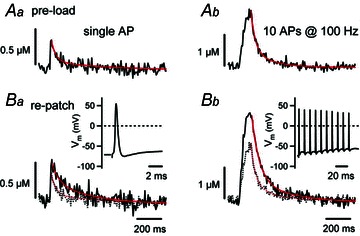
A, [Ca2+]i transients recorded from an intact terminal ‘preloaded’ with about 100 μm Fura-6F during a brief episode of whole-cell recording. Afterwards the pipette was withdrawn and the terminal was stimulated by afferent fibre stimulation. Both the single stimulus response and the [Ca2+]i transient following burst stimulation display a double exponential decay. B, responses to the same stimuli during whole-cell recording, after washout of a slow endogenous buffer. The decays can be fitted with single exponentials with average time constants of about 80 ms. Reproduced with permission from Müller et al. (2007).
While these results provide valuable insights regarding the rapid decay phase of the global [Ca2+]i transients, the problem remains that current models of Ca2+ buffering and sequestration do not reproduce the slow phase of [Ca2+]i decay following single APs. This is already evident in Fig. 7 of Müller et al. (2007) in which the influence of 50 μm parvalbumin on the [Ca2+]i transient is calculated on the basis of a single compartment model. Inclusion of this slow buffer in the model calculation does reproduce a biphasic decay of the [Ca2+]i transient with the correct time constant of the fast component. However, the amplitude of the slow component is about a factor of 10 smaller than the experimental value and its time constant is several-fold too long. The discrepancies between the predictions of this model and experiments are expected to become even more severe during AP trains, when [Ca2+]i transients summate (see Fig. 2 for a simulation of this effect). Faithful reproduction of the global [Ca2+]i changes are, however, essential for the modelling of short-term plasticity (see above), since they may influence release probability and vesicle recruitment. We therefore summarize here efforts to further define the properties of [Ca2+]i transients and we explore what properties of Ca2+ buffers and Ca2+ sequestration mechanisms have to be postulated in order to reproduce the observed [Ca2+]i waveforms.
Figure 2. The influence of Ca2+ dissociation rate on action potential (AP)-induced [Ca2+]i transients.
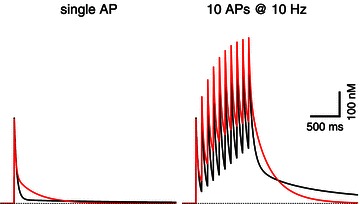
The red traces represent [Ca2+] responses to a single AP (left) and a 10 Hz train of APs (right). Traces represent model calculations, using most parameters as suggested in the text (κs= 26; γ= 260 s−1; modified parvalbumin: 100 μm binding sites with k+= 7.8 × 106 m−1 s−1). The dissociation rate k−, for which a minimum value of 5.1 s-1 was derived earlier in the text, was set to 14.4 s−1 for a better illustration of its effect on the decay of [Ca2+]i. The two black traces are the corresponding model responses when k− of parvalbumin is changed to the low literature value (0.95 s−1). Note that the slow component of the [Ca2+]i transient after the single AP (left, black trace), is hardly visible on the scale of the display. Its amplitude is about 14 nm. For the 10 Hz trains, slow components add up, leading to a substantial very slow component lasting several seconds in the case of the slow k−. The fast dissociation rate, which faithfully reproduces the slow component of the single AP response, causes a stronger build-up of [Ca2+] during the train (due to dissociation of Ca2+ from the buffer) and a biphasic decay with a larger slow component, which, however, decays within a second. The simulations also include 100 μm of a fast buffer with 15 μm dissociation constant representing the indicator dye Fura-6F.
Model assumptions and constraints
‘Single compartment models’ assume that within a given compartment the molecular species of interest are in diffusional equilibrium, i.e. they are homogeneously distributed. At the calyx of Held this is most likely the case for Ca2+ ions, small diffusible Ca2+ chelators, and Ca2+ indicator dyes shortly after an AP. The thickness of the calyx is typically around 0.5 μm, as are nearest neighbour distances between active zones. With apparent diffusion coefficients for chelators and Ca2+ ions in the range 15–100 μm2 s-1 (Gabso et al. 1997; Naraghi et al. 1998), such distances are covered at times shorter 6 ms.
Numerous studies have been performed on the kinetics of Ca2+ buffering and sequestration (Neher & Augustine, 1992; Roberts, 1994; Lee et al. 2000; Chuhma & Ohmori, 2001; Billups & Forsythe, 2002; Kim et al. 2005; Habets & Borst, 2006; Aponte et al. 2008). When accounting for these experimental observations in numerical simulations, the global [Ca2+]i is typically calculated on the basis of a balance of Ca2+ influx (specific Ca2+ currents), Ca2+ efflux (Ca2+ pumps, Ca2+ exchangers and sequestration into organelles), rapid binding to endogenous buffers as well as to added buffers (indicator dyes), and binding to slow buffers, such as parvalbumin or EGTA. A very detailed study on the calyx of Held by Helmchen et al. (1997) revealed that the presynaptic cytosol behaves like a low affinity immobile Ca2+ buffer with a Ca2+-binding ratio κs of 40. κs describes the ratio of Ca2+ ions bound to buffer over free ones at equilibrium. During prolonged whole-cell recording, the decay of [Ca2+]i following an AP was mono-exponential and well described by a linear extrusion mechanism with a rate constant γ of 400 s−1 at 23°C. γ is the slope of the relationship between Ca2+ extrusion rate (in m s−1) and [Ca2+]i (in m). The simplest possible single compartment model, which only considers influx, extrusion and a single low affinity buffer (Neher & Augustine, 1992) predicts an exponentially decaying [Ca2+]i transient with a time constant τ= (1+κ)/γ≍ 100 ms, in agreement with the measurements.
The recent study by Müller et al. (2007) confirmed these findings for the calyx of Held in the whole-cell configuration. However, as mentioned above, it also provided evidence for a slow mobile buffer in the unperturbed calyx, which is probably lost during prolonged whole-cell recording. Extending the simplest model by including an EGTA-like buffer with slow kinetics Müller et al. (2007) could reproduce the experimentally observed, fast [Ca2+]i transient with a decay time constant of about 25 ms, caused by the binding of Ca2+ ions to this buffer.
In the following sections we will demonstrate that for a satisfactory reproduction of the slow decay component and for larger [Ca2+]i transients the model had to be changed in several respects: We have to assume (i) a lower value for κs (≍25), (ii) a lower slope for the low concentration limit of the extrusion rate, equivalent to a γ of 200–300 s−1, (iii) a non-linear increase in extrusion rate, and (iv) a relatively low affinity for the endogenous mobile buffer.
Our calculations are based on the single compartment model of Neher & Augustine (1992), as extended for inclusion of a slow buffer by Lee et al. (2000). In the latter model the time constant τf of the fast decay component is given by
| (1) |
Here, the second term on the right side is very small for [Ca2+] below 5 μm and the last term, k−, representing dissociation of calcium from buffer, is reasonably small relative to the first term (see below). γ is the rate constant of the extrusion mechanism and k+ is an apparent second order rate constant of Ca2+ binding to a parvalbumin-like buffer with concentration [P]. It should be noted that parvalbumin also binds Mg2+, and that in the presence of free Mg2+ the apparent rate constant is lower than the true one (Lee et al. 2000). Furthermore, eqn (1) is only valid if Mg2+ binding and unbinding is fast with respect to the time courses observed. ∑κ is the sum of Ca2+-binding ratios of all rapid buffers. Usually ∑κ is approximated by a term, κs, representing the endogenous fast buffer, and by κB representing the Ca2+ indicator dye (Neher & Augustine, 1992). If measurements are performed with a low concentration of a low affinity dye, κB is small and we can write, for simplicity:
| (2) |
In fact, it turns out that the small errors made by neglecting k− and those by neglecting κB partially compensate each other (see below). The term k+[P] represents the binding of Ca2+ to free buffer (i.e. parvalbumin), which, together with the extrusion mechanism, determines the [Ca2+]i decay. Below, we will also offer an alternative interpretation of the fast decay component.
Müller et al. (2007) found a fast time constant τf of 25 ms for the relatively unperturbed calyx and, in agreement with Helmchen et al. (1997), a single time constant of around 100 ms after washout of the slow buffer parvalbumin. For the latter condition eqn (2) simplifies to:
| (3) |
From eqn (3) and τ= 0.1 s we can conclude:
| (4) |
Using eqns (2) and (4), and τf= 25 ms we obtain:
| (5) |
Equations (4) and (5) fix the ratios between γ, κs, and the product k+[P]0. Absolute values for κ, γ and k+ can only be obtained with additional information.
Helmchen et al. (1997) used several methods to calculate values for κs and γ. κs ranged from 27–71 with a grand mean of approximately 40 and correspondingly γ= 400 s−1 (see eqn (4)). These values are based on extrapolation to zero indicator dye concentration. Unfortunately, this extrapolation is not very robust. However, one can also analyse other aspects of their data. For instance, when terminals were loaded with 100 μm fura-2, a decay time constant of 1 s and an amplitude of 50 nm was measured. In this situation κB, the Ca2+-binding ratio of fura-2, is 200 and dominates in eqns (1)–(3). Assuming κs= 40 one obtains 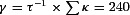 s−1. From this κs= 24 is calculated, which is not consistent with the assumption of κs= 40. However, the values for γ= 220 s−1 and κs= 22 are consistent with each other. They may be more reliable.
s−1. From this κs= 24 is calculated, which is not consistent with the assumption of κs= 40. However, the values for γ= 220 s−1 and κs= 22 are consistent with each other. They may be more reliable.
Another indication for a lower value for κs (and γ) comes from an estimate of the terminal's volume and Ca2+ inflow during an AP. Helmchen et al. (1997) report that 0.96 pC flows into an accessible volume of 0.4 pl, resulting in a total additional [Ca2+] of 12 μm. With their estimate of the amplitude of the AP-induced [Ca2+]i transient of 0.4 μm (extrapolated to zero indicator dye), κs is estimated as 29. Müller et al. (2007) found an AP-induced [Ca2+]i transient amplitude of 0.5 μm under similar conditions, using 100 μm Fura-6F. From that we obtain ∑κ= 24. Here, the dye contributes a value of 7 to the ∑κ, such that a value for κs as low as 17 might be applicable. Taken together, we will consider in our simulations below the range 17 < κs <40 and correspondingly 170 < γ < 400 s−1. For further analytical estimates we will take the geometric means of these values:
| (6) |
Returning to the problem of the slow component of [Ca2+]i decay, we now ask the question of how the properties of a slow buffer have to be tailored so that the model predicts such a decay component with sufficiently large amplitude (Müller et al. (2007) report that the slow component accounts for ≥15% of the decay of [Ca2+]i transient). We first ask what criteria have to be fulfilled for observing a double exponential decay in a single compartment model. Lee et al. (2000) found that this is the case, if (using our terminology):
| (7) |
With k+ [P] = 3γ (eqn (5)) this criterion is marginally fulfilled, considering the last term on the right side only. The first term contributes further, such that there is some basis for a two component fit (see below for an alternative interpretation). The amplitude of the slow component, As, represents the equilibrium of Ca2+ with all buffers, including the slow one:
| (8) |
Here, [Ca]tot is the total [Ca2+] entering the cell during the AP and κp is the Ca2+-binding ratio of the slow buffer, which for a low affinity, non-saturated buffer is simply the ratio of its total concentration over its dissociation constant KD,P
| (9) |
Considering that the initial amplitude At before equilibration of the slow buffer is given by:
| (10) |
we readily derive:
| (11) |
For a ≥15% contribution of the slow component to the decay of [Ca2+]i (As/Af=As/(At-As) ≥ 0.176) and considering eqns (5) and (6), we find a value for k− of ≥5.1 s−1. This lower bound to the actual value of k− is substantially higher than previous estimates for the Ca2+ unbinding rates of ‘slow’ buffers such as EGTA with k−,EGTA= 0.45 s−1 (Naraghi, 1997) or 0.75 s−1 (Nägerl et al. 2000). It is also larger than published Ca2+ dissociation rates from parvalbumin (0.95 s−1; Lee et al. 2000). Unfortunately, the single compartment model, provided all the underlying assumptions are correct, leaves little freedom in the choice of parameters when trying to obtain combinations of parameters, which would result in a lower value for k−. Considering eqn (11), one can see that for a given relative amplitude of the slow decay component, k− changes very little when simultaneously changing k+[P] and κs. According to eqn (5), the latter two quantities have to be changed in proportion, if one wants to maintain the experimentally determined relationship between the fast decay time constants measured in the presence (25 ms) or absence (100 ms) of endogenous mobile buffer.
Given the values found for k− (≥5.1 s−1) and the product k+[P] (780 s−1) we can ask what the binding rate constant and the dissociation constant of an endogenous buffer would be if it were present at a total concentration of 100 μm (as assumed by Müller et al. (2007)). The answer is: k+= 7.8 × 106 s−1 m−1 and KD= 0.65 μm. It should be noted though that the considerations up to this point only specify the product k+[P]. Any low affinity buffer that has a k− of ≥5.1 s−1 and is added at a concentration to yield the same product will reproduce the kinetics described so far.
Regarding the discrepancy between our postulate of a relative fast Ca2+ dissociation rate of this buffer and the published value for parvalbumin of 0.95 s-1 (Lee et al. 2000), we should emphasize that Ca2+ competes with Mg2+ for binding to parvalbumin. Thus, both our values for k+ and k− should be considered apparent ones, reflecting the presence of an unknown amount of Mg2+ in the unperturbed cytosol. In the study of Lee et al. (2000), free Mg2+ was buffered to about 140 μm which is an order of magnitude lower than can be expected for the cytosol. Therefore, their k− value should not be compromised by the effects of Mg2+ binding. Thus, either the endogenous buffer may not be parvalbumin or else, more likely, one of the assumptions in the derivation of eqn (11) is not correct. We will discuss an alternative below.
Finally we can ask what slow time constant τs would be expected for the parameters arrived at so far:
| (12) |
This is somewhat faster than measured. The discrepancy can be traced back to the neglect of the contribution of the indicator dye to Ca2+ buffering and to the approximations implied in eqn (8).
Deviations from linearity for larger [Ca2+]i transients
All the arguments about the kinetics of global [Ca2+]i transients so far were based on signals, which were on average smaller than 500 nm. This is expected to justify the numerous assumptions regarding linearity (with [Ca2+]i):
a low affinity endogenous buffer, represented by κs
a low affinity indicator dye, represented by κB
a linear extrusion mechanism, represented by the extrusion rate constant γ
a low affinity endogenous buffer.
The question remains: do these approximations also hold for larger signals? Literature data on a variety of cell types confirm this for the case of the fast endogenous buffer. Xu et al. (1997) titrated endogenous buffers in adrenal chromaffin cells and found a dissociation constant of about 100 μm. Habets & Borst (2006) concluded on the basis of kinetic modelling that the KD should be >5 μm in the calyx of Held. Sabatini & Regehr (1998) found a low affinity endogenous buffer in cerebellar granule cells, similar to that of magnesium green and mag-fura-5.
Ca2+ removal is less likely to be linear over a large [Ca2+]i range. Although Habets & Borst (2006) conclude from modelling that removal should be linear up to about 1 μm in the calyx of Held, Kim et al. (2005) found a square-law dependence of Ca2+ removal on [Ca2+]i, which saturated with high [Ca2+]i at a K½ of about 2 μm (Fig. 3). The maximum slope of this relationship was around 1000 s−1, which is fourfold higher than the low-concentration limit.
Figure 3. Ca2+ removal after strong stimulation.
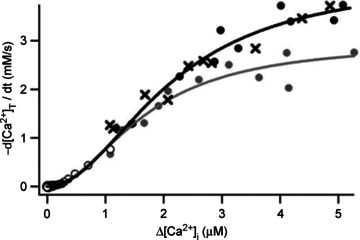
[Ca2+]i transients were elicited by 50 ms depolarizing pulses of various amplitudes and the initial slopes of the decays were plotted against peak [Ca2+] values. Whole-cell recording with a K+-containing pipette solution, either with 50 μm Fura-2FF (filled symbols and crosses) or with Fura-4F (open symbols). Grey symbols are data points from measurements in the presence of the mitochondrial Ca2+-uptake blocker carbonyl cyanide m-chlorophenylhydrazone (CCCP). Reproduced with permission from Kim et al. (2005).
Kim et al. (2005) attributed this non-linear Ca2+ removal largely to the contribution of Na+–Ca2+ exchangers. They derived their flux-data by measuring decay rates of [Ca2+]i after depolarizing pulses and converting these into fluxes under the assumption of a constant κs of 40. This leaves, in principle, the possibility that the non-linear rise in removal rate is an apparent one due to saturation of the endogenous buffer. In any case, the study by Kim et al. (2005) indicates that one or both of our assumptions (linear buffer and linear removal) are violated and that substantial deviations from the prediction of the linear model are expected, if [Ca2+]i signals of 1 μm or larger are to be analysed.
Conclusions and open questions
A giant nerve terminal, such as the calyx of Held, offers unique possibilities to determine the mechanisms and parameters that govern presynaptic intracellular Ca2+ signals during and after AP firing. Literature data, based on terminals loaded with low concentrations of low affinity Ca2+ indicator dyes, suggest the following components to control [Ca2+]i signals:
A low affinity fixed buffer with a Ca2+-binding ratio κs between 15 and 40.
An endogenous mobile buffer with properties similar to those of parvalbumin, but with an apparent dissociation rate constant of 10–15 s−1, which is much higher than previously suggested.
A Ca2+- removal mechanism with a [Ca2+]i-dependent rate that has a limiting low-concentration slope of 150 to 400 s−1 and a sigmoid shape for concentrations above 500 nm.
These elements provide a ‘phenomenological’ description of the diffusionally equilibrated [Ca2+]i signal in the framework of the single compartment model. However, due to the intrinsic properties of Ca2+ buffering and removal, parameters derived using the single compartment model are not well defined in terms of their absolute values:
for the Ca2+-binding ratio κ and the strength of the Ca2+-removal mechanism γ, only their ratio is well-defined
the exact shape of the Ca2+-removal curve may deviate from that of Fig. 3 if the Ca2+-binding ratio κ is not constant for [Ca2+]i values higher than 1 μm.
To date, it seems, no measurements have been performed that rigorously test the assumption of a constant Ca2+-binding ratio (or else the low affinity assumption) of the endogenous fixed buffer. More precise measurements of the absolute value of κs with low-affinity buffers after complete washout of mobile buffers are required.
The discrepancy between our estimates for the apparent dissociation rate constant for the endogenous mobile buffer and published values for parvalbumin, which is the best candidate for this buffer, call for reconsideration of the assumptions underlying the characterization of the endogenous buffer. Most critical in this respect is the assumption of a rapid binding and unbinding kinetics of parvalbumin for Mg2+, or, more precisely, that this reaction is fast. If this is not the case, the fast decay of the [Ca2+]i signal (characterized by τf) may, indeed, be largely shaped by the dissociation of Mg2+ from parvalbumin. In such a scenario, Ca2+ would initially rapidly ‘consume’ free paravalbumin. Subsequently, the decay of [Ca2+]i would be determined by the dissociation of Mg2+ from Mg2+-bound parvalbumin, liberating a ‘sink’ for Ca2+ (see also the concept of ‘buffer regeneration’ recently proposed by Eggermann & Jonas, 2012). The gradual exchange of bound Mg2+ by Ca2+, which may happen on the time scale of the experimentally observed rapid [Ca2+]i decay, would eventually lower [Ca2+]i to the point at which Mg2+ again competes with Ca2+ for binding to parvalbumin. This steady state, in turn, may be strongly influenced by ATP which acts as a Mg2+ buffer. The final decay of the [Ca2+]i transient will subsequently be caused by Ca2+ removal mechanisms such as Ca2+ pumps and exchangers.
These considerations indicate that [Ca2+]i dynamics may be critically dependent on [Mg2+]i and [ATP2−]i, parameters which have so far received little attention when interpreting [Ca2+]i transients in unperturbed terminals. Both parameters may vary considerably, depending on the metabolic state of cells. In addition, free ATP2−, a very fast Ca2+ buffer, may dominate the concentration of free Ca2+ in the immediate vicinity of Ca2+ channels (Naraghi & Neher, 1997). Thus, despite of the favourable experimental conditions provided by the calyx of Held and the numerous detailed studies on the mechanisms of global [Ca2+]i modulation at this synapse, one has to conclude that many aspects of [Ca2+]i signalling in unperturbed nerve terminals remain unresolved.
Acknowledgments
We would like to thank our colleagues Ralf Schneggenburger and Suk-Ho Lee for valuable discussions.
References
- Aponte Y, Bischofberger J, Jonas P. Efficient Ca2+ buffering in fast-spiking basket cells of rat hippocampus. J Physiol. 2008;586:2061–2075. doi: 10.1113/jphysiol.2007.147298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucurenciu I, Kulik A, Schwaller B, Frotscher M, Jonas P. Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron. 2008;57:536–545. doi: 10.1016/j.neuron.2007.12.026. [DOI] [PubMed] [Google Scholar]
- Chuhma N, Ohmori H. Differential development of Ca2+ dynamics in presynaptic terminal and postsynaptic neuron of the rat auditory synapse. Brain Res. 2001;904:341–344. doi: 10.1016/s0006-8993(01)02506-9. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Peskoff A, Vergara JL. Measurement of action potential-induced presynaptic calcium domains at a cultured neuromuscular junction. J Neurosci. 1999;19:7846–7859. doi: 10.1523/JNEUROSCI.19-18-07846.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci. 2012;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Jonas P. How the ‘slow’ Ca2+ buffer parvalbumin affects transmitter release in nanodomain-coupling regimes. Nat Neurosci. 2012;15:20–22. doi: 10.1038/nn.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabso M, Neher E, Spira ME. Low mobility of the Ca2+ buffers in axons of cultured Aplysia neurons. Neuron. 1997;18:473–481. doi: 10.1016/s0896-6273(00)81247-7. [DOI] [PubMed] [Google Scholar]
- Habets RL, Borst JG. An increase in calcium influx contributes to post-tetanic potentiation at the rat calyx of Held synapse. J Neurophysiol. 2006;96:2868–2876. doi: 10.1152/jn.00427.2006. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Borst JG, Sakmann B. Calcium dynamics associated with a single action potential in a CNS presynaptic terminal. Biophys J. 1997;72:1458–1471. doi: 10.1016/S0006-3495(97)78792-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi N, Sakaba T, Neher E. Quantitative analysis of calcium-dependent vesicle recruitment and its functional role at the calyx of Held synapse. J Neurosci. 2007;27:14286–14298. doi: 10.1523/JNEUROSCI.4122-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Korogod N, Schneggenburger R, Ho WK, Lee SH. Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J Neurosci. 2005;25:6057–6065. doi: 10.1523/JNEUROSCI.0454-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of Held. J Neurosci. 2005;25:5127–5137. doi: 10.1523/JNEUROSCI.1295-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Kim MH, Ho WK, Lee SH. Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of Held synapse. J Neurosci. 2008;28:7945–7953. doi: 10.1523/JNEUROSCI.2165-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Schwaller B, Neher E. Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for [Ca2+] transients of neuronal dendrites. J Physiol. 2000;525:419–432. doi: 10.1111/j.1469-7793.2000.t01-2-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Felmy F, Schwaller B, Schneggenburger R. Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of held that accelerates the decay of Ca2+ and short-term facilitation. J Neurosci. 2007;27:2261–2271. doi: 10.1523/JNEUROSCI.5582-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Novo D, Mody I, Vergara JL. Binding kinetics of calbindin-D(28k) determined by flash photolysis of caged Ca2+ Biophys J. 2000;79:3009–3018. doi: 10.1016/S0006-3495(00)76537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- Naraghi M, Muller TH, Neher E. Two-dimensional determination of the cellular Ca2+ binding in bovine chromaffin cells. Biophys J. 1998;75:1635–1647. doi: 10.1016/S0006-3495(98)77606-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Pan B, Zucker RS. A general model of synaptic transmission and short-term plasticity. Neuron. 2009;62:539–554. doi: 10.1016/j.neuron.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WM. Localization of calcium signals by a mobile calcium buffer in frog saccular hair cells. J Neurosci. 1994;14:3246–3262. doi: 10.1523/JNEUROSCI.14-05-03246.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Optical measurement of presynaptic calcium currents. Biophys J. 1998;74:1549–1563. doi: 10.1016/S0006-3495(98)77867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Xu T, Naraghi M, Kang H, Neher E. Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophys J. 1997;73:532–545. doi: 10.1016/S0006-3495(97)78091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]