

Abstract
The cAMP/protein kinase A (PKA) signalling cascade is ubiquitous, and each step in this cascade involves enzymes that are expressed in multiple isoforms. We investigated the effects of this diversity on the integration of the pathway in the target cell by comparing prefrontal cortical neurones with striatal neurones which express a very specific set of signalling proteins. The prefrontal cortex and striatum both receive dopaminergic inputs and we analysed the dynamics of the cAMP/PKA signal triggered by dopamine D1 receptors in these two brain structures. Biosensor imaging in mouse brain slice preparations showed profound differences in the D1 response between pyramidal cortical neurones and striatal medium spiny neurones: the cAMP/PKA response was much stronger, faster and longer lasting in striatal neurones than in pyramidal cortical neurones. We identified three molecular determinants underlying these differences: different activities of phosphodiesterases, particularly those of type 4, which strongly damp the cAMP signal in the cortex but not in the striatum; stronger adenylyl cyclase activity in the striatum, generating responses with a faster onset than in the cortex; and DARPP-32, a phosphatase inhibitor which prolongs PKA action in the striatum. Striatal neurones were also highly responsive in terms of gene expression since a single sub-second dopamine stimulation is sufficient to trigger c-Fos expression in the striatum, but not in the cortex. Our data show how specific molecular elements of the cAMP/PKA signalling cascade selectively enable the principal striatal neurones to respond to brief dopamine stimuli, a critical process in incentive learning.
Key points
Dopamine D1 receptors activate the cAMP/protein kinase A (PKA) pathway in both cortex and striatum, with different types of signalling enzymes being involved in cAMP/PKA signal integration. We investigated the functional implications of such differences.
Biosensor imaging in mouse brain slice preparations revealed that the cAMP/PKA signal increases faster, reaches higher levels and lasts longer in striatal neurones than in cortical neurones.
These differences result from faster cAMP production and lower degradation by type 4 phosphodiesterase activities in the striatum than in the cortex. In addition, DARPP-32 in the striatum prolongs the PKA response by inhibiting phosphatases.
These molecular features confers on striatal neurones a particular ability to temporally decode sub-second dopamine signals associated with reward and learning.
Introduction
The cAMP–protein kinase A (PKA) pathway is one of the most common and versatile signalling pathways in eukaryotic cells, playing a role in the regulation of cell functions in virtually all mammalian tissues. Real-time changes in cAMP concentration are difficult to address by biochemical approaches, and the advent of fluorescent biosensors has made it possible to analyse the dynamics of the signalling cascade directly in specific cell types and, thus, to determine differences in integration profile between cell types.
Biosensors commonly consist of a domain sensitive to the biological signal of interest sandwiched between a pair of fluorophores that provide an optical readout of the conformational change triggered by the biological signal (Zhang et al. 2002b). They include Epac1-camps (Nikolaev et al. 2004), which is moderately sensitive (EC50= 2.4 μm) to cAMP, and Epac2-camps300 (Norris et al. 2009), which is more sensitive (EC50= 320 nm) to cAMP. The AKAR3 sensor consists of a phospho-amino acid binding domain and a PKA-specific substrate: it reports the equilibrium between PKA-mediated phosphorylation and phosphatase activity (Allen & Zhang, 2006).
Biosensor imaging has shown large cAMP responses to receptor stimulation in various preparations. For example, strong responses to β-adrenergic stimulation have been recorded with Epac1-camps in primary cultures of cardiomyocytes, fibroblasts, macrophages and thyroid cells (Leroy et al. 2008; Calebiro et al. 2009). Strong responses have also been observed in cell lines such as the MIN6 pancreatic cell line, after stimulation of the glucagon receptor (Landa et al. 2005), and HEK-293 cells after stimulation of β2-adrenergic receptors, in which cAMP concentration increases up to 10 μm (Violin et al. 2008). Spontaneous cAMP oscillations have also been reported in cultured retinal ganglion cells with a low-sensitivity cAMP sensor (Dunn et al. 2006). Similarly, strong cAMP responses to β-adrenergic stimulation have been recorded with Epac1-camps in primary cultures of hippocampal neurones (Nikolaev et al. 2004).
We used these biosensors in brain slice preparations in which mature living neurones with a preserved morphology were experimentally accessible. In these conditions, the activation of several Gs-type G protein-coupled receptors (β1-adrenergic, 5-HT7, VPAC1, CRF1 and PAC1) triggered partial PKA responses, as reported by AKAR sensors (Gervasi et al. 2007; Castro et al. 2010; Hu et al. 2011; Vincent et al. 2012), while, surprisingly, little or no response was detected at the level of cAMP with Epac1-camps (Castro et al. 2010; Hu et al. 2011). Using the Epac2-camps300 biosensor with higher cAMP sensitivity, we have recorded β1-adrenergic responses in the cortex (Castro et al. 2010), demonstrating very low cAMP levels in response to neuromodulators in our preparation. Among the large number of phosphodiesterases (PDEs), type 4 phosphodiesterase (PDE4) plays a critical role in many organs (Houslay, 2010). In the cortex, PDE4 inhibition with rolipram is associated with procognitive and antidepressant effects (Ramos et al. 2003; O’Donnell & Zhang, 2004). We have shown that this PDE4 activity is required to maintain low cAMP concentrations and thus allow for the subcellular compartmentation of the cAMP signal in pyramidal neurones (Castro et al. 2010).
The medium-sized spiny neurones (MSNs) of the striatum are endowed with a very unusual set of signalling proteins that are much less abundant or even absent in most other cell types. For example, adenylyl cyclase stimulation is mediated by the Golf-type G protein rather than the usual Gs, and striatal neurones express large amounts of type 5 adenylyl cyclase (AC5) (Matsuoka et al. 1997), and dopamine- and cAMP-dependent phosphoprotein 32 kDa (DARPP-32) (Hervé & Girault, 2005). We investigated whether these biochemical differences were related to differences in the dynamics of signal integration. As both the cortex and striatum are targets of midbrain dopaminergic neurones, we compared the cAMP and PKA responses to dopamine D1 receptor activation. We found that these responses in striatal neurones were stronger and quicker than in cortical neurones, particularly when the dopamine stimulus was brief. This unique property appears to result from a higher level of adenylyl cyclase activity, a lower level of type 4 phosphodiesterase activity and the presence of the phosphatase inhibitor DARPP-32.
Methods
Brain slice preparation
Wild-type C57Bl/6J mice were obtained from Janvier (Le Genest Saint Isle, France). Heterozygous mice with one disrupted allele of Gαolf (Belluscio et al. 1998) were crossed to produce heterozygous Gαolf mutant mice and wild-type litter mates. Homozygous mice expressing DARPP-32 with the T34A mutation have been described elsewhere (Svenningsson et al. 2003). Mice were maintained in a 12 h light–12 h dark cycle, in stable conditions of temperature (22°C), with food and water available ad libitum. All the experiments were performed according to French Ministry of Agriculture and Forestry guidelines for handling animals (decree 87–848).
Brain slices were prepared from male mice aged from 8 to 12 days, as previously described (Castro et al. 2010). Briefly, mice were decapitated, their brain removed, and coronal and sagittal brain slices were cut with a VT1200S microtome (Leica, Germany). Slices were prepared in an ice-cold solution of the following composition: 125 mm NaCl, 0.4 mm CaCl2, 1 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, 25 mm glucose and 1 mm kynurenic acid, saturated with 5% CO2 and 95% O2. The slices were incubated in this solution for 30 min and then placed on a Millicell-CM membrane (Millipore) in culture medium (50% minimum essential medium, 50% Hanks’ balanced salt solution, 6.5 g l−1 glucose, penicillin–streptomycin; Invitrogen, San Diego, CA, USA). We used the Sindbis virus as a vector to induce expression of the various probes (Ehrengruber et al. 1999). The coding sequences of AKAR3, Epac1-camps and Epac2-camps300 were inserted into the viral vector pSinRep5 (Invitrogen), as previously described (Gervasi et al. 2007). The viral vector (∼5 × 105 particles per slice) was added and slices were incubated overnight at 35°C under an atmosphere containing 5% CO2. Before the experiment, slices were incubated for 30 min in the recording solution (identical to the solution used for cutting, except that the calcium concentration was 2 mm and kynurenic acid was omitted). During recordings, brain slices were continuously perfused with this solution saturated with 5% CO2–95% O2 in a recording chamber maintained at 32°C. The viability of the neurones in these experimental conditions has been checked by patch-clamp recording, which showed electrical activity to be normal (Gervasi et al. 2007; Castro et al. 2010). MSNs expressing Epac1-camps were easily patched in our preparation and displayed normal electrophysiological features (Supplemental Fig. 1; supplemental material is available online only).
Optical recordings on brain slices
Recordings were made on visually identified pyramidal neurones in layer V of the mouse prefrontal cortex. Neurones were selected for a diameter less than 14 μm, comparable to the size of medium spiny neurones. MSNs constitute 95% of neurones in the striatum. Large neurones, presumably cholinergic interneurones, were excluded (i.e. diameter larger than 14 μm). Wide-field images were obtained with an Olympus BX50WI or BX51WI upright microscope with a ×20 0.5 NA or a ×40 0.8 NA water-immersion objective and an ORCA-AG camera (Hamamatsu Photonics, Massy, France). Images were acquired with iVision (Biovision, Exton, PA, USA). The excitation and dichroic filters were D436/20 and 455dcxt. Signals were acquired by alternating the emission filters, HQ480/40 for CFP (Cyan Fluorescent Protein), and D535/40 for YFP(Yellow-shifted Fluorescent Protein), with a filter wheel (Sutter Instruments, Novato, CA, USA). All filters were obtained from Chroma Technology (Brattleboro, VT, USA).
Two-photon images were obtained with a custom-built two-photon laser-scanning microscope based on an Olympus BX51WI upright microscope with a ×60 0.9 NA water-immersion objective and a Ti:sapphire laser (MaiTai HP; Spectra Physics, Mountain View, CA, USA) tuned to 850 nm for CFP excitation. Galvanometric scanners (model 6210, Cambridge Technology, Cambridge, MA, USA) were used for raster scanning, and a piezo-driven objective scanner (P-721 PIFOC, Physik Instrumente GmbH, Karlsruhe, Germany) was used for Z-stack image acquisition. The system was controlled by MPscope software (Nguyen et al. 2006). A two-photon emission filter was used to reject residual excitation light (E700 SP, Chroma Technology). A fluorescence cube containing 479/40 and 542/50 emission filters and a 506 nm dichroic beamsplitter (FF01-479/40, FF01-542/50 and FF506-Di02-25x36 Brightline Filters, Semrock, Rochester, NY, USA) was used for the orthogonal separation of the two fluorescence signals. Two imaging channels (H9305 photomultipliers, Hamamatsu) were used for simultaneous detection of the two types of fluorescence emission.
Images were analysed with custom routines written in the IGOR Pro environment (Wavemetrics, Lake Oswego, OR, USA). The emission ratio as calculated for each pixel: emission at 535 nm divided by the emission at 480 nm (F535/F480) for AKAR3 and emission at 480 nm divided by the emission at 535 nm (F480/F535) for Epac2-camps300 and Epac1-camps. The pseudocolour images were calculated so as to display the ratio value coded in hue and the fluorescence of the preparation coded in intensity. A calibration square indicates the intensity values from left to right and the ratio values from bottom to top. The size of the square indicates the scale of the image in micrometres.
No correction for bleed-through or direct excitation of the acceptor was applied (Börner et al. 2011) because we considered the correction coefficients to be potentially unreliable in the brain slice preparations as a result of differences in optical properties between slices. Bleed-through and direct excitation corrections increase the apparent ratio changes but without improving signal-to-noise ratio (Ducros et al. 2009). The ratio changes in our conditions therefore appear smaller than those reported by other studies in which such corrections were applied.
The biosensor chromophores are sensitive to non-specific environmental disturbances. We used a mutated version of AKAR3 in which the threonine residue of the PKA phosphorylation site was replaced with an alanine residue (T391A) as a control. Consistent with the previously reported lack of response of AKAR2mut to β-adrenergic stimulation in the cortex, this AKAR3(T391A) control sensor reported no ratio change in response to the D1-like agonist SKF38393 (1 μm) and forskolin (13 μm) in the cortex (Supplemental Fig. 2A; n= 3). It also reported no change in the striatum in response to 1 μm SKF38393, 10 μm CGS21680 and 13 μm forskolin (Supplemental Fig. 2B; n= 3). The baseline ratio with this control AKAR3(T391A) biosensor differed between cells, so the baseline value cannot be considered to reflect cell-to-cell variations in PKA activity level. Similar cell-to-cell variation in baseline ratio has also been reported in various other preparations and/or with other biosensors (Hepp et al. 2007; van der Krogt et al. 2008).
Fast drug application
A fast focal application system was used for kinetic studies (Gervasi et al. 2007). A glass pipette (80–120 μm tip diameter) was placed 300 μm to the side of and 200 μm above the brain slice and ejected the drug contained in the same solution as the bath. The same device was used for the sustained or transient (10 s) application of 1 μm SKF38393 or 13 μm forskolin. Dopamine uncaging was performed with UV light at 360 nm applied in wide-field mode (UVILED, Rapp OptoElectronic, Hamburg, Germany).
Image acquisition in these fast wide-field recordings was automatic at a frequency ranging from 0.2 to 0.3 Hz. This temporal resolution was sufficient to temporally resolve the onset and decay of AKAR3 responses to dopamine or cAMP uncaging (Supplemental Fig. 3). Image acquisition was otherwise triggered manually by the user.
Immunohistochemistry
We carried out c-Fos immunohistochemistry with a standard peroxidase-based method (Vectastain Elite ABC Kit, Vector Laboratories, UK), using 3,3′-diaminobenzidine (Sigma-Aldrich) as the chromogenic substrate. The primary c-Fos antisera was used at a dilution of 1:1000 (rabbit polyclonal antiserum from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Before immunohistochemistry, brain slices were stimulated with dopamine which was released either by local flash photolysis of NPEC-dopamine (0.1 or 1 s, in the cortex or striatum) or bath application of SKF38393 (1 μm, 5 min) or forskolin (13 μm, 5 min) in the recording solution. After stimulation and incubation for 90 min at 32°C, brain slices were fixed by overnight immersion in phosphate-buffered 4% paraformaldehyde at 4°C, pH 7.4. Slices were then rinsed three times in ice-cold phosphate buffer and incubated with an avidin–biotin blocking kit. Positive nuclei were counted blind, with a ×20 water-immersion objective, on four adjacent regions of 1400 μm2 each, on at least three slices per condition.
Data analysis and statistics
Two-photon imaging (Figs 1 and 2) can be used to separate individual neurones and was used for precise quantification of the amplitude of the response. The nucleus was excluded from the measurement. Ratiometric quantification was performed with a ratio value, indicative of the signal of interest, being between the Rmin and Rmax values, which correspond to the minimal ratio value (no biological signal) and maximal response (saturated biosensor) (Grynkiewicz et al. 1985; Börner et al. 2011). The baseline ratio in control conditions was considered to be equal to Rmin because adenylyl cyclase inhibition with 50 μm SQ22536 yielded no ratio decrease with any of the three biosensors in the cortex (Epac2-camps300: n= 5) and in the striatum (AKAR3: n= 11; Epac2-camps300: n= 6; Epac1-camps: n= 4). Tetrodotoxin (1 μm), CNQX (10 μm), APV (25 μm), gabazine (1 μm) and Cd2+ (200 μm) had no effect on basal AKAR3 ratio (n= 5), indicating that the basal PKA activity is not affected by network activity and synaptic release of neuromodulators.
Figure 1. The PKA response to D1 stimulation was stronger in the striatum than in the cortex.
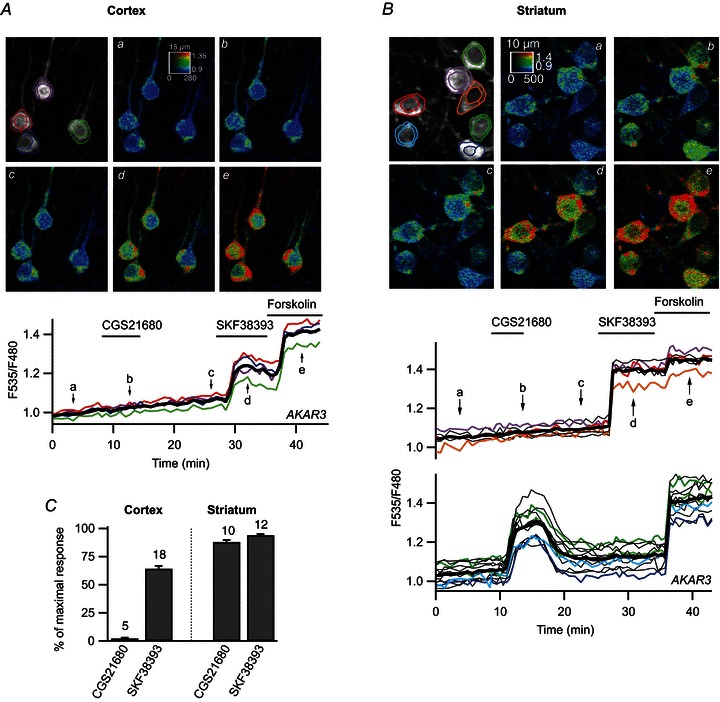
Prefrontal cortical neurones (A) and medium spiny neurones (B) in a mouse brain slice transduced for AKAR3 expression and imaged by two-photon laser-scanning microscopy. Images show the raw fluorescence at 535 nm (left, in grey scale) and the ratio (in pseudocolour), indicating levels of PKA-dependent phosphorylation at the times indicated by the corresponding arrows on the graph below. The calibration square on the pseudocolour image indicates from left to right increasing intensity levels, from bottom to top increasing ratio values. The size of the square is indicated above, in micrometres. Each trace on the graphs indicates the F535/F480 emission ratio measurements on the regions indicated by the colour contour on the grey-scale image; thin traces in black correspond to cells outside the displayed region; the thick black line represents the average of all the traces. The nucleus was excluded from the measurement. Each neurone responded to SKF38393 by an increase in F535/F480 ratio. Forskolin further increased the ratio to a maximal steady-state level. Note that the basal ratio differed between cells, and that these differences remained unchanged throughout the recording and during the response to forskolin; similar differences between cells were observed with the mutant probe (see Methods) and these differences do not reflect genuine differences in PKA activities. B, traces are separated into two plots on the basis of their response to the A2A agonist CGS21680 or the D1 agonist SKF38393. SKF38393 (1 μm), CGS21680 (1 μm) and forskolin (13 μm) were added to the bath during the time represented by the horizontal bar. C, amplitude of the AKAR3 response (normalised to the response to forskolin; see Methods) to CGS21680 and SKF38393. Striatal neurones are separated into two groups depending on their responsiveness to either CGS21680 or SKF38393; numbers above columns indicate the number of experiments.
Figure 2. D1 stimulation increases free cAMP concentration to higher levels in striatal neurones than in cortical neurones.
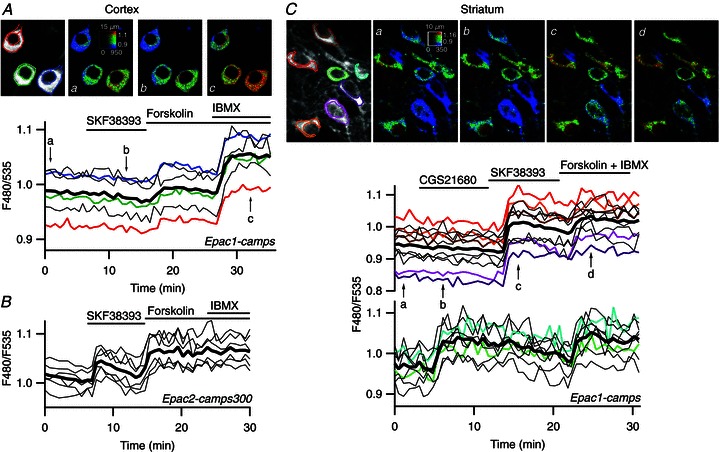
A, prefrontal cortical neurones in a mouse brain slice were transduced for expression of Epac1-camps and imaged by two-photon laser-scanning microscopy. Images show the raw fluorescence at 535 nm (left, in grey scale) and the ratio (in pseudocolour) indicating the change in conformation of Epac1-camps following binding to cAMP, at the times indicated by the arrows on the graph below. Each trace on the graphs indicates F480/F535 emission ratio measurements on regions indicated by the colour contour drawn on the raw images; thin traces in black correspond to cells outside the displayed region; the thick black line represents the average of all the traces. Forskolin increased the ratio to a moderate level and the non-specific inhibition of phosphodiesterases with IBMX resulted in a stronger response. B, as for A, except that the pyramidal cortical neurones were transduced with Epac2-camps300. SKF38393 triggered a clear response, which was further increased by forskolin. C, medium spiny neurones of the striatum transduced for Epac1-camps expression were imaged by two-photon laser-scanning microscopy. Traces are separated into two plots on the basis of the response to either the A2A agonist CGS21680 or to the D1 agonist SKF38393. SKF38393 (1 μm), CGS21680 (1 μm), forskolin (13 μm) and forskolin (13 μm) + IBMX (200 μm) were added to the bath during the time represented by the horizontal bar.
The maximal response (Rmax, corresponding to biosensor saturation) was determined for each neurone at the end of the recording. This level was determined by applying 13 μm forskolin, sufficient to maximally phosphorylate the highly sensitive probe AKAR3 in cortical neurones (Gervasi et al. 2007). In the striatum, the response to 13 μm forskolin recorded with AKAR3 reached 98.2 ± 0.6% (n= 4) of the response obtained when the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX; 200 μm) was added to forskolin, indicating that forskolin was also sufficient to reach the AKAR3 maximal response in the striatum. With the less sensitive Epac1-camps and Epac2-camps300, forskolin was not sufficient to reach sensor saturation and Rmax was taken as the ratio value reached in the presence of both 13 μm forskolin and 200 μm IBMX. IBMX may have some antagonist effect on purinergic receptors and we verified that a cocktail of various PDE inhibitors produced the same response as IBMX (Supplemental Fig. 4). Absolute ratio values differed between cells (as shown with the mutant biosensor, see above), so the amplitude of the response to receptor stimulation was quantified for each neurone as the fractional change in ratio from its own baseline (Rmin) and maximal final ratio response (Rmax). Measurements were performed on regions of interest and means were determined for 7–9 consecutive image planes in the vertical stack (i.e. 3.5–4.5 μm-thick slices centred on the cell body).
In wide-field imaging experiments (Figs 5–8), some of the signal measured on a region of interest comes from out-of-focus neurones. This has no effect on the kinetics features of the ratio signal, but affects the absolute amplitude of the response when out-of-focus neurones have a pharmacological response that is different from that of the in-focus neurone. Indeed, responses to SKF38393 or CGS21680 in the striatum appeared smaller in amplitude when measured with wide-field imaging as compared to two-photon imaging because of the fluorescence background contributed by the non-responding neurones. Still, for a given region of interest, the steady-state response to a bath-applied saturating dose (1 μm) of SKF38393 was genuinely representative of the response of all D1-expressing neurones contributing to the signal of this region of interest, and this value was used for normalisation in kinetic analyses.
Figure 5. The onset of the PKA response is faster in the striatum than in the cortex.
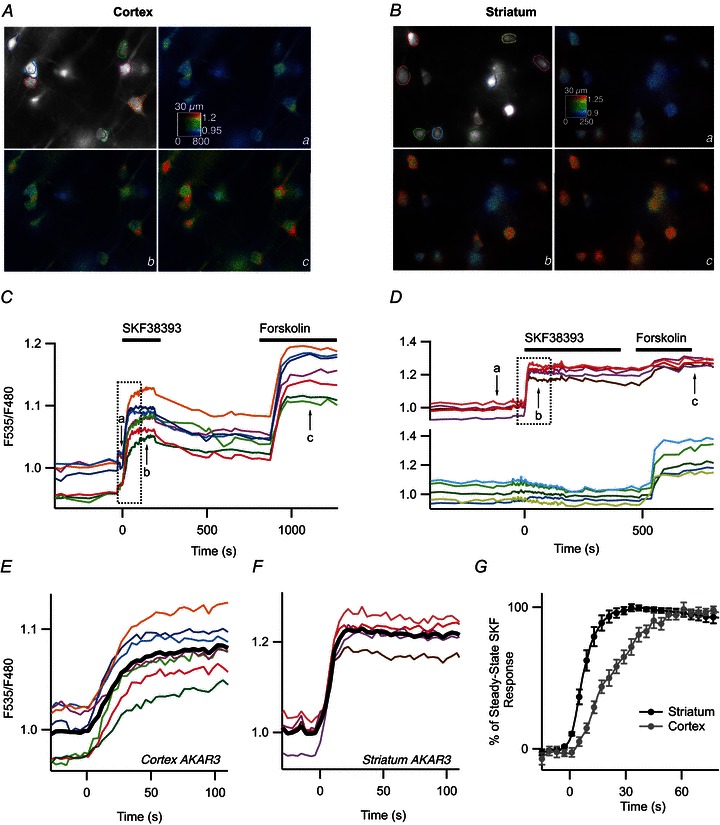
SKF38393 (1 μm) was applied with a fast perfusion system. Pyramidal cortical neurones (A) and MSNs in the dorsal striatum (B) were transduced for AKAR3 expression and imaged by wide-field microscopy. Images show the raw fluorescence at 535 nm (left, in grey scale) and the ratio (in pseudocolour), indicating levels of PKA-dependent phosphorylation in control condition (a), during the responses to 1 μm SKF38393 (b) and 13 μm forskolin (c). C and D, time-course of the F535/F480 emission ratio measured in the regions indicated by the colour contour on the grey-scale image in the cortex (A) and striatum (B), respectively. E and F, expansion indicated by the dotted rectangle in of C and D, respectively, showing the response onset. The black trace represents the average. G, average similar experiments in the cortex (n= 8) and striatum (n= 10). Error bars represent SEM.
Figure 8. Striatal neurones respond more efficiently than pyramidal cortical neurones to brief dopamine uncaging.
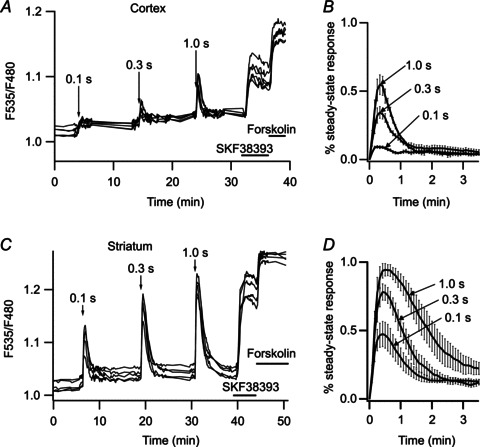
A, pyramidal cortical neurones expressing AKAR3 were imaged in wide-field fluorescence mode and the ratio obtained was plotted against time. Arrows indicate the UV flash, for the duration indicated on the arrow (0.1, 0.3 and 1 s). Increases in the duration of flash photolysis of caged dopamine (NPEC-dopamine, 5 μm, in the bath) were associated with increases in the amplitude of the ratio response. For each flash duration, responses are normalised with respect to the steady-state response to bath-applied SKF38393. Mean responses are presented in B, with error bars indicating the SEM, n= 6. C, as in A, except that the recording was performed in the striatum. D, as in B, but for the striatum, n= 5. Amplitudes differed between uncaging times (P < 0.001) (B and D). Unpaired two-tailed t tests were used for statistical comparisons.
We analysed at least four neurones per brain slice, averaging their responses, with n indicating the number of independent experiments (i.e. brain slices). Unpaired two-tailed Student's t tests were used for statistical comparisons. Differences were considered significant when P < 0.05.
Drugs
SKF38393 hydrobromide, CGS21680 hydrochloride, 3-isobutyl-1-methylxanthine (IBMX), rolipram, (N)- 1-(2-nitrophenyl) ethylcarboxy-3, 4-dihydroxyphenethylamine (NPEC-caged dopamine) and forskolin were obtained from Tocris Cookson (Bristol, UK); R(+)-SCH23390 hydrochoride was obtained from Sigma-Aldrich (Saint-Quentin Fallavier, France).
Results
D1 stimulation results in strong PKA signalling in the striatum
The genetically encoded sensor for PKA activity AKAR3 (Allen & Zhang, 2006) was used for real-time imaging of PKA activation in living cells. Viral vectors encoding this biosensor were used to express the biosensor in brain slice preparations from neonatal mice and the PKA response was measured in real time, by ratiometric (F535/F480) two-photon microscopy. In pyramidal neurones of the prefrontal cortex, activation of the adenosine A2A receptors with CGS21680 (1 μm) has no effect on the emission ratio (n= 5 independent experiments; see Methods), whereas stimulation of the D1 receptors with a saturating dose (1 μm) of SKF38393 yielded a large, but submaximal, increase in the F535/F480 emission ratio of more than 90% of AKAR3-expressing neurones (Fig. 1A). On average, SKF38393 increased the emission ratio to 64.4 ± 2.6% (n= 18) of the maximal response induced with the direct adenylyl cyclase activator forskolin (13 μm; Fig. 1C; see Methods for details of the determination of maximum response). In the dorsal striatum, the neurones that responded to the D1 stimulation exhibited a much larger ratio change with AKAR3, with the response to 1 μm SKF38393 reaching 94.1 ± 1.0% (n= 12) of the maximal level (Fig. 1B and C). Thus, the stimulation of D1 receptors resulted in higher levels of PKA target phosphorylation in MSNs than in pyramidal cortical neurones.
Only 53% (n= 85 of 160) of striatal cells responded to SKF38393; 40% strongly responded to the A2A receptor agonist CGS21680 (1 μm); 5% responded to neither agonist and 2% responded to both agonists (Fig. 1B). This is consistent with previous studies showing that D1 and A2A receptors are segregated in the two major subsets of MSNs corresponding to the direct and indirect pathways, respectively (Bateup et al. 2008; Bertran-Gonzalez et al. 2008).
D1 stimulation results in high cAMP levels in the striatum
We then measured the cAMP signal underlying the PKA responses with the Epac1-camps cAMP sensor (Nikolaev et al. 2004), which is moderately sensitive to cAMP (EC50= 2.4 μm). In the cortex, we observed no significant response to SKF38393 (5.0 ± 1.9%, n= 11). When adenylyl cyclases were directly stimulated with forskolin, the emission ratio increased to 35.4 ± 3% (n= 11) of the maximal response obtained with forskolin plus 200 μm of the phosphodiesterase inhibitor IBMX (Fig. 2A).
Degradation by PDEs is an important determinant of the steady-state cAMP concentration. Figure 2A shows that in the cortex, PDE inhibition with the non-specific drug IBMX strongly potentiates the response to forskolin, showing that PDE activities contribute to the negative control of cAMP.
We then used the Epac2-camps300 sensor (Norris et al. 2009), which is more sensitive to cAMP (EC50= 0.32 μm). This sensor reported a clear response to SKF38393 in cortical neurones (29.1 ± 4.4%, n= 9 of the maximal level reached in the presence of forskolin and IBMX; Fig. 2B). The activation level of the biosensor remained below 50%, showing that the free cAMP concentration during the D1 response remained below the EC50 of the Epac2-camps300 biosensor, i.e. below 0.3 μm.
In marked contrast to the weak responses measured in the cortex, the MSNs displayed very strong cAMP responses: in the MSNs sensitive to D1 agonist, the response to SKF38393 measured with the low-sensitivity cAMP sensor, Epac1-camps, was close to the maximal response (79.1 ± 3%; n= 17) obtained with forskolin and IBMX (Fig. 2C). In the MSNs responsive to A2A agonist, the response to 1 μm CGS21680 was also strong, at 68.9 ± 6.8% of maximal levels (n= 10), and was smaller (unpaired two-tailed Student's t test, P < 0.01) but of the same order of magnitude as the response to SKF38393. The large responses obtained after D1 stimulation indicate a free cAMP concentration during these responses well above the EC50 of the Epac1-camps biosensor, i.e. above 2.4 μm.
In contrast to what was observed in the cortex, IBMX did not increase the response to forskolin in the striatum: the response to forskolin alone was 94 ± 2%n= 3 of the level reached with forskolin and IBMX. This indicates that PDEs in the striatum are not sufficient to counteract the cAMP production elicited by forskolin.
The striatum is composed of the dorsal caudate nucleus and the ventral nucleus accumbens. We investigated whether the cAMP/PKA response differed between these two regions. As in the dorsal striatum, strong cAMP signals were obtained in the shell of the nucleus accumbens, in which Epac1-camps responses to SKF38393 reached 58.7 ± 2.9% maximal levels (n= 6). These signals were lower than those in the dorsal striatum (unpaired two-tailed Student's t test, P < 0.01), but nonetheless reflected much higher cAMP levels than in the prefrontal cortex.
Buffering effect of the biosensor
The small cAMP responses obtained in the cortex may result from cAMP being buffered by a large concentration of Epac-based biosensors. This buffering effect should become more visible and reduce the amplitude of the response as the biosensor concentration increases. In the cortex, we performed this analysis on the responses that were detectable, i.e. with the Epac2-camps300 sensor. We evaluated this effect by analysing the relationship between biosensor concentration, estimated by the absolute fluorescence intensity measured in the cytosol by two-photon imaging, and the amplitude of the response to SKF38393. No downward trend was observed (Fig. 3A), demonstrating that our measurements were not affected by this type of buffering effect.
Figure 3. The intracellular biosensor concentration does not affect the amplitude of the cAMP response.
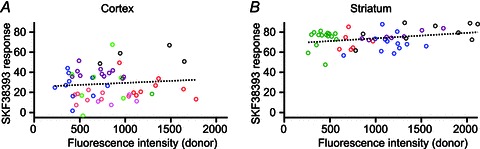
The amplitude of the response to SKF38393 was quantified in individual neurones as a percentage of the maximal forskolin + IBMX response and was plotted against fluorescence intensity. Each data point corresponds to a region of interest drawn on a single neurone. Neurones recorded during the same experiment were plotted in the same colour. A, pyramidal cortical neurones expressing Epac2-camps300; 11 experiments. B, striatal neurones expressing Epac1-camps; 5 experiments. The linear regression was calculated for all the data points on the graph with slopes of 0.0040% AU−1 (A) and 0.0054% AU−1 (B), where AU represents arbitrary units.
We also investigated whether buffering of cAMP by the biosensor affected the amplitude of the D1 response recorded in the dorsal striatum. We performed this analysis on Epac1-camps responses, as the Epac2-camps300 are most likely saturated. Again, we found no relationship between biosensor concentration and response amplitude (Fig. 3B), ruling out a potential buffering effect of the biosensor on the cAMP signal. No difference in the range of intensities was observed between the striatum and the cortex, showing that the biosensor is expressed at similar levels in both brain structures.
In summary, for all three biosensors with different sensitivities (AKAR3 > Epac2-camps300 > Epac1-camps), and independently of biosensor concentration, the responses in the striatum were much stronger than those in the cortex (P < 10−8 unpaired two-tailed t test; Fig. 4), demonstrating the occurrence of a much higher cAMP level and stronger PKA response in striatal neurones than in pyramidal neurones.
Figure 4. The cAMP/PKA response to D1 stimulation was stronger in the striatum than in the cortex, for all three biosensors, Epac1-camps, Epac2-camps300 and AKAR3.
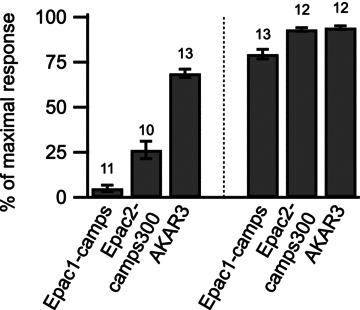
The response amplitude measured with each biosensor was normalised with respect to the maximal biosensor response (see Methods) and plotted as a histogram. Error bars indicate the SEM; the number of brain slices used is indicated above each bar. The difference between cortex and striatum was significant for each of the three biosensors (P < 10−8 unpaired two-tailed t test).
Faster onset kinetics in the striatum than in the cortex
We then looked at the kinetics of the cAMP/PKA response and we increased the temporal resolution of our recordings by using focal applications of 1 μm SKF38393 with a fast onset while monitoring the response of AKAR3 with a wide-field CCD camera (Fig. 5A–D). This increase in temporal resolution was valid for kinetic measurements, but was obtained at the expense of a loss of spatial resolution and the inclusion of autofluorescence and fluorescence from out-of-focus cells in the ratio signal measured over cell bodies. Kinetic responses were therefore normalised to the level obtained at steady state with the same agonist (see Methods). Time 0 was defined as the intercept of the baseline with the line fitted to the 10–90% incremental phase of the response (Fig. 5E and F). T1/2on and t1/2off were defined as the time to reach the half of the maximal response, respectively of the onset and the decay.
The increase in AKAR3 ratio was three times faster in the striatum than in the cortex (unpaired two-tailed t test, P < 0.001), with a t1/2on of 7.9 ± 1.1 s, n= 10 and 21.4 ± 2.6 s, n= 8, respectively (Fig. 5G). This kinetic difference suggests that PKA signalling upon D1 receptor stimulation is stronger in the striatum than in the cortex and we tried to determine the molecular basis underlying this difference.
It is well known that one distinguishing feature of the striatum is the expression of high levels of D1 receptors compared to other brain regions, including the cortex (Boyson et al. 1986; Fremeau et al. 1991). While this difference in receptor density probably plays a role in the differences between striatum and cortex, we wanted to determine the contribution of the enzymes downstream of D1 receptors in the cAMP/PKA responses. To this end, we monitored the onset of the response upon direct stimulation of adenylyl cyclase by forskolin. Application of forskolin increased the PKA signal much faster in the striatum than in the cortex (t1/2on= 12.5 ± 1.8 s, n= 5 in the striatum and 30.4 ± 5.9 s, n= 5 in the cortex, unpaired two-tailed t test, P < 0.05; Fig. 6A). These results clearly show that beyond the difference in D1 receptor expression level, cortex and striatum differently integrate the PKA signal.
Figure 6. The increase in cAMP/PKA signal in response to D1 or adenylyl cyclase stimulation is faster in the striatum than in the cortex.
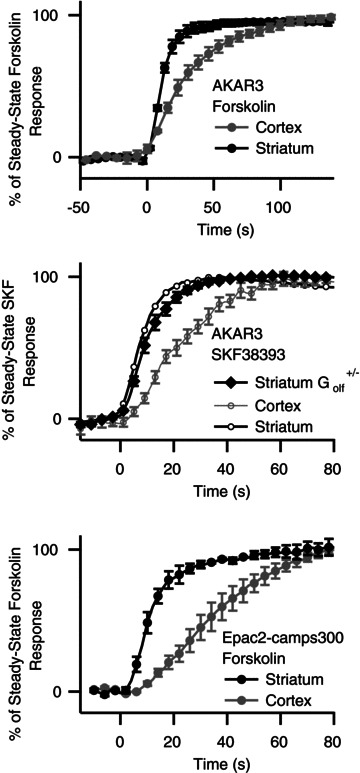
A, striatal neurones responded more rapidly than cortical neurones to a D1 stimulation. The onset of the PKA response was monitored with AKAR3 in the cortex and striatum of C57Bl/6J mice during the fast application of 1 μm SKF38393, as described in Fig. 5. This protocol was repeated (cortex: n= 8; striatum: n= 10) and the responses were averaged. B, as for A, but for the striatum of heterozygous Golf+/− mice (n= 17). Traces from A are overlaid for comparison. C, onset of the cAMP response to 13 μm forskolin, measured with Epac2-camps300, in the cortex and striatum of C57Bl/6J mice. A, B and C, bars indicate the SEM.
The striatum also expresses high levels of Golf, rather than the usual Gs, which has been shown to couple the D1 receptors to adenylyl cyclase (Corvol et al. 2001). We studied the onset kinetics of the AKAR3 response to D1-like stimulation in heterozygous Golf+/− mice, which have levels of Golf about 50% of those in wild-type mice (Corvol et al. 2001, 2007). In the striatum of Golf+/− mice, PKA activity onset was still significantly faster than in the cortex showing that there remains enough Golf protein in the striatum of Golf+/− mice to ensure a quick onset of the D1 response (Fig. 6B).
The striatum contains large amounts of adenylyl cyclases, in particular AC5 (Gehlert et al. 1984; Matsuoka et al. 1997), potentially accounting for the faster onset of the cAMP/PKA signal in striatal neurones. We tested this hypothesis by using forskolin to activate all adenylyl cyclases directly, while monitoring the increase in cAMP concentration with the Epac2-camps300 sensor: forskolin treatment led to an increase in cAMP concentration that was three times faster in the striatum than in the cortex (t1/2on= 10 ± 1 s, n= 4 and 35 ± 4 s, n= 4, respectively, unpaired two-tailed t test, P < 0.001; Fig. 6C). The type 4 phosphodiesterase inhibitor rolipram (100 nm) had no effect on onset kinetics in cortical neurones, indicating that the slower response in the cortex did not result from cAMP degradation by PDE4. Altogether, these results show that, independently of the expression level of the D1 receptor and Golf protein, cAMP production and PKA response is faster in the striatum than in the cortex.
Stronger negative control of the PKA signal in the cortex than in the striatum
We then studied the negative control exerted on the cAMP/PKA signalling cascade by examining the decay phase of the D1-like responses. We used the fast focal application system to apply a 10 s puff of SKF38393 while imaging the preparation by wide-field fluorescence microscopy (Fig. 7). For both cortex and striatum, brief SKF38393 stimulation resulted in a transient increase in AKAR3 emission ratio (Fig. 7A and B), which was reproducible, with no significant change in amplitude or kinetics. However, the responses differed considerably between the two brain regions: cortical neurones generated a small-amplitude response (69 ± 5%, n= 6, of the steady-state SKF38393 response), whereas striatal neurones displayed an almost maximal response (93 ± 2%, n= 7, of the steady-state SKF38393 response) that lasted three times longer than the response in the cortex (t1/2off= 6.9 ± 1.2 min, n= 7 vs. t1/2off= 1.9 ± 0.3 min, n= 6, respectively; unpaired two-tailed t test, P < 0.005).
Figure 7. Brief (10 s) applications of SKF38393 produced stronger and longer-lasting PKA responses in the striatum than in the cortex.
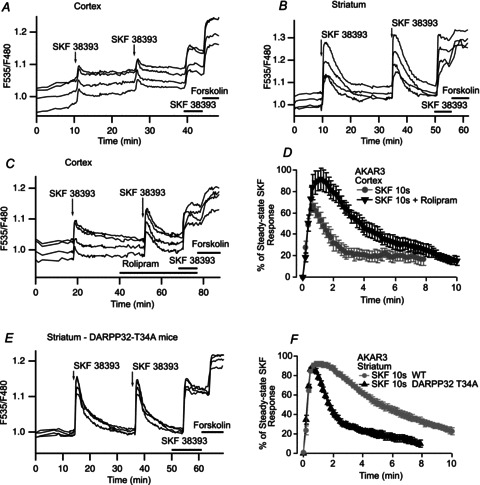
PKA activation was measured with AKAR3 during the application of a 10 s pulse of 1 μm SKF38393 in the cortex (A) and striatum (B). Each trace on the graphs indicates the AKAR3 emission ratio of an individual neurone in wide-field imaging. Responses were reproducible, with no significant difference in amplitude or kinetics. At the end of each experiment, SKF38393 and forskolin (13 μm) were added to the bath for the times indicated by the horizontal bars, and the response to the sustained presence of SKF38393 was used for normalisation. C, PKA response to a brief (10 s) application of SKF38393 (1 μm) alone and in the presence of the PDE4 inhibitor rolipram (100 nm), in the cortex: rolipram strongly increased the amplitude and prolonged the duration of the PKA response. D, mean responses in the cortex to brief SKF38393 stimulation alone (n= 6) or in the presence of rolipram (n= 4); bars indicate the SEM. E, PKA response to a brief (10 s) application of SKF38393 (1 μm) in the striatum of DARPP-32 T34A mice. F, mean responses to brief SKF38393 stimulation, in the striatum of wild-type (n= 7) and DARPP-32 T34A mice (n= 7). The higher level of phosphatase 1 activity in DARPP-32 T34A mice had no effect on the amplitude of the PKA response, but strongly reduced the duration of the PKA response.
PDE4 activity determines cAMP degradation in the cortex
Since PDE4 activity in the cortex efficiently contributes to cAMP degradation and maintains low cAMP concentration (Castro et al. 2010), we tested whether PDE4 was involved in the negative control of the transient D1 response. In the presence of 100 nm rolipram, the amplitude of the PKA response to brief D1 stimulation in the cortex was 30% greater in amplitude (96 ± 5%, n= 4, of the steady-state SKF38393 response; unpaired two-tailed t test, P < 0.002) and the decay was twice as slow (t1/2off= 4.2 ± 0.3 min, n= 4; unpaired two-tailed t test, P < 0.001) as in the absence of this compound (Fig. 7C and D). Thus, PDE4 effectively decreases cAMP levels and thereby PKA activity in the cortex, consistent with previous biochemical data (Kuroiwa et al. 2012).
In the striatum, we searched for possible effects of PDE4 on responses to SKF38393. First, we looked at a potential involvement of PDE4 in the steady-state PKA activation level reached upon sustained receptor stimulation. Since the response to 1 μm SKF38393 produced a maximal response, we used a lower concentration of 3 nm SKF38393, which does not yield maximal PKA activation. In these conditions, 100 nm rolipram had no effect on the response (20 ± 8%, n= 5 vs. 29 ± 6%, n= 14, expressed as percentage of the maximal response obtained with forskolin). Thus, PDE4 was not involved in regulating the cAMP/PKA signalling in the MSNs of the striatum, consistent with previous reports (Nishi et al. 2008).
Phosphorylation of the threonine 34 (T34) residue of DARPP-32 prolongs the effect of PKA
PDE4 inhibition in the cortex delayed the recovery of the PKA signal considerably after transient D1-like stimulation, but this recovery was still more rapid than in the striatum, suggesting that another mechanism may underlie the slower dephosphorylation occurring in the striatum. Striatal neurones express high levels of DARPP-32, which becomes a potent inhibitor of protein phosphatase 1 (PP1) following the phosphorylation of its T34 residue by PKA (Svenningsson et al. 2004). We used a knock-in mutant mouse in which this threonine residue was replaced with an alanine residue (DARPP-32 T34A) (Svenningsson et al. 2003) to determine the functional implication of DARPP-32 in PP1-mediated regulation of the PKA response.
In DARPP-32 T34A mice without DARPP-32-dependent PP1 inhibition, the recovery rate following the PKA response to 10 s of stimulation with SKF38393 was more than three times faster than that in wild-type mice (t1/2off= 2.0 ± 0.2 min, n= 7 vs. t1/2off= 6.9 ± 1.2 min, n= 7, respectively; unpaired two-tailed t test, P < 0.002; Fig. 7E and F), demonstrating that DARPP-32 plays a critical role in keeping PKA targets phosphorylated in the striatum. In these mutant mice, neither the kinetics of increase nor the amplitude of the transient response with respect to the steady-state SKF38393 response was modified. This shows that DARPP-32 in the striatum plays a critical role in inhibiting PP1 resulting in a slower recovery at the end of the D1 stimulation.
We also evaluated the role of DARPP-32 on the PKA responses in the cortex (Supplemental Fig. 5). The kinetics of the response to 10 s stimulation with SKF38393 was identical in DARPP-32 T34A and wild-type mice.
A sub-second dopamine signal is sufficient to activate PKA
Physiological activation of D1-like receptors is commonly assumed to occur after a burst of action potentials in dopaminergic neurones in response to a reward signal (Schultz & Dickinson, 2000). These action potential bursts are associated with phasic dopamine release for less than one second in the cortex or striatum (Garris & Wightman, 1994; Gonon, 1997). We mimicked phasic dopamine signals by uncaging NPEC-dopamine (5 μm) for various durations (0.1, 0.3 and 1 s), while monitoring PKA activity with AKAR3 by wide-field imaging (see Methods; Fig. 8). Cortical neurones displayed little or no response to uncaging for 0.1 s, and uncaging for 1 s yielded responses about half the maximal steady-state response to SKF38393 (Fig. 8A and B). By contrast, striatal neurones responded to NPEC-dopamine uncaging for 0.1 s with a signal of up to ∼50% of the maximal response, with the maximal response reached following uncaging for 1 s (Fig. 8C and D). The difference in response amplitude was significantly different between cortex and striatum for the responses to 0.3 and 1 s uncaging (unpaired two-tailed t test, P < 0.001). Thus, striatal neurones can detect a sub-second dopamine signal, whereas the cortex is much less sensitive.
Transient dopamine activates c-Fos expression in the striatum
We then investigated whether this brief dopamine signal was sufficient to induce gene expression and we monitored the induction of c-Fos expression by a transient dopamine stimulation. Brief NPEC-dopamine uncaging (0.1 or 1 s UV) elicited a robust induction of c-Fos immunoreactivity in the striatum after 1.5 h of incubation (Fig. 9): the number of c-Fos-labelled nuclei per field was 32 ± 3, n= 4 after uncaging for 0.1 s and 33 ± 4, n= 4 after uncaging for 1 s, both these results being significantly different from the control without caged dopamine (11.3 ± 7, n= 3, unpaired two-tailed Student's t test, P < 0.05). No significant effect was detected when the same protocol was applied to the prefrontal cortex (6 ± 3, n= 4 for 0.1 s and 7 ± 2, n= 4 for 1 s vs. 9 ± 7, n= 3 without caged dopamine). As a positive control, a bath application of SKF38393 for 5 min induced c-Fos expression in the cortex (23 ± 1, n= 5, P < 0.05). Thus, a single sub-second dopaminergic stimulation is sufficient to trigger c-Fos expression in the striatum, an effect often associated with long-term changes related to plasticity and learning, whereas the cortex remains unresponsive.
Figure 9. A sub-second dopaminergic stimulation was sufficient to induce c-Fos expression in the striatum, but not in the cortex.
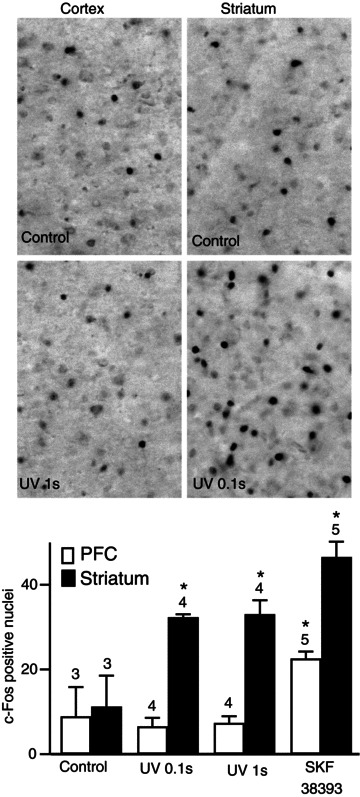
PFC, prefrontal cortex. Brain slices were stimulated with dopamine released by the local flash photolysis of NPEC-dopamine (5 μm, 0.1 or 1 s) or bath application of SKF38393 (1 μm, 5 min). The brain slice was fixed 90 min after the stimulus and c-Fos-immunoreactive nuclei were quantified in both structures. Grey-scale images show brain slices stained for c-Fos in the conditions indicated in the panel. Positive nuclei were counted and the counts were plotted on the histogram: each bar indicates the mean number of c-Fos-positive nuclei in the corresponding conditions and error bars indicate the SEM. Significant responses were obtained for all uncaging durations for the striatum, whereas the cortex responded only to bath-applied SKF38393. Unpaired two-tailed t tests were carried out for comparisons with control conditions, and differences were considered significant when P < 0.05, indicated by an asterisk on the graph.
Discussion
With the high resolution provided by biosensors, we were able to demonstrate profound differences in the responses of cortical and striatal neurones to a dopamine signal: cAMP levels remain very low in cortical neurones, whereas striatal neurones display faster, stronger and longer-lasting responses, even to sub-second dopamine stimulation.
The small increase in cAMP concentration in pyramidal cortical neurones after the activation of Gs-coupled receptors was particularly striking. Epac-based sensors have been used to report cAMP responses in various cellular systems, but the Epac1-camps biosensor, which has a micromolar affinity for cAMP, reported little or no response to stimulation of the neuropeptide receptors VPAC1, CRF1 and PAC1 in pyramidal neurones of the somatosensory cortex (Hu et al. 2011). Similarly, β1-adrenergic stimulation led to little Epac1-camps activation, whereas the more sensitive Epac2-camps300 sensor revealed a moderate increase in the cAMP signal, together with partial PKA activation (Castro et al. 2010). Similarly, only partial PKA activation was reported in response to 5-HT7 receptor agonist in the intralaminar thalamic nuclei (Gervasi et al. 2007). The D1 responses in the cerebral cortex reported here are clearly consistent with these previous findings, suggesting that cAMP concentrations in the sub-micromolar range and partial PKA activation in response to neuromodulatory stimulation are common features of various mature neurones in brain slice preparations. This observation contrasts strongly with the strong (micromolar) spontaneous or stimulated cAMP signals reported by Epac sensors in cultured embryonic neurones (Gorbunova & Spitzer, 2002; Nikolaev et al. 2004; Dunn et al. 2006; Dunn & Feller, 2008; Calebiro et al. 2009; Shelly et al. 2010; Nicol et al. 2011). These large cAMP oscillations or strong responses to neuromodulators in embryonic neurones might be a normal feature of growth, path-finding and maturation processes, whereas mature neurones need to maintain tight control over cAMP concentration, possibly to ensure spatial compartmentation of the cAMP signal. PDE4 is a key player in the negative control of cAMP concentration in cortical neurones (Yamashita et al. 1997; O’Donnell & Zhang, 2004) and its activity is required to maintain the subcellular compartmentation of the cAMP signal in various cells (Conti & Beavo, 2007), including neurones (Neves et al. 2008; Castro et al. 2010). Our results demonstrate that PDE4 is indeed essential for the clearance of cAMP after D1 receptor activation in the cortex.
Surprisingly, we found that striatal neurones in brain slice preparations behaved very differently from this model, in that they displayed faster, stronger and longer-lasting cAMP/PKA signals. We were able to identify three molecular elements responsible for this particularly strong cAMP/PKA response in the striatum. In addition to these, the high expression level of D1 receptors (Boyson et al. 1986; Dearry et al. 1990; Monsma et al. 1990; Sunahara et al. 1990; Zhou et al. 1990; Fremeau et al. 1991) may also contribute to strengthen the response, and there may be other signalling components that have not been identified yet.
By contrast to the situation in the cortex, in which PDE4 plays a critical role in cAMP degradation, our experiments show a lack of PDE4 activity in the striatum, as previously described (Nishi et al. 2008). This may reflect a lower level of PDE4 in the striatum, as only moderate levels of PDE4B are found in the striatum, whereas the cortex and many other brain regions express large amounts of the PDE4A and D isoforms (Cherry & Davis, 1999; Perez-Torres et al. 2000; Zhang et al. 2002a; Nishi et al. 2008). Indeed, other PDEs are expressed in the striatum, such as PDE1B (Polli & Kincaid, 1994), PDE2 (Van Staveren et al. 2003) and PDE10 (Seeger et al. 2003). These phosphodiesterases certainly contribute to cAMP clearance, and their respective implication in the control of cAMP dynamics remains to be analysed with this approach.
Our results also indicate that cAMP production is more efficient in the striatum than in the cortex, resulting in a faster increase in cAMP concentration and PKA activation. Direct activation of adenylyl cyclases with forskolin showed that the faster response in the striatum was a specific feature arising downstream from the receptor. Our results thus provide a functional relevance for the high levels of AC5 expression and activity reported in the striatum (Gehlert et al. 1984; Matsuoka et al. 1997; Lane-Ladd et al. 1997). In addition, it is possible that the functional coupling between Golf and AC5 in the striatum is intrinsically more efficient than the coupling between Gs and the other adenylyl cyclases in the cortex.
Positive coupling to adenylyl cyclases is mediated by Golf rather than Gs in the striatum. However, Golf+/− mice which express half as much Golf as wild-type mice still exhibit faster onset kinetics than in the cortex, suggesting that, in our experimental design, Golf is not critical for the faster onset kinetics of cAMP production. Finally, we found that the T34 residue of DARPP-32 was essential for a sustained phosphorylation response in striatal neurones. A role for DARPP-32 in amplifying and extending the duration of phosphorylation of PKA substrates dephosphorylated by PP1 has long been suggested, based on the results of classical biochemical experiments, but the kinetic aspects of these effects in neurones are largely unknown (Svenningsson et al. 2004). While there was little effect on the onset and maximum level of phosphorylation of a substrate following a transient stimulus as compared to the steady-state D1 response, we show here, by dynamic measurements in live striatal neurones, that phosphatase inhibition plays a major role in extending the duration of the response. The details probably differ between substrates, but PP1 inhibition by phosphorylated DARPP-32 is probably critical for fixing the duration of a ‘window of dopamine action’ following transient dopamine release. This may be an essential aspect of the role of DARPP-32 in multiple aspects of striatal function (Yger & Girault, 2011).
Dopamine neurones appear to be much less homogeneous than previously thought (Lammel et al. 2011; Henny et al. 2012), but they have many features in common, including their capacity to switch from tonic to burst firing (Grace & Bunney, 1980). The brief increase in dopamine release resulting from phasic activity is thought to be of critical functional importance, regulating acute striatum-dependent behaviour and its long term adaptation, presumably through the stimulation of D1 receptors (Schultz, 1998; Bromberg-Martin et al. 2010). However, there is very little direct evidence for the hypothesised ability of striatal neurones to detect short pulses of dopamine (Gonon, 1997; Tsai et al. 2009). We show here that MSNs are much more sensitive than cortical neurones to a brief exposure to dopamine. The functional relevance of this high sensitivity was revealed by the ability of a single sub-second exposure to dopamine to induce c-Fos expression in striatal, but not cortical neurones. Thus, a phasic dopamine signal encoding a strongly meaningful reward or salience signal is sufficient to have long-lasting selective consequences on MSNs. Other factors, such as the anatomical arrangement of neurotransmitter release sites and re-uptake mechanisms, may contribute to the sensitivity of the striatum to phasic dopamine release, but we demonstrate here the important contribution of the dynamics of postsynaptic signalling mechanisms to this remarkable property of the striatum. We suggest that the fitness provided by this selective sensitivity of striatal neurones to phasic dopamine release may account for the evolutionary selection of a unique set of signalling proteins in striatal neurones.
Acknowledgments
This project was supported by Université Pierre et Marie Curie, Centre National pour la Recherche Scientifique, NeRF, Fondation pour la Recherche Médicale (FDT20100920258), Fundaçao para a Ciencia e Tecnologia (Portugal) and the European Research Council. The participating groups are affiliated to the Paris School of Neuroscience (ENP) and the Bio-Psy Labex. We thank Paul Greengard for kindly providing us with the DARPP-32 mutant mice. We thank Laure Rondi-Reig and Bénédicte Babayan for expert help in c-Fos immunohistochemistry and Philippe Faure for helpful comments.
Glossary
- AC5
type 5 adenylyl cyclase
- DARPP-32
dopamine- and cAMP-dependent phosphoprotein 32 kDa
- MSNs
medium-sized spiny neurones
- PDE
phosphodiesterase
- PDE4
type 4 phosphodiesterase
- PKA
protein kinase A
- PP1
protein phosphatase 1
- Rmax
maximal ratio
- Rmin
minimal ratio
Author contributions
D.P.-T. and P.V. supervised the project. L.R.V.C., M.B., E.G. and M.P. performed the experiments. M.B. and C.W.K. did the initial observations. J.-A.G. and D.H. provided mouse lines and participated in interpretation and writing. L.R.V.C., D.P-T and P.V. wrote the manuscript. All authors have approved the submission of this version.
Author's present address
C. W. Korn: Department of Education and Psychology, Freie Universität Berlin and Berlin School of Mind and Brain, Humboldt Universität zu Berlin, Berlin, Germany.
Supplementary material
Supplemental Fig. 1
Supplemental Fig. 2A
Supplemental Fig. 2B
Supplemental Fig. 3
Supplemental Fig. 4
Supplemental Fig. 5
References
- Allen MD, Zhang J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun. 2006;348:716–721. doi: 10.1016/j.bbrc.2006.07.136. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Svenningsson P, Kuroiwa M, Gong S, Nishi A, Heintz N, Greengard P. Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat Neurosci. 2008;11:932–939. doi: 10.1038/nn.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluscio L, Gold GH, Nemes A, Axel R. Mice deficient in Golf are anosmic. Neuron. 1998;20:69–81. doi: 10.1016/s0896-6273(00)80435-3. [DOI] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Herve D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börner S, Schwede F, Schlipp A, Berisha F, Calebiro D, Lohse MJ, Nikolaev VO. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat Protoc. 2011;6:427–438. doi: 10.1038/nprot.2010.198. [DOI] [PubMed] [Google Scholar]
- Boyson SJ, McGonigle P, Molinoff PB. Quantitative autoradiographic localization of the D1 and D2 subtypes of dopamine receptors in rat brain. J Neurosci. 1986;6:3177–3188. doi: 10.1523/JNEUROSCI.06-11-03177.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron. 2010;68:815–834. doi: 10.1016/j.neuron.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7:e1000172. doi: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro LR, Gervasi N, Guiot E, Cavellini L, Nikolaev VO, Paupardin-Tritsch D, Vincent P. Type 4 phosphodiesterase plays different integrating roles in different cellular domains in pyramidal cortical neurons. J Neurosci. 2010;30:6143–6151. doi: 10.1523/JNEUROSCI.5851-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JA, Davis RL. Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement, and affect. J Comp Neurol. 1999;407:287–301. [PubMed] [Google Scholar]
- Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- Corvol JC, Studler JM, Schonn JS, Girault JA, Herve D. Gαolf is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J Neurochem. 2001;76:1585–1588. doi: 10.1046/j.1471-4159.2001.00201.x. [DOI] [PubMed] [Google Scholar]
- Corvol JC, Valjent E, Pascoli V, Robin A, Stipanovich A, Luedtke RR, Belluscio L, Girault JA, Herve D. Quantitative changes in Gαolf protein levels, but not D1 receptor, alter specifically acute responses to psychostimulants. Neuropsychopharmacology. 2007;32:1109–1121. doi: 10.1038/sj.npp.1301230. [DOI] [PubMed] [Google Scholar]
- Dearry A, Gingrich JA, Falardeau P, Fremeau RTJ, Bates MD, Caron MG. Molecular cloning and expression of the gene for a human D1 dopamine receptor. Nature. 1990;347:72–76. doi: 10.1038/347072a0. [DOI] [PubMed] [Google Scholar]
- Ducros M, Moreaux L, Bradley J, Tiret P, Griesbeck O, Charpak S. Spectral unmixing: analysis of performance in the olfactory bulb in vivo. PLoS One. 2009;4:e4418. doi: 10.1371/journal.pone.0004418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn TA, Feller MB. Imaging second messenger dynamics in developing neural circuits. Dev Neurobiol. 2008;68:835–844. doi: 10.1002/dneu.20619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn TA, Wang CT, Colicos MA, Zaccolo M, DiPilato LM, Zhang J, Tsien RY, Feller MB. Imaging of cAMP levels and protein kinase A activity reveals that retinal waves drive oscillations in second-messenger cascades. J Neurosci. 2006;26:12807–12815. doi: 10.1523/JNEUROSCI.3238-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU, Lundstrom K, Schweitzer C, Heuss C, Schlesinger S, Gähwiler BH. Recombinant Semliki Forest virus and Sindbis virus efficiently infect neurons in hippocampal slice cultures. Proc Natl Acad Sci U S A. 1999;96:7041–7046. doi: 10.1073/pnas.96.12.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RTJ, Duncan GE, Fornaretto MG, Dearry A, Gingrich JA, Breese GR, Caron MG. Localization of D1 dopamine receptor mRNA in brain supports a role in cognitive, affective, and neuroendocrine aspects of dopaminergic neurotransmission. Proc Natl Acad Sci U S A. 1991;88:3772–3776. doi: 10.1073/pnas.88.9.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris PA, Wightman RM. Different kinetics govern dopaminergic transmission in the amygdala, prefrontal cortex, and striatum: an in vivo voltammetric study. J Neurosci. 1994;14:442–450. doi: 10.1523/JNEUROSCI.14-01-00442.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlert DR, Dawson TM, Yamamura HI, Wamsley JK. Localization of [3H]forskolin binding sites in the rat brain using quantitative autoradiography. Eur J Pharmacol. 1984;106:223–225. doi: 10.1016/0014-2999(84)90707-6. [DOI] [PubMed] [Google Scholar]
- Gervasi N, Hepp R, Tricoire L, Zhang J, Lambolez B, Paupardin-Tritsch D, Vincent P. Dynamics of protein kinase A signaling at the membrane, in the cytosol, and in the nucleus of neurons in mouse brain slices. J Neurosci. 2007;27:2744–2750. doi: 10.1523/JNEUROSCI.5352-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonon F. Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum in vivo. J Neurosci. 1997;17:5972–5978. doi: 10.1523/JNEUROSCI.17-15-05972.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova YV, Spitzer NC. Dynamic interactions of cyclic AMP transients and spontaneous Ca2+ spikes. Nature. 2002;418:93–96. doi: 10.1038/nature00835. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Nigral dopamine neurons: intracellular recording and identification with L-dopa injection and histofluorescence. Science. 1980;210:654–656. doi: 10.1126/science.7433992. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Henny P, Brown MT, Northrop A, Faunes M, Ungless MA, Magill PJ, Bolam JP. Structural correlates of heterogeneous in vivo activity of midbrain dopaminergic neurons. Nat Neurosci. 2012;15:613–619. doi: 10.1038/nn.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepp R, Tricoire L, Hu E, Gervasi N, Paupardin-Tritsch D, Lambolez B, Vincent P. Phosphodiesterase type 2 and the homeostasis of cyclic GMP in living thalamic neurons. J Neurochem. 2007;102:1875–1886. doi: 10.1111/j.1471-4159.2007.04657.x. [DOI] [PubMed] [Google Scholar]
- Hervé D, Girault J-A. Signal transduction of dopamine receptors. In: Dunnett SB, Bentivoglio M, Björklund A, editors. Dopamine. Elsevier; 2005. pp. 109–151. [Google Scholar]
- Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Hu E, Demmou L, Cauli B, Gallopin T, Geoffroy H, Harris-Warrick RM, Paupardin-Tritsch D, Lambolez B, Vincent P, Hepp R. VIP, CRF, and PACAP act at distinct receptors to elicit different cAMP/PKA dynamics in the neocortex. Cereb Cortex. 2011;21:708–718. doi: 10.1093/cercor/bhq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroiwa M, Snyder GL, Shuto T, Fukuda A, Yanagawa Y, Benavides DR, Nairn AC, Bibb JA, Greengard P, Nishi A. Phosphodiesterase 4 inhibition enhances the dopamine D1 receptor/PKA/DARPP-32 signaling cascade in frontal cortex. Psychopharmacology (Berl) 2012;219:1065–1079. doi: 10.1007/s00213-011-2436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70:855–862. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landa LRJ, Harbeck M, Kaihara K, Chepurny O, Kitiphongspattana K, Graf O, Nikolaev VO, Lohse MJ, Holz GG, Roe MW. Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 β-cell line. J Biol Chem. 2005;280:31294–31302. doi: 10.1074/jbc.M505657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane-Ladd SB, Pineda J, Boundy VA, Pfeuffer T, Krupinski J, Aghajanian GK, Nestler EJ. CREB (cAMP response element-binding protein) in the locus coeruleus: biochemical, physiological, and behavioral evidence for a role in opiate dependence. J Neurosci. 1997;17:7890–7901. doi: 10.1523/JNEUROSCI.17-20-07890.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, Conti M, Fischmeister R, Vandecasteele G. Spatiotemporal dynamics of β-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res. 2008;102:1091–1100. doi: 10.1161/CIRCRESAHA.107.167817. [DOI] [PubMed] [Google Scholar]
- Matsuoka I, Suzuki Y, Defer N, Nakanishi H, Hanoune J. Differential expression of type I, II, and V adenylyl cyclase gene in the postnatal developing rat brain. J Neurochem. 1997;68:498–506. doi: 10.1046/j.1471-4159.1997.68020498.x. [DOI] [PubMed] [Google Scholar]
- Monsma FJJ, Mahan LC, McVittie LD, Gerfen CR, Sibley DR. Molecular cloning and expression of a D1 dopamine receptor linked to adenylyl cyclase activation. Proc Natl Acad Sci U S A. 1990;87:6723–6727. doi: 10.1073/pnas.87.17.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves SR, Tsokas P, Sarkar A, Grace EA, Rangamani P, Taubenfeld SM, Alberini CM, Schaff JC, Blitzer RD, Moraru II, Iyengar R. Cell shape and negative links in regulatory motifs together control spatial information flow in signaling networks. Cell. 2008;133:666–680. doi: 10.1016/j.cell.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QT, Tsai PS, Kleinfeld D. MPScope: a versatile software suite for multiphoton microscopy. J Neurosci Methods. 2006;156:351–359. doi: 10.1016/j.jneumeth.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Nicol X, Hong KP, Spitzer NC. Spatial and temporal second messenger codes for growth cone turning. Proc Natl Acad Sci U S A. 2011;108:13776–13781. doi: 10.1073/pnas.1100247108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem. 2004;279:37215–37218. doi: 10.1074/jbc.C400302200. [DOI] [PubMed] [Google Scholar]
- Nishi A, Kuroiwa M, Miller DB, O’Callaghan JP, Bateup HS, Shuto T, Sotogaku N, Fukuda T, Heintz N, Greengard P, Snyder GL. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J Neurosci. 2008;28:10460–10471. doi: 10.1523/JNEUROSCI.2518-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris RP, Ratzan WJ, Freudzon M, Mehlmann LM, Krall J, Movsesian MA, Wang H, Ke H, Nikolaev VO, Jaffe LA. Cyclic GMP from the surrounding somatic cells regulates cyclic AMP and meiosis in the mouse oocyte. Development. 2009;136:1869–1878. doi: 10.1242/dev.035238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell JM, Zhang HT. Antidepressant effects of inhibitors of cAMP phosphodiesterase (PDE4) Trends Pharmacol Sci. 2004;25:158–163. doi: 10.1016/j.tips.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Perez-Torres S, Miro X, Palacios JM, Cortes R, Puigdomenech P, Mengod G. Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and[3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J Chem Neuroanat. 2000;20:349–374. doi: 10.1016/s0891-0618(00)00097-1. [DOI] [PubMed] [Google Scholar]
- Polli JW, Kincaid RL. Expression of a calmodulin-dependent phosphodiesterase isoform (PDE1B1) correlates with brain regions having extensive dopaminergic innervation. J Neurosci. 1994;14:1251–1261. doi: 10.1523/JNEUROSCI.14-03-01251.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos BP, Birnbaum SG, Lindenmayer I, Newton SS, Duman RS, Arnsten AF. Dysregulation of protein kinase A signaling in the aged prefrontal cortex: new strategy for treating age-related cognitive decline. Neuron. 2003;40:835–845. doi: 10.1016/s0896-6273(03)00694-9. [DOI] [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dickinson A. Neuronal coding of prediction errors. Annu Rev Neurosci. 2000;23:473–500. doi: 10.1146/annurev.neuro.23.1.473. [DOI] [PubMed] [Google Scholar]
- Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL, Lanfear J, Ryan AM, Schmidt CJ, Strick CA, Varghese AH, Williams RD, Wylie PG, Menniti FS. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003;985:113–126. doi: 10.1016/s0006-8993(03)02754-9. [DOI] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, Poo MM. Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science. 2010;327:547–552. doi: 10.1126/science.1179735. [DOI] [PubMed] [Google Scholar]
- Sunahara RK, Niznik HB, Weiner DM, Stormann TM, Brann MR, Kennedy JL, Gelernter JE, Rozmahel R, Yang YL, Israel Y, Seeman P, O’Dowd BF. Human dopamine D1 receptor encoded by an intronless gene on chromosome 5. Nature. 1990;347:80–83. doi: 10.1038/347080a0. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Tzavara ET, Carruthers R, Rachleff I, Wattler S, Nehls M, McKinzie DL, Fienberg AA, Nomikos GG, Greengard P. Diverse psychotomimetics act through a common signaling pathway. Science. 2003;302:1412–1415. doi: 10.1126/science.1089681. [DOI] [PubMed] [Google Scholar]
- Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, Deisseroth K. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324:1080–1084. doi: 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Krogt GN, Ogink J, Ponsioen B, Jalink K. A comparison of donor-acceptor pairs for genetically encoded FRET sensors: application to the Epac cAMP sensor as an example. PLoS One. 2008;3:e1916. doi: 10.1371/journal.pone.0001916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Staveren WC, Steinbusch HW, Markerink-Van Ittersum M, Repaske DR, Goy MF, Kotera J, Omori K, Beavo JA, De Vente J. mRNA expression patterns of the cGMP-hydrolyzing phosphodiesterases types 2, 5, and 9 during development of the rat brain. J Comp Neurol. 2003;467:566–580. doi: 10.1002/cne.10955. [DOI] [PubMed] [Google Scholar]
- Vincent P, Castro LR, Gervasi N, Guiot E, Brito M, Paupardin-Tritsch D. PDE4 control on cAMP/PKA compartmentation revealed by biosensor imaging in neurons. Horm Metab Res. 2012;44:786–789. doi: 10.1055/s-0032-1311631. [DOI] [PubMed] [Google Scholar]
- Violin JD, Dipilato LM, Yildirim N, Elston TC, Zhang J, Lefkowitz RJ. β2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J Biol Chem. 2008;283:2949–2961. doi: 10.1074/jbc.M707009200. [DOI] [PubMed] [Google Scholar]
- Yamashita N, Yamauchi M, Baba J, Sawa A. Phosphodiesterase type 4 that regulates cAMP level in cortical neurons shows high sensitivity to rolipram. Eur J Pharmacol. 1997;337:95–102. doi: 10.1016/s0014-2999(97)01285-5. [DOI] [PubMed] [Google Scholar]
- Yger M, Girault JA. DARPP-32, Jack of All Trades. Master of Which. Front Behav Neurosci. 2011;5:56. doi: 10.3389/fnbeh.2011.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M, O’Donnell JM. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology. 2002a;27:587–595. doi: 10.1016/S0893-133X(02)00344-5. [DOI] [PubMed] [Google Scholar]
- Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002b;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Grandy DK, Thambi L, Kushner JA, Van Tol HH, Cone R, Pribnow D, Salon J, Bunzow JR, Civelli O. Cloning and expression of human and rat D1 dopamine receptors. Nature. 1990;347:76–80. doi: 10.1038/347076a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.