

Abstract
Human genetic diversity in southern Europe is higher than in other regions of the continent. This difference has been attributed to postglacial expansions, the demic diffusion of agriculture from the Near East, and gene flow from Africa. Using SNP data from 2,099 individuals in 43 populations, we show that estimates of recent shared ancestry between Europe and Africa are substantially increased when gene flow from North Africans, rather than Sub-Saharan Africans, is considered. The gradient of North African ancestry accounts for previous observations of low levels of sharing with Sub-Saharan Africa and is independent of recent gene flow from the Near East. The source of genetic diversity in southern Europe has important biomedical implications; we find that most disease risk alleles from genome-wide association studies follow expected patterns of divergence between Europe and North Africa, with the principal exception of multiple sclerosis.
Keywords: admixture, IBD segments, Maghreb, population genetics, Iberia
Multiple models have been proposed to explain clinal gradients of human genetic diversity in Europe including directional migration, climate, natural selection, and isolation by distance (1–4). A particular pattern of interest is the higher level of genetic diversity in southern European populations compared with those in northern latitudes. Three main hypotheses have been proposed to explain this phenomenon. Under the first hypothesis, populations retreated to glacial refugia in southern Europe about 20,000 y ago (ya), but when these populations later recolonized the continent, only a subset of the genetic diversity was carried into northern regions (5). The second hypothesis is that gene flow from the Near East, associated with the demic diffusion of agriculture, differentially affected geographic regions and in particular introduced additional genetic diversity to southeastern Europe (6, 7). The third hypothesis suggests that increased genetic diversity is the result of migrations from the African continent into southern Europe (8, 9). These hypotheses are not mutually exclusive; however, we focus on testing a hypothesis of gene flow from Africa to Europe, which has received the least amount of attention and may be the easiest to detect due to the recent time frame of the proposed demographic event.
About 20,000 ya during the Last Glacial Maximum, populations in Europe retreated into the glacial refugia located in the Mediterranean peninsulas, where climate conditions were milder. Differences in genetic diversity in extant European populations have been explained by a recolonization from these glacial refugia at the end of the glacial period, a process during which only a subset of the genetic diversity from the refugia would expand into the rest of the continent. For instance, radiocarbon dates suggest that recolonization of Britain took place around 14,700 ya (10). The geographic distribution and ages of mtDNA haplogroup HV0, V, H1, and H3 in European populations reflect that pattern of postglacial human recolonization from the Franco-Cantabrian refugia (11–13), and a similar pattern has also been detected in Y chromosomes as in the case of haplogroup I (14). Differential gradients of genetic diversity in many other species within Europe (e.g., grasshoppers, brown bears, and oak trees) have also been attributed to postglacial expansions during this time (15).
Changes in genetic diversity in European populations have also been associated with the Neolithic expansion from the Near East (7). The relative effect of demic diffusion of early agriculture on the genetic composition of European populations remains a hotly contested topic (16–18). It has been suggested that Near Eastern Neolithic mtDNA lineages comprise almost one quarter of the extant European haplogroups (19) and Y chromosome genetic diversity also retains a strong signal from the Near East (20). Extensive archaeological data document the spread of the Neolithic across southern Europe beginning about 8,000 ya; for example, at this time similar Neolithic pottery is found in both Europe and the Near East. However, strong similarities in pottery production are also found between southern Iberia and Northwest Africa 7,500 ya. The existence of “maritime pioneers” in the Mediterranean Sea during this period has been hypothesized (21). As a consequence, some authors support the existence of Neolithic networks joining the European and African shores of the western Mediterranean Sea (22).
Lastly, three recent studies highlight the possibility of genetic exchange between Europe and Africa. Moorjani et al. (9) estimated that about 1–3% of recent Sub-Saharan African ancestry is present in multiple southern European populations; Cerezo et al. (23) find evidence of older (11,000 ya) Sub-Saharan gene flow toward Europe based on mtDNA genomes; and Auton et al. (8) found that short haplotypes were shared between the Yoruban Nigerians and southwestern Europeans. However, given the geographic barrier imposed by the Sahara Desert between North Africa and Sub-Saharan Africa, and the proximity of North Africa to Europe, it is plausible that gene flow from Africa to Europe actually originated in North Africa. North Africans are significantly genetically diverged from Sub-Saharan populations (24, 25), and hence previous studies may not have accurately estimated the proportion or range of admixture in Europe by using a Sub-Saharan sample as a source population. For example, the Moorish Berber conquest in Iberia began in the 8th century common era and lasted for more than 500 y; this conquest has been suggested as a potential source of gene flow from North Africa toward the Iberian Peninsula. The Y chromosome haplogroup E3b2-M81 distribution is in agreement with recent North African gene flow at that period (26).
Here we analyze recently published SNP data from seven North African populations (25), together with data from 30 European populations (25, 27) (including new Affymetrix 6.0 data for three Spanish populations: Galician, Andalusian, and Canary Islands), two European Jewish populations (28), one Near Eastern population (29), and HapMap3 Sub-Saharan African populations (SI Appendix, Table S1). We aim to quantify the extent and pattern of recent gene flow between European and African populations. We use allele frequencies to estimate North African ancestry proportions in European populations. To quantify the variance in ancestry in European populations and obtain bounds on the time since admixture, we use a quantitative model for the decrease in ancestry variance with the time since admixture (30). We additionally detect gene flow between populations by analyzing long haplotypes shared identically by descent (IBD) with high-density SNP genotyping data (31, 32). We investigate regional patterns of haplotype sharing between North Africa, Sub-Saharan Africa, the Near East, and Europe in detail, and observe a significant latitudinal gradient of North African ancestry within Europe characterized by a dramatic difference between the Iberian Peninsula and the neighboring regions.
Results
Estimating Gene Flow Between Africa and Europe.
Ancestry proportions.
Previous work suggests that European and North African human populations exhibit moderate to substantial population differentiation (Fst = 0.06) (25). The degree to which admixture vs. population divergence contributes to this genetic differentiation remains largely unexplored.
To estimate allele-based sharing between Africans and Europeans, we applied an unsupervised clustering algorithm, ADMIXTURE (33), to data from all populations (SI Appendix, Table S1). We explored k = 2–10 ancestral populations and performed 10 iterations for each k (SI Appendix, Figs. S1 and S2). Our analysis does not assume that source populations are unadmixed; that is, since the analysis is run unsupervised, Sub-Saharan African ancestry, for example, can be detected in both North Africans and Europeans. Furthermore, estimates of admixture based on hundreds of thousands of markers (as we use here) show little bias using an unsupervised approach when the ancestral populations are significantly diverged (34). As the number of k ancestral clusters increased, we observed several well-supported population-specific ancestry clusters. We conservatively present k = 3 through 6 (Fig. 1) but additional results are presented in the SI Appendix.
Fig. 1.
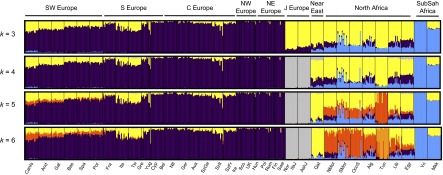
Allele-based estimates of ancestry in Europe and for European Jews, the Near East, North Africa, and Sub-Saharan Africa. Unsupervised ADMIXTURE results for k = 3–6. Cross-validation indicated k = 4 as the best fit, but higher density datasets (25) and higher values of k continue to identify population-specific ancestries (SI Appendix, Fig. S2); we therefore conservatively focused on k = 3:6 ancestral populations.
At k = 4, the ancestry assignment differentiated between non-Jewish European populations (from now on referred to as “European”), European Jews, Sub-Saharan Africans, and a group formed by Near Eastern and North African populations. At k = 5,6 components mainly assigned to North African populations and Tunisian Berbers, respectively, clearly appear. European populations sharing this North African ancestral component are almost exclusively in southern Europe (Fig. 1 and SI Appendix, Fig. S3). Southern European populations have a high proportion (5–35%) of joint Near Eastern | North African ancestry assigned at k = 4. However, identification of distinct Near Eastern and North African ancestries in k ≥ 5 differentiates southeastern from southwestern Europe. Southwestern European populations average between 4% and 20% of their genomes assigned to a North African ancestral cluster (SI Appendix, Fig. S3), whereas this value does not exceed 2% in southeastern European populations. Contrary to past observations, Sub-Saharan ancestry is detected at <1% in Europe, with the exception of the Canary Islands. In summary, when North African populations are included as a source, allele frequency-based clustering indicates better assignment to North African than to Sub-Saharan ancestry, and estimates of African ancestry in European populations increase relative to previous studies. European ancestry is also detected in North African populations. At k = 6 it ranges between 4% and 16% in the rest of North Africa, with notable intrapopulation variation (35) and is absent in most Maghrebi (western North African) individuals from Tunisia and Western Sahara.
To test whether our results were robust to the inference procedure in ADMIXTURE, we compared the ADMIXTURE results to those from a supervised machine learning algorithm, RFMix (36). Our analysis assumed three putative source populations for ancestry in Europeans: German, Saharawi, and Qatari. Estimates of North African ancestry range between 5% and 14% in the European populations and trends of the overall ancestry clines are concordant with ADMIXTURE (SI Appendix, Table S2 and Fig. S4). We tested whether ADMIXTURE could accurately infer North African ancestry proportions in Europeans via simulation of historical admixture scenarios; we find that k = 4,5 gave more accurate admixture estimates of North African ancestry. The correlation between the simulated North African ancestry and the one inferred with ADMIXTURE dramatically increases from k = 3,4 in all simulated populations (SI Appendix, Fig. S4) and the average difference in ancestry proportions at the individual level decreases from 0.04 to 0.02 when 4 or 5 ancestral components are considered.
Isolation by distance.
It has been shown that ADMIXTURE may misidentify ancestral components when the populations tested follow an isolation by distance model (37). To test whether the North African component detected by ADMIXTURE reflects admixture from distinct source populations or is a consequence of an isolation-by-distance process, we performed a Mantel test comparing pairwise genetic and geographic distances among European and North African populations. The great circle geographic distances between populations were calculated including a western waypoint located at the Gibraltar Strait for North Africa, following ref. 38. A Mantel test was performed using the software Isolation by Distance, Web Service v3.23 (39). When all European and North African populations are included in the analysis, there is a positive correlation between genetic and geographic distances of r2 = 0.268. However, this result is driven by isolation by distance within the European population (SI Appendix). When we compared genetic and geographic distances focusing only on pairwise European vs. North African comparisons, no correlation between genetic and geographic distance is found, r2 < 0.001 (P = 0.931), ruling out the hypothesis that gene flow between North Africa and Europe follows an isolation-by-distance model (SI Appendix, Fig. S5).
Long identical-by-descent haplotypes.
Recent gene flow among populations results in haplotypes shared identical by descent. To investigate differences in African ancestry among European populations, we identified genomic segments inferred to be IBD among samples from Sub-Saharan Africa, North Africa, Europe, and the Near East (SI Appendix). Migration from one endogamous population to another generates genetic segments that share a recent common ancestor (and over short time spans are IBD) between the two populations; the distribution and length of IBD segments are informative of recent migration. We restrict our analysis to IBD segments greater than 1.5 cM identified using fastIBD (40). Long IBD segments can be reliably detected even if there is substantial ascertainment bias in the SNPs used to calculate IBD state. Furthermore, by analyzing inferred IBD segments greater than 1.5 cM, we minimize background linkage disequilibrium, which affects inference of short shared haplotypes (41).
We calculated a summary statistic informative of the level of gene flow (although not the directionality) between two populations: “WEA” is the sum of lengths (in centimorgans) of all DNA segments inferred to be shared identical by descent between a given European population “E” and North African or Sub-Saharan African populations “A” normalized by the average sample size and scaled here by 100 (28). We note that extensive IBD sharing in a given genomic region may be a signal of positive selection shared among populations (42), but we do not expect extensive genome-wide sharing except through extensive gene flow. To confirm that the IBD geographic pattern was not due to natural selection, we examined excess sharing across the genome for all IBD segments (SI Appendix) in European and North African IBD individuals.
A gradient of shared IBD segments is observed from southern to northern Europe (based on WEA; Fig. 2 and SI Appendix, Table S3). This sharing is highest in the Iberian Peninsula for both North Africa and Sub-Saharan African IBD segments. Interestingly, the Basques are an exception to this pattern because they show similar levels of sharing to other European populations, but inhabit the Iberian Peninsula. Additionally, IBD sharing between North Africa and Europe is nearly an order of magnitude higher than that between Sub-Saharan Africa and Europe, of which a total of 30% of its IBD segments are also shared between North Africa and Europe. Interestingly, these segments represent only 2% of the bulk of IBD segments shared between North Africa and Europe, a proportion similar to that found in previous studies based only on Sub-Saharan populations (9). Considering that only 2% of the segments shared between North Africa and Europe have a Sub-Saharan origin, it is not likely that the gradients observed in Fig. 2B is driven primarily by theSub-Saharan segments. Finally, high correlation (0.83) exists among the values of WEA between Sub-Saharan Africa and Europe, and North Africa and Europe. Overall, these results support the hypothesis that Sub-Saharan gene flow detected in Europe entered with North African gene flow. We regressed the North African–European IBD metric (WEA) on the sine of latitude to evaluate the strength of this gradient and find a significant relationship across southern-to-northern Europe, P = 7.4 × 10−8 (Fig. 2D).
Fig. 2.
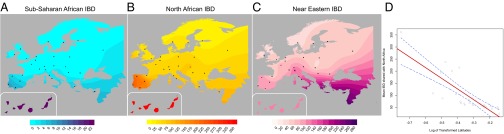
Haplotype-based estimates of genetic sharing between Europe and Africa show a significant latitudinal gradient where the highest sharing is in the Iberian Peninsula. Genetic sharing between geographic regions is represented as a density map of WEA estimates for 30 European populations where haplotypes are IBD with (A) Sub-Saharan Africa, (B) North Africa and (C) the Near East. The Canary Islands are shown in the Lower Left. (D) To determine the relationship between latitude and mean IBD count (WEA) within Europe, we regressed WEA on log(sin(latitude)). The sine of the latitude was used to obtain distance-appropriate vertical values; then we log-transformed these value to obtain the expected decay of allele sharing in 2-dimensional habitats (52). The P value of the regression for IBD shared between North Africa and Europe is 7.4 × 10−8.
To pinpoint which specific North African regions exchanged migrants with Europe, we calculated WEA between a given European population and each of the seven North African and Near Eastern populations (Fig. 3 and SI Appendix, Table S3). Southwestern European populations, and in particular the Canary Islands, show the highest levels of IBD sharing with northwestern African populations (i.e., the Maghreb: Morocco, Western Sahara, Algeria, and Tunisia), whereas southeastern European populations share more IBD segments with Egypt and the Near East (SI Appendix, Fig. S7). Whereas inferred IBD sharing does not indicate directionality, the North African samples that have highest IBD sharing with Iberian populations also tend to have the lowest proportion of the European cluster in ADMIXTURE (Fig. 1), e.g., Saharawi, Tunisian Berbers, and South Moroccans. For example, the Andalucians share many IBD segments with the Tunisians (Fig. 3), who present extremely minimal levels of European ancestry. This suggests that gene flow occurred from Africa to Europe rather than the other way around.
Fig. 3.

Population-specific estimates of haplotype sharing (in centimorgans) between North Africa and Europe. Estimates of WEA (scaled by 100 for ease of presentation) between each European population (x axis) and each North African population and the Qatari are represented by colors and symbols. A substantial increase in haplotype sharing is detected between southwestern European populations and Maghrebi populations in comparison with the remainder of the European continent. The excess of sharing between the Near East and southern central and Eastern Europe is also noteworthy.
These results also rule out a model where observed sharing between Europe and North Africa is the result of recent gene flow from the Near East into both regions. We compared IBD between Qatari (the best Near Eastern representatives genotyped with the Affymetrix platform currently available, SI Appendix, Fig. S8), Europe, and North Africa. As shown in Fig. 3 and SI Appendix, Fig. S7), southwestern Europe has more IBD segments shared with the Maghreb than Qatar, whereas eastern Mediterranean populations share more segments IBD with the Near East than with western North Africa. On the other hand, northern European populations show only limited IBD sharing with both North Africa and the Near East (Figs. 2C and 3 and SI Appendix, Fig. S7). The southwest-to-northeast gradient of North African IBD sharing (Fig. 2B) and the distinct peak in sharing between Iberia and the Maghreb (Fig. 3) indicate that sharing in southwestern Europe is independent of gene flow from the Near East. It is possible that this sharp peak of North African IBD sharing in Iberia contributes to the apparent isolation of Iberian populations from other Europeans (43).
Implications of Gene Flow from North Africa to Europe.
Time since admixture estimates.
The variance in ancestry assignments for individuals within a population depends on the total ancestry proportions, the timing and duration of gene flow, population structure and/or assortative mating within the population, and errors in assignment (30, 44). We used variance in ancestry proportions across individuals estimated with ADMIXTURE to infer effective admixture times, i.e., the times required to achieve the observed variance in the population given a single gene flow event in a randomly mating population (see model from ref. 30). Focusing on the North African component at k = 6, we found that a migration event from North Africa to Europe would have occurred at least 6–10 generations ago (∼240–300 ya) in Spain, and at least 5–7 generations ago in France and Italy (Fig. 4). The pattern of North African ancestry at k = 7 remains very similar to the pattern at k = 6 with the estimate of admixture time decreasing 1 generation on average for Iberian populations (SI Appendix, Fig. S9). Because population structure, continuous gene flow, assortative mating, and errors in assignments may considerably increase the variance (and thus reduce the effective migration time), we consider these time estimates to be lower bounds: under all of the proposed variance-increasing scenarios, there must be a substantial proportion of migration that has occurred before the effective migration time, possibly much earlier. We additionally compare the estimate variance in ancestry from simulated populations to that predicted by a pulse model of migration. We found that the estimates were consistent with the actual number of generations since migration began, within confidence intervals obtained from bootstrapping over simulations (SI Appendix, Figs. S10 and S11). Additionally, these estimates were robust to imperfect inference of the North African ancestry or source population when the pulse of gene flow occurred less than 15 generations ago.
Fig. 4.

Variance in ancestry proportions within populations depends on the overall ancestry proportions in the population and the time of gene flow. Using the proportion of North African ancestry inferred at k = 6 with ADMIXTURE, we estimated the variance in ancestry within each of 11 European populations. The gray lines show the expected relationship between ancestry proportions (x axis) and variances (Left y axis), under a single pulse model occurring at generation g (Right y axis). Departures from single-pulse models tend to increase the variance in ancestry and so the corresponding effective times should be thought of as lower bounds: significant migration must have occurred before the effective times (see text).
Disease risks.
We asked whether the migrations between North Africa and Europe affected the pattern of alleles associated with disease risk in these regions (45). By drawing on a database of genome-wide association study (GWAS) risk alleles, we determined the cumulative risk for 134 diseases in each European and African population for which we had high-density SNP data (SI Appendix). We studied the deviations from random drift for all diseases with a false discovery rate (FDR) <0.05. Pairwise q-values controlling for the FDR of all possible population comparisons within each disease (not across all diseases) were also calculated. The vast majority of disease alleles reflect expected patterns of neutral divergence (assessed with Fst) among populations. Interestingly, we found that the multiple sclerosis (MS) risk calculated from 53 independent loci displayed a significant deviation from random drift for several North African populations. Maghrebi populations (e.g., Moroccans, SI Appendix, Fig. S12) had a significantly elevated predicted genetic risk for MS, whereas the Canary Islanders, the population with highest inferred North African ancestry, had a significantly decreased risk for MS. We computed the cumulative genetic risk of each population using the 53 known SNPs associated with MS that intersect our dataset. The Northern and Southern Moroccan populations have a cumulative risk allele frequency of 0.55 and 0.52, respectively. The Canary Islanders have a cumulative risk allele frequency of 0.44. This is beyond what is expected under genetic drift (FDR < 0.05). Whereas MS prevalence is thought to increase along south-to-north latitudinal gradients in the northern hemisphere, prevalence data for North Africans are extremely limited (46). Our results suggest that North African Maghrebi have a greater genetic risk than expected under a neutral model, although presentation of MS could be attenuated by environmental variables such as UV exposure (47).
Discussion
Using genome-wide SNP data from over 2,000 individuals, we characterize broad clinal patterns of recent gene flow between Europe and Africa that have a substantial effect on genetic diversity of European populations. We have shown that recent North African ancestry is highest in southwestern Europe and decreases in northern latitudes, with a sharp difference between the Iberian Peninsula and France, where Basques are less influenced by North Africa (as suggested in ref. 48). Our estimates of shared ancestry are much higher than previously reported (up to 20% of the European individuals’ genomes). This increase in inferred African ancestry in Europe is due to our inclusion of seven North African, rather than Sub-Saharan African populations. Specifically, elevated shared African ancestry in Iberia and the Canary Islands can be traced to populations in the North African Maghreb such as Moroccans, Western Saharans, and the Tunisian Berbers. Our results, based on both allele frequencies and long shared haplotypes, support the hypothesis that recent migrations from North Africa contributed substantially to the higher genetic diversity in southwestern Europe. Previous Y chromosome data have highlighted examples of male-biased gene flow from Africa to Europe, such as the eastern African slave ancestry in Yorkshire, England (49) and the legacy of Moors in Iberia (26). Here we show that gene flow from Africa to Europe is not merely reflected on the Y chromosome but corresponds to a much broader effect.
An alternative model is that the patterns of allele sharing among North Africans and Europeans are actually due to shared ancestry among Southern Europeans and the Near East. Whereas migration(s) from the Near East have likely had an effect on genetic diversity between southern and northern Europe, they do not appear to explain the gradients of African ancestry in Europe. We detect low levels of IBD and allele sharing between the Near East and the majority of the European continent. Both IBD and allele sharing with the Near East appear elevated in southeastern Europe (e.g., Italy, Yugoslavia, and Cyprus). It is possible that these patterns reflect more ancient migrations, perhaps dating back to the Neolithic, which resulted in a low level of short Near Eastern haplotypes across much of Europe. A model of gene flow from the Near East into both Europe and North Africa, such as a strong demic wave during the Neolithic, could result in shared haplotypes between Europe and North Africa. However, the haplotype sharing we observe between Europe and the Near East follows a southeast to southwest gradient, whereas sharing between Europe and the Maghreb follows the opposite pattern (Fig. 2); this suggests that gene flow from the Near East cannot account for the sharing with North Africa.
The observation that the majority of disease risk alleles in this study follow an expected pattern of neutral drift among populations is consistent with the interpretation that these common alleles are not strongly affected by natural selection. We note that alleles identified in GWASs of individuals of largely northern European descent have limited portability to neighboring populations because the tagged GWAS SNPs may no longer be in linkage disequilibrium with the causative variant. Thus, estimates of genetic risk for these diseases in North Africans are likely inaccurate because North African-specific risk SNPs are missing. With these caveats, we note that one disease, multiple sclerosis, does not conform to a pattern of neutral genetic drift and this raises the hypothesis that natural selection affects the frequency of these risk variants that may also be linked to phenotypes other than MS. Our results show an increased genetic risk for multiple sclerosis in North African populations. West Saharans and North Moroccans carry higher frequencies of MS alleles that deviate from neutral expectations of divergence among European and African populations. Based on our model, we would predict individuals with high North African ancestry living in Europe to have a higher genetic risk for MS (see supporting evidence for North African immigrants in France in ref. 50). However, the Canary Islands, although displaying the highest amount of North African ancestry, have the lowest predicted genetic risk for MS. The complexity of these results serves to emphasize the importance of conducting disease associations in many diverse populations (51). The significant gene flow from North Africa into southern Europe will result in a miscalculation of genetic disease risk in certain European populations, if North African-specific risk variants are not taken into account.
Materials and Methods
Data.
Recently published and new single nucleotide polymorphism (SNP) data were used to build a database of 43 populations and 2,099 individuals. The database includes seven North African populations (25), together with data from 27 European populations (25, 27), two European Jewish populations (28), one Near Eastern population (29), and HapMap3 Sub-Saharan African populations (SI Appendix, Table S1). Additionally, new data for three Spanish populations [Galician (NW Spain), Andalusian (S Spain), and the Canary Islands] were included in the database. Informed consent was obtained from all newly collected Spanish populations and research was approved by the Comitè Ètic d'Investigació Clínica - Institut Municipal d'Assistència Sanitària (CEIC-IMAS), Barcelona. Samples were genotyped on the Affymetrix 6.0 chip, and quality control filtered for missing loci and close relatives. Data from these new populations can be found at http://bhusers.upf.edu/dcomas/.
ADMIXTURE Analysis.
An unsupervised clustering algorithm ADMIXTURE 1.21 (33) was used to determine allele-based sharing in a dataset of 243,000 markers formed by a total of 41 populations. For the sake of equal representation, a random subset of 15 individuals was chosen for any population having a much larger sample size. Ten ancestral clusters (k = 2 through 10) in total were tested successively, running 10 iterations for each ancestral cluster (SI Appendix, Fig. S1) and calculating cross-validation errors for every run (SI Appendix, Fig. S2). Moreover, for k = 4 through 6, 200 bootstraps were performed by resampling subsets of each chromosome, so that SEs for each ancestral cluster estimate could be obtained (33).
IBD Detection.
The analysis of IBD sharing was conducted using all of the populations in the dataset (SI Appendix, Table S1) with the exclusion of the European Jewish populations. We note that in the ADMIXTURE analysis at k = 3, there is shared ancestry between Europeans and Jewish populations; however, this could represent either shared ancestral variation or gene flow. Levels of k > 3 showed very little recent Jewish ancestry in European populations and North African populations show negligible ancestry from North African Jews (35). The removal of Jewish populations from the dataset increased the number of common markers from 243,000 to 274,000 and to a total of 41 populations.
Correction for Sample Size.
To compare between the different statistics calculated from the IBD results, we correct for sample size, given that in European populations there are differences in sample size of two orders of magnitude. We follow Atzmon et al. (28)’s calculation of the average pairwise population IBD sharing metrics:
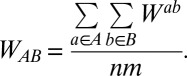 |
SD from WAB statistic was obtained for each pairwise comparison and scaled by 100 for ease of presentation.
Supplementary Material
Acknowledgments
We are grateful to Oscar Lao, Atul Butte, and Graham Coop for helpful suggestions, Txema Heredia for Information Technology help, the North African and Spanish participants for their generous contributions of DNA, and the Banco Nacional de ADN (DNA) for providing the Galician and Andalusian samples. B.M.H. and C.D.B. were supported by National Institutes of Health Grant 3R01HG003229. L.R.B. and D.C. were supported by Ministerio de Ciencia e Innovación Grant CGL2010-14944/BOS and Generalitat de Catalunya Grant 2009SGR1101. C.F. was supported by Instituto de Salud Carlos III Grant PI11/00623. The Spanish National Institute for Bioinformatics supported this project.
Footnotes
Conflict of interest statement. C.D.B. is on the Scientific and/or Medical Advisory Boards of Personalis, InVitae, Ancestry.com, MedTek, and the 23andMe, Inc. “Roots into the future” project. None of these entities played any role in the experimental design, data collection, or analysis of the project data.
This article is a PNAS Direct Submission.
Data deposition: The data from new populations has been made available from the Human Genome Diversity Panel at the IBE (Institut de Biologia Evolutiva), http://bhusers.upf.edu/dcomas.
See Commentary on page 11668.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1306223110/-/DCSupplemental.
References
- 1.Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nat Genet. 2008;40(5):646–649. doi: 10.1038/ng.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pickrell JK, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19(5):826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lao O, et al. Correlation between genetic and geographic structure in Europe. Curr Biol. 2008;18(16):1241–1248. doi: 10.1016/j.cub.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 4.Novembre J, et al. Genes mirror geography within Europe. Nature. 2008;456(7218):98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forster P. Ice Ages and the mitochondrial DNA chronology of human dispersals: A review. Philos Trans R Soc Lond B Biol Sci. 2004;359(1442):255–264, discussion 264. doi: 10.1098/rstb.2003.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Currat M, Excoffier L. The effect of the Neolithic expansion on European molecular diversity. Proc Biol Sci. 2005;272(1564):679–688. doi: 10.1098/rspb.2004.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cavalli-Sforza LL, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton, NJ: Princeton Univ Press; 1994. [Google Scholar]
- 8.Auton A, et al. Global distribution of genomic diversity underscores rich complex history of continental human populations. Genome Res. 2009;19(5):795–803. doi: 10.1101/gr.088898.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moorjani P, et al. The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet. 2011;7(4):e1001373. doi: 10.1371/journal.pgen.1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobi RM, Higham TFG. The early Late glacial re-colonization of Britain: New radiocarbon evidence from Gough's Cave, southwest England. Quat Sci Rev. 2009;28(19–20):1895–1913. [Google Scholar]
- 11.Torroni A, et al. A signal, from human mtDNA, of postglacial recolonization in Europe. Am J Hum Genet. 2001;69(4):844–852. doi: 10.1086/323485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Achilli A, et al. The molecular dissection of mtDNA haplogroup H confirms that the Franco-Cantabrian glacial refuge was a major source for the European gene pool. Am J Hum Genet. 2004;75(5):910–918. doi: 10.1086/425590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pereira L, et al. High-resolution mtDNA evidence for the late-glacial resettlement of Europe from an Iberian refugium. Genome Res. 2005;15(1):19–24. doi: 10.1101/gr.3182305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rootsi S, et al. Phylogeography of Y-chromosome haplogroup I reveals distinct domains of prehistoric gene flow in europe. Am J Hum Genet. 2004;75(1):128–137. doi: 10.1086/422196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hewitt GM. Post-glacial re-colonization of European biota. Biol J Linn Soc Lond. 1999;68(1-2):87–112. [Google Scholar]
- 16.Haak W, et al. Ancient DNA from the first European farmers in 7500-year-old Neolithic sites. Science. 2005;310(5750):1016–1018. doi: 10.1126/science.1118725. [DOI] [PubMed] [Google Scholar]
- 17.Bramanti B, et al. Genetic discontinuity between local hunter-gatherers and central Europe’s first farmers. Science. 2009;326(5949):137–140. doi: 10.1126/science.1176869. [DOI] [PubMed] [Google Scholar]
- 18.Gignoux CR, Henn BM, Mountain JL. Rapid, global demographic expansions after the origins of agriculture. Proc Natl Acad Sci USA. 2011;108(15):6044–6049. doi: 10.1073/pnas.0914274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards M, et al. Tracing European founder lineages in the Near Eastern mtDNA pool. Am J Hum Genet. 2000;67(5):1251–1276. [PMC free article] [PubMed] [Google Scholar]
- 20.Rosser ZH, et al. Y-chromosomal diversity in Europe is clinal and influenced primarily by geography, rather than by language. Am J Hum Genet. 2000;67(6):1526–1543. doi: 10.1086/316890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zilhão J. Radiocarbon evidence for maritime pioneer colonization at the origins of farming in west Mediterranean Europe. Proc Natl Acad Sci USA. 2001;98(24):14180–14185. doi: 10.1073/pnas.241522898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Linstädter J, Medved I, Solich M, Weniger G-C (2012) Neolithisation process within the Alboran territory: Models and possible African impact. Quaternary International 274:219–232.
- 23.Cerezo M, et al. Reconstructing ancient mitochondrial DNA links between Africa and Europe. Genome Res. 2012;22(5):821–826. doi: 10.1101/gr.134452.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fadhlaoui-Zid K, et al. Mitochondrial DNA structure in North Africa reveals a genetic discontinuity in the Nile Valley. Am J Phys Anthropol. 2011;145(1):107–117. doi: 10.1002/ajpa.21472. [DOI] [PubMed] [Google Scholar]
- 25.Henn BM, et al. Genomic ancestry of North Africans supports back-to-Africa migrations. PLoS Genet. 2012;8(1):e1002397. doi: 10.1371/journal.pgen.1002397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams SM, et al. The genetic legacy of religious diversity and intolerance: Paternal lineages of Christians, Jews, and Muslims in the Iberian Peninsula. Am J Hum Genet. 2008;83(6):725–736. doi: 10.1016/j.ajhg.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson MR, et al. The Population Reference Sample, POPRES: A resource for population, disease, and pharmacological genetics research. Am J Hum Genet. 2008;83(3):347–358. doi: 10.1016/j.ajhg.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Atzmon G, et al. Abraham’s children in the genome era: Major Jewish diaspora populations comprise distinct genetic clusters with shared Middle Eastern Ancestry. Am J Hum Genet. 2010;86(6):850–859. doi: 10.1016/j.ajhg.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunter-Zinck H, et al. Population genetic structure of the people of Qatar. Am J Hum Genet. 2010;87(1):17–25. doi: 10.1016/j.ajhg.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gravel S. Population genetics models of local ancestry. Genetics. 2012;191(2):607–619. doi: 10.1534/genetics.112.139808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Browning SR, Browning BL. High-resolution detection of identity by descent in unrelated individuals. Am J Hum Genet. 2010;86(4):526–539. doi: 10.1016/j.ajhg.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gusev A, et al. The architecture of long-range haplotypes shared within and across populations. Mol Biol Evol. 2012;29(2):473–486. doi: 10.1093/molbev/msr133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexander DH, Lange K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics. 2011;12:246. doi: 10.1186/1471-2105-12-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell CL, et al. North African Jewish and non-Jewish populations form distinctive, orthogonal clusters. Proc Natl Acad Sci USA. 2012;109(34):13865–13870. doi: 10.1073/pnas.1204840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abecasis GR, et al. 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Safner T, Miller MP, McRae BH, Fortin MJ, Manel S. Comparison of bayesian clustering and edge detection methods for inferring boundaries in landscape genetics. Int J Mol Sci. 2011;12(2):865–889. doi: 10.3390/ijms12020865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramachandran S, et al. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci USA. 2005;102(44):15942–15947. doi: 10.1073/pnas.0507611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jensen JL, Bohonak AJ, Kelley ST. Isolation by distance, web service. BMC Genet. 2005;6:13. doi: 10.1186/1471-2156-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Browning BL, Browning SR. A fast, powerful method for detecting identity by descent. Am J Hum Genet. 2011;88(2):173–182. doi: 10.1016/j.ajhg.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conrad DF, et al. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet. 2006;38(11):1251–1260. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- 42.Albrechtsen A, Moltke I, Nielsen R. Natural selection and the distribution of identity-by-descent in the human genome. Genetics. 2010;186(1):295–308. doi: 10.1534/genetics.110.113977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ralph P, Coop G (2013) The geography of recent genetic ancestry across Europe. PLoS Biol 11(5):e1001555. [DOI] [PMC free article] [PubMed]
- 44.Pool JE, Nielsen R. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics. 2009;181(2):711–719. doi: 10.1534/genetics.108.098095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corona E, Dudley JT, Butte AJ. Extreme evolutionary disparities seen in positive selection across seven complex diseases. PLoS ONE. 2010;5(8):e12236. doi: 10.1371/journal.pone.0012236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosati G. The prevalence of multiple sclerosis in the world: An update. Neurol Sci. 2001;22(2):117–139. doi: 10.1007/s100720170011. [DOI] [PubMed] [Google Scholar]
- 47.Handel AE, Giovannoni G, Ebers GC, Ramagopalan SV. Environmental factors and their timing in adult-onset multiple sclerosis. Nat Rev Neurol. 2010;6(3):156–166. doi: 10.1038/nrneurol.2010.1. [DOI] [PubMed] [Google Scholar]
- 48.Martínez-Cruz B, et al. Genographic Consortium Evidence of pre-Roman tribal genetic structure in Basques from uniparentally inherited markers. Mol Biol Evol. 2012;29(9):2211–2222. doi: 10.1093/molbev/mss091. [DOI] [PubMed] [Google Scholar]
- 49.King TE, et al. Africans in Yorkshire? The deepest-rooting clade of the Y phylogeny within an English genealogy. Eur J Hum Genet. 2007;15(3):288–293. doi: 10.1038/sj.ejhg.5201771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurtzke JF, Delasnerie-Lauprêtre N, Wallin MT. Multiple sclerosis in North African migrants to France. Acta Neurol Scand. 1998;98(5):302–309. doi: 10.1111/j.1600-0404.1998.tb01738.x. [DOI] [PubMed] [Google Scholar]
- 51.Bustamante CD, Burchard EG, De la Vega FM. Genomics for the world. Nature. 2011;475(7355):163–165. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rousset F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997;145(4):1219–1228. doi: 10.1093/genetics/145.4.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.