

Abstract
Sickle cell disease can lead to hepatic complications ranging from acute hepatic crises to chronic liver disease including intrahepatic cholestasis, and iron overload. Although uncommon, intrahepatic cholestasis may be severe and medical treatment of this complication is often ineffective. We report a case of a 37 year-old male patient with sickle cell anemia, who developed liver failure and underwent successful orthotopic liver transplantation. Both pre and post-operatively, he was maintained on red cell transfusions. He remains stable with improved liver function 42 months post transplant. The role for orthotopic liver transplantation is not well defined in patients with sickle cell disease, and the experience remains limited. Although considerable challenges of post-transplant graft complications remain, orthotopic liver transplantation should be considered as a treatment option for sickle cell disease patients with end-stage liver disease who have progressed despite conventional medical therapy. An extended period of red cell transfusion support may lessen the post-operative complications.
Key words: sickle cell, liver transplantation, cholestasis, transfusions
Introduction
Sickle cell disease (SCD) is an inherited disorder of hemoglobin (HbS) leading to complications in multiple organ systems. Many complications are due to either recurrent vasoocclusion or chronic hemolysis. Frequently observed manifestations of hepatic and biliary tract disease in sickle cell disease patients include vaso-occlusion of hepatic sinusoids due to the sickling process, hemosiderosis from chronic transfusions, viral hepatitis, and cholelithiasis due to chronic hemolysis.1 In many patients the cause of liver dysfunction is multi-factorial.
Acute vaso-occlusion is a common event resulting from polymerization of deoxygenated HbS leading to decreased pliability of red blood cell (RBC) membrane leading to sickle shaped cells, and increasing the adhesiveness of the RBC membrane to the endothelium of small vessels. When sickling occurs in the bone, these changes result in microvascular occlusions that cause ischemia which manifest as vaso-occlusive pain crises. In the liver, vasoocclusion of sickled RBCs occurs in the hepatic sinusoids, perhaps due to low oxygen tension. 1 The obstructed hepatic sinusoids generate hepatic congestion leading to hepatomegaly and hepatocyte necrosis. In addition, Kupffer cell hypertrophy from increased phagocytosis of the sickled RBCs contributes to further sinusoidal obstruction.2 This phenomenon of sickle RBC induced sinusoidal obstruction has been termed sickle cell hepatopathy.1,3,4 Sinusoidal congestion and ischemic necrosis may lead to cirrhosis. In one study by Traina et al., among 20 SCD patients with liver dysfunction who underwent liver biopsy 19 patients demonstrated sickle cell hepatopathy from intra-sinusoidal congestion and vaso-occlusion.1
Congestion of hepatic sinusoids may manifest as a transient acute hepatic crisis associated with right upper quadrant pain, jaundice, fever, leukocytosis, increased serum aminotransferases and elevated bilirubin.3 This form of sickle cell crisis often resolves without clinical consequences. However, rarely the sequestration of sickle cells in the hepatic sinusoids may lead to more severe and potentially fatal sickle cell intrahepatic cholestasis (SCIC), which usually presents as acute hepatic failure from local ischemia.3,5,6 Untreated SCIC has been associated with a mortality rate of up to 40%.6 While treatment with red cell transfusions improves survival over supportive management alone, it is often ineffective with a mortality of 17% in chronic intrahepatic cholestasis.6,7 The role for orthotopic liver transplant (OLT) in patients with SCD and liver disease is not well defined. There have only been several cases of OLT for sickle cell disease patients with liver failure. We describe a case of a patient with sickle cell anemia and end-stage liver disease from sickle cell cholestasis who underwent a successful OLT and was maintained on perioperative transfusions with a long-term survival.
Case Report
Our patient is a 37 year old African American male with homozygous sickle cell disease whose course included intermittent vaso-occlusive crises resulting in 1-2 hospitalizations per year. He has a history of cholecystectomy in 1991 and hospitalizations for acute pulmonary infiltrates in 1992 and 1998. His estimated RBC transfusion burden was ~20 units prior to his OLT. Throughout his adult course aminotransferases (AST, ALT) and bilirubin were elevated (Figure 1). Viral serologies for hepatitis B, hepatitis C and human immunodeficiency virus were negative and ferritin was 129 ng/mL. In 2005, treatment with hydroxyurea 1g daily was started and he used occasional opioids as needed.
Figure 1.
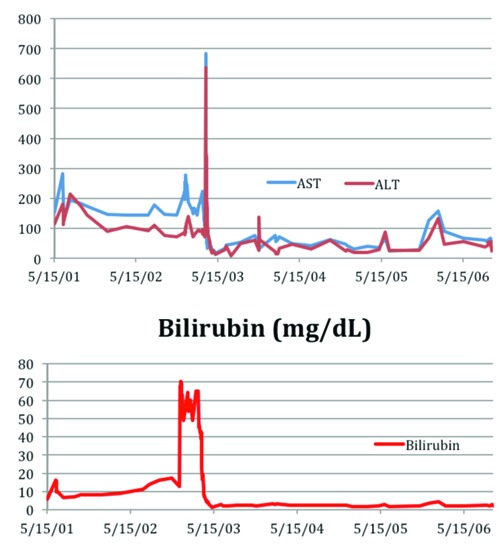
Aspartate aminotransferase/alanine aminotransferase (AST/ALT) (IU/L), and bilirubin (mg/dL).
In August 2006, 8 months prior to orthotopic liver transplant, he developed jaundice. His total bilirubin was 14.0 ng/dL, alkaline phosphatase 614, AST 176, ALT 110, and ferritin 237 ng/mL. Liver biopsy in November 2006 showed severe cholestasis, bile ductular proliferation and cirrhosis (Figure 2). RBC exchange transfusions were begun and continued every 4-6 weeks to achieve Hgb S concentrations <30%. One month prior to his transplant, he was maintained on simple RBC transfusions to maintain Hgb >7 g/dL.
Figure 2.
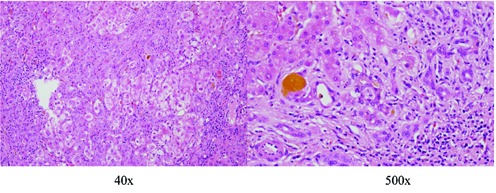
Liver biopsy in 2006 - severe cholestasis with fibrosis, ductular proliferation and cirrhosis (Haematoxilyn and Eosin stain).
In March 2007 he was admitted to the Intensive Care Unit with severe anemia, acute renal insufficiency, and deepening jaundice. Laboratory data included Hgb 5.8 g/dL, Hct 17.1, Hgb S 15.7%, bilirubin 46.5, alk phos 387, AST 223, ALT 93, and creatinine 4.5. He was evaluated and listed for OLT with a Model for End-Stage Liver Disease score of 40. Fourteen days later he underwent an orthotopic liver transplantation from an ABO matched adult cadaveric donor.
The transplantation was accomplished with minimal blood loss. During surgery the patient was transfused with 3 units of packed RBCs, 1000 mL of 5% albumin and 3 liters of crystalloid. The donor graft appeared normal; the cold ischemia time was 9 hours and the warm ischemia time was 28 minutes. The graft liver was placed in a standard piggyback fashion with bile duct-to-duct anastomosis. The liver explant showed cirrhosis with marked autolysis, and cholestasis with bile duct proliferation.
The post-operative course was complicated by seizures and acute respiratory distress, which required intubation during the post-operative course. His Hgb was maintained throughout the hospital course between 7-10 and Hgb S <2% with RBC transfusions. His urine output, lung function and mental status improved gradually, and he was discharged on the postoperative day 31 in stable condition.
The patient received immunosuppression with mycophenolate mofetil, prednisone, and tacrolimus. The prednisone was gradually tapered and he was maintained on mycophenolate mofetil and tacrolimus long-term. He was maintained on 2 units RBC transfusions every 4 weeks for target of Hgb of 10 g/dL for one year. His AST and ALT have decreased since the transplantation and liver function has improved (Figures 1 and 2). He had no post-operative sickle cell related complications and 12 months after transplant RBC treatments were stopped and he has been maintained on hydroxyurea.
Discussion and Conclusions
SCIC is a highly fatal complication of sickle cell disease. It has been documented that intrahepatic cholestasis has a mortality of 40% without the use of exchange transfusions.6,8 In a review of 26 reported patients, Khurshid et al. found that with exchange transfusion therapy, the mortality from intrahepatic cholestasis was 17% suggesting that it remains a potentially fatal condition despite medical treatment.7 In the patient reported here, RBC exchange transfusion therapy every 4-6 weeks for 3 months prior to liver transplantation was successful to maintain a target Hgb S <30%, but his liver function continued to decline. In these patients, OLT becomes the only therapeutic option.
There have been few reported cases of OLT in patients with sickle cell disease. A review of the literature identified 10 adult and 5 children with sickle cell disease (Table 1).2,5,6,9-17 In 6 of the 10 reported adult cases, liver transplantation resulted in improved function and long-term survival. Of the 4 that resulted in post-OLT death, 2 were from causes unrelated to the OLT or liver dysfunction (1 from intracranial bleed 5 months post-op due to pre-existing vascular anomaly and 1 from pulmonary embolism 22 months post-op). Nevertheless, sickle cell related allograft complications occurred in 5 of the 6 survived cases. Among 3 of 5 pediatric cases, transplantation was successful.
Table 1.
Review of orthotopic liver transplant cases in sickle cell disease patients.
| Study citation | Sex | Age | HbType | Liver pathologies | Transfusion management (pre-op goal) | Transfusion management (post-op goal) | Outcome |
|---|---|---|---|---|---|---|---|
| Perini 20109 | M | 37 | HbS beta thalassemia | Hepatitis C; hemosiderosis | Not stated | Post-op target HbS<30% with exchange transfusions | Death after 5 months 3 months: acute sickle hepatic crisis with elevated LFTs and RUQ pain-resolved with exchange transfusions and hydroxycarbamide 5 months: died from intracranial hemorrhage from Moyamoya disease |
| Greenberg 201010 | F | 30 | HbSS | Extensive hepatocyte necrosis but no signs of SC1C or pregnancy related hepatopathy | RBC exchange from presenting HbS=80% to HbS=26% |
Post-op: HbS<30% | Survival POD 1-3:ICU requiring mechanical ventilation POD 28: discharge home |
| Baichi 20056 | F | 27 | HbSS | SC1C; autoimmune hepatitis; cirrhosis | Presented with HbS=69.6%, exchange transfusion to keep HbS<10% | Exchange transfusion to keep HbS<10% | Death on POD 35 from peritoneal bleed and MOF |
| Baichi 20056 | F | 26 | HbSS | Sclerosing cholangitis; periductal fibrosis consistent with intrahepatic cholestasis | Presented initially with HbS=15.1%; exact amount of transfusion not stated | HbS<10% without any transfusions during OLT hospitalization | Death on POD 85 from septic shock and MOF |
| Kindscher 19952 | F | 47 | HbSS | Chronic hepatitis C; cirrhosis | Exchanged 4 units to reduce HbS from 52% to 27% | Target HbS<30% | Survival: discharge home on POD 40; no signs of rejection Complication: cerebral hemorrhage POD 12 managed conservatively w/ CNS improvement after 3 months f/u |
| Ross 200211 | M | 49 | HbSS | SC1C | Initially presented with Hb=5.2 and HbS=52%, which prompted 6 U RBC transfusion | Target HbS<20% | Death: discharge POD 33 Complication: biliary anastomosis leak repair POD 7 22 months Post-op: death from pulmonary embolus; autopsy reveals no cholestasis or rejection |
| Gilli 20025 | M | 22 | HbS beta thalassemia | SC1C; cirrhosis | Target HbS<30% | Target HbS<20% | Survival: mild intrahepatic sickling 3 months post-op; no signs of rejection 2 years post-op |
| Delis 200612 | F | 19 | HbSD | Hepatitis B; cirrhosis | Not stated | Exchange transfusion at 17 months post-op for elevated LFTs: decreased HbS from 33.8 to 7.8% | Survival: no rejection Complication: intrahepatic cholestasis of allograft resolved after transfusion |
| Van den Hazel 200313 | M | 23 | HbSS | Hemochromatosis | 5 U RBCs exchange decreased HbS from 32% to 19% | Intra-op: transfused 44 U RBCs HbS=4.9% 1 wk post-op (w/o tranfusion) | Survival: no rejection; liver functions intact at 5.5 yrs post-OLT |
| Lerut 199914 | F | 42 | HbS beta thalassemia | Cryptogenic cirrhosis | Target not stated | Target HbS<10% | Survival Complication: partial graft infarction/necrosis at post-op 6 month-spontaneous recovery |
| Emre200015 | M | 6 | HbSS | Dilated sinusoids filled with sickled RBCs | HbS=21.6% at presentation prior to first OLT; pre-op target not stated | Target HbS<20% | Death from sepsis after 3rd transplant; 1st graft failed due to veno-occlusive disease; 2nd graft failed from hepatic artery thrombosis |
| Lang 199516 | M | 11 | HbSS | Biliary cirrhosis | Not stated | HbS<20% | Survival; intact graft function |
| Meekel 200717 | N/A | 8,17,17 | Not specified | 3 had intrahepatic cholestasis; 1 had chronic hepatitis C | Exchange transfusions for target HbS<25% and Hb>9 | Exchange transfusions for target HbS<25% and Hb>9 | 2 Survived 1 Death: recurrent intrahepatic cholestasis and (3 patients) chronic graft failure, and died 6 yrs post-op from subdural hematoma from fall |
Hb, hemoglobin; SCIC, sickle cell intrahepatic cholestasis; RBC, red blood cell; OLT, orthotopic liver transplant; MOF, multiorgan failure.
There are a number of challenges in considering OLT for sickle cell disease patients. First, surgery itself presents as a procedure with high morbidity and mortality risk. Early studies show on average 10% mortality and >50% risk of perioperative complications in sickle cell disease patients undergoing a variety of surgical procedures but this can be reduced substantially with preoperative RBC transfusion. 17,18 Sickle cell related complications such as intrahepatic sickling and vaso-occlusion can recur in the graft causing deterioration in function.5,12,14,15,17 Preoperative transfusion therapy can be used to mitigate the post-op complications. The National Preoperative Transfusion Study evaluating the risk of cholecystectomy in SCD patients found that post-op complications of SCD were higher in the nontransfused than the transfused group.19 Our patient was heavily transfused in preparation for his liver transplant to decrease sickle cell related complications. Notably, post-operative transfusion therapy has been utilized by a majority of the OLT cases to reduce risk of long-term graft sickle cell related complications. Nevertheless, the intensity and duration of transfusion therapy is not well defined and varies between studies. The experience with our patient suggests that aggressive RBC transfusion is warranted; however, chronic transfusion therapy places patients at risk for transfusion related iron overload.
Liver failure secondary to sickle cell intraheptic cholestasis is a complicated clinical problem and in our case, medical management with hydroxyurea, and aggressive RBC transfusions were not able to reverse his declining liver function. The current experience and role for OLT in sickle cell disease patients is limited. This case had a successful outcome with OLT plus RBC support. OLT should be considered as treatment option for sickle cell disease patients with advanced liver failure.
References
- 1.Traina F, Jorge SG, Yamanaka A, et al. Chronic liver abnormalities in sickle cell disease: a clinicopathological study in 70 living patients. Acta Haematol. 2007; 118:129-35 [DOI] [PubMed] [Google Scholar]
- 2.Kindscher JD, Laurin J, Delcore R, Forster J.Liver transplantation in a patient with sickle cell anemia. Transplantation. 1995; 60:762-4 [DOI] [PubMed] [Google Scholar]
- 3.Banerjee S, Owen C, Chopra S. Sickle cell hepatopathy. Heptology. 2001; 33:1021-8 [DOI] [PubMed] [Google Scholar]
- 4.Shao S, Orringer E. Sickle cell intrahepatic cholestasis: approach to a difficult problem. Gastroenterology 1995;90:2048. [PubMed] [Google Scholar]
- 5.Gilli SCO, Boin IFS, Leonardi LS, et al. Liver transplantation in a patient with S/beta-thalassemia. Transplantation. 2002; 74:896-8 [DOI] [PubMed] [Google Scholar]
- 6.Baichi MM, Arifuddin RM, Mantry PS, et al. Liver transplanatation in sickle cell anemia: a case of acute sickle cell intrahepatic cholestasis and a case of sclerosing cholangitis. Transplantation. 2005; 80:1630-2 [DOI] [PubMed] [Google Scholar]
- 7.Khurshid I, Anderson L, Downie GH, Pape GS. Sickle cell disease, extreme hyperbilirubinemia, and pericardial tamponade: case report and review of the literature. Crit Care Med. 2002; 30:2363-7 [DOI] [PubMed] [Google Scholar]
- 8.Sheehy TW, Law DE, Wade BH. Exchange transfusion for sickle cell intrahepatic cholestasis. Arch Intern Med. 1980; 140:1354-66 [PubMed] [Google Scholar]
- 9.Perini GF, Santos FPS, Ferraz Neto JBH, et al. Acute sickle hepatic crisis after liver transplantation in a patient with sickle beta-thalassemia. Transplantation. 2010; 90:463-4 [DOI] [PubMed] [Google Scholar]
- 10.Greenberg M, Daugherty TJ, Elihu A, et al. Acute liver failure at 26 weeks’ gestation in a patient with sickle cell disease. Liver Transplantation. 2009; 15:1236-41 [DOI] [PubMed] [Google Scholar]
- 11.Ross AS, Graeme-Cook F, Cosimi BA, Chung R. Combined liver and kidney transplantation in a patient with sickle cell disease. Transplantation Feb. 2002; 73:605-8 [DOI] [PubMed] [Google Scholar]
- 12.Delis SG, Dervenis C. Is there a role of exchange transfusions in patients with sickle cell anemia and major liver surgery? European Society for Organ Transplantation. 2007; 20:299-300 [DOI] [PubMed] [Google Scholar]
- 13.van den Hazel SJ, Metselaar HJ, Tilanus HW, et al. Successful liver transplantation in a patient with sickle-cell anaemia. Transpl Int. 2003; 16:434-6 [DOI] [PubMed] [Google Scholar]
- 14.Lerut JP, Claeys N, Laterre PF, et al. Hepatic sickling: an unusual cause of liver allograft dysfunction. Transplantation. 1999; 67:65-8 [DOI] [PubMed] [Google Scholar]
- 15.Emre S, Kitibayashi K, Schwartz ME, et al. Liver transplantation in a patient with acute liver failure due to sickle cell intrahepatic cholestasis. Transplantation. 2000; 69:675-7 [DOI] [PubMed] [Google Scholar]
- 16.Lang T, Berquist WE, So SK, et al. Liver transplantation in a child with sickle cell anemia. Transplantation. 1995; 59:1490-2 [DOI] [PubMed] [Google Scholar]
- 17.Mekeel KL, Langham MR, Gonzalez-Peralta R, et al. Liver transplantation in children with sickle-cell disease. Liver Transplantation. 2007; 13:505-8 [DOI] [PubMed] [Google Scholar]
- 18.Vichinsky EP, Haberkern CM, Neumayr L, et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. N Engl J Med. 1995; 333:206-13 [DOI] [PubMed] [Google Scholar]
- 19.Haberkern CM, Neumayr LD, Orringer EP, et al. Cholecystecomy in sickle cell anemia patients: perioperative outcome of 364 cases from the National Preoperative Transfusion Study. Blood. 1997; 89:1533-42 [PubMed] [Google Scholar]