

Abstract
Background & Aims
Proprotein convertase 1/3 (PC1/3) deficiency, an autosomal recessive disorder caused by rare mutations in the PCSK1 gene, has been associated with obesity, severe malabsorptive diarrhea, and certain endocrine abnormalities. Common variants in PCSK1 have also been associated with obesity in heterozygotes in several population studies. PC1/3 is an endoprotease that processes many prohormones expressed in endocrine and neuronal cells. We investigated clinical and molecular features of PC1/3 deficiency.
Methods
We studied the clinical features of 13 children with PC1/3 deficiency and performed sequence analysis of PCSK1. We measured enzymatic activity of recombinant PC1/3 proteins.
Results
We identified a pattern of endocrinopathies that develop in an age-dependent manner. Eight of the mutations had severe biochemical consequences in vitro. Neonates had severe malabsorptive diarrhea and failure to thrive, required prolonged parenteral nutrition support, and had high mortality. Additional endocrine abnormalities developed as the disease progressed, including diabetes insipidus, growth hormone deficiency, primary hypogonadism, adrenal insufficiency, and hypothyroidism. We identified growth hormone deficiency, central diabetes insipidus, and male hypogonadism as new features of PCSK1 insufficiency. Interestingly, despite early growth abnormalities, moderate obesity, associated with severe polyphagia, generally appears.
Conclusion
In a study of 13 children with PC1/3 deficiency caused by disruption of PCSK1, failure of enteroendocrine cells to produce functional hormones resulted in generalized malabsorption. These findings indicate that PC1/3 is involved in processing of one or more enteric hormones that are required for nutrient absorption.
Keywords: genetic, congenital diarrheal disorder, enteroendocrine development, NEUROGENIN
Introduction
Congenital diarrheal disorders are rare and caused by a diverse group of inherited mutations that have been identified over the last several years 1. Generally these disorders can be classified into either a secretory or malabsorptive form by whether the diarrhea improves with fasting. Malabsorption disorders, in turn, can be grouped by response to dietary challenges into either selective or generalized impairment of nutrient digestion or transport. Histologic assessment of the small intestine is particularly useful in elucidating the generalized diarrheal form, as abnormalities of inflammation, enterocyte subcellular structures, and the relative size of the crypt-villus axis are helpful in distinguishing the various disorders.
Three genes, neurogenin-3 (NEUROG3), autoimmune regulator (AIRE) and proprotein convertase subtilisin/kexin type 1 (PCSK1) are known to be involved in disorders characterized by abnormal enteroendocrine development or function that manifest in generalized malabsorption 2–5. We previously described a cohort of children with missense mutations of NEUROG3, neonatal onset of diarrhea, and a severe paucity of enteroendocrine cells (enteric anendocrinosis) as assessed by an absence of staining for chromogranin and gut hormones [MIM:610370] 2. Mutations in AIRE have been found in patients with autoimmune polyglandular syndrome type 1, a disorder associated with various endocrinopathies including a severe noncongenital form of generalized malabsorptive diarrhea associated with a depletion of enteroendocrine cells [MIM:240300] 5.
The enzyme encoded by PCSK1, prohormone convertase 1/3 (PC1/3), is responsible for peptide hormone processing within the enteroendocrine cell. Deleterious homozygote mutations of PCSK1 have been described in three cases, two of which involved neonatal diarrhea [MIM:600955] 3, 4, 6. Prohormone convertase 1/3 is a calcium-dependent serine endoprotease essential for the conversion of a variety of prohormones into their bioactive forms. PC1/3 is richly expressed in endocrine cells in the gut, in the arcuate and paraventricular nuclei of the hypothalamus, and in β cells of the pancreas, where it has a well-defined role in processing proinsulin 7.
The first reported case of severe PC1/3 deficiency was assessed in a woman who presented in her 40s with postprandial hypoglycemia, obesity, primary hypogonadism, and adrenal insufficiency 3, 4. In a follow-up report, this patient recollected having severe diarrhea in childhood 6. A second case exhibited hypocortisolemia and a generalized malabsorptive diarrhea that required prolonged parenteral nutrition, but this patient died at 18 months 6. A recent case described a six-year-old who had a similar form of congenital diarrhea who became severely obese 8. This child also had central hypocortisolemia, believed to be secondary to defective processing of proopiomelanocortin (POMC) proprotein to ACTH, and polyuria and polydipsia that could not be attributed to diabetes insipidus (DI).
In addition, two common heterozygote variants of PCSK1, rs6232 and rs6235, have been associated with obesity and/or diabetes in the general population despite reducing the catalytic activity of PC1/3 by less than 10% 9. Several murine models of Pcsk1 depletion have been reported with complex and varied phenotypes depending on the severity of the mutation, diet, and likely the genetic background of the mouse strain 10, 11.
Since only three cases have been reported to date, the clinical course of patients with severe homozygous mutations of PCSK1 is still unclear. In the report below, we present evidence that the severe malabsorptive diarrhea of early infancy is followed by many endocrine abnormalities that have not yet been described. We describe 13 children from 11 families with 12 novel mutations (five missense, five nonsense, one frame shift, and two splice site) of PCSK1. Each family had a unique homozygous mutation, and one family had a second homozygous mutation in the same gene. We assessed 9 mutations for processing, secretion, and enzymatic activity using established in vitro assays. These results suggest that impairment of processing of prohormones secreted by enteroendocrine cells likely accounts for the generalized malabsorptive diarrhea, which dominates the early clinical course of this disorder.
Material and Methods
Subjects
Samples for mutation screening were identified from the UCLA Pediatric Diarrhea Research Database, which includes samples referred for clinical diagnosis or research since 2004, and was approved by our institutional review board. Inclusion criteria for the database was a history of chronic (>2 mo) severe diarrhea during the Pediatric ages (<18 yo), while subjects with various causes of short bowel syndrome, inflammatory bowel disease, Celiac disease and pancreatic insufficiency were excluded. The database contains over 172 kindreds composed of 194 children with chronic diarrhea, 163 of which were classified as congenital in origin. Approximately 25 of the subjects were identified with various forms of selective malabsorptive diarrhea, while 133 were classified with the generalized form malabsorption. Within this latter group, 45 had normal an otherwise normal histology based on pathologic (e.g. H&E) assessment, and 35 were sequenced by Sanger methods for significant variants of PCSK1.
Genomic DNA Isolation, PCR and Sequencing
Genomic DNA was extracted from blood or saliva by standard procedures, and measured by Qubit (Invitrogen). The 14 exons of PCSK1 were PCR-amplified and sequenced using oligonucleotides based on adjacent intronic sequence. Oligonucleotide pairs used to amplify genomic DNA and PCR conditions are presented in Supplement 1.
Wild-type and mutant PC1/3 expression clones
Flag-tagged human wild-type PC1/3 (kindly provided by John Creemers, University of Leuven) 9 was modified by site-directed mutagenesis using the Stratagene QuikChange method to introduce the mutations shown in Figure 1 and Table 2. All final clones were confirmed to contain only the designated mutation by sequencing of the entire cDNA insert.
Figure 1. Pedigrees and mutations in PCSK1.

Pedigrees of families studied, showing genotypes (./. NA, 0/0 WT, 0/1 heterozygous, 1/1 homozygous) and Sanger sequencing results for proband, and unaffected control. The proband(s) that were sequenced are indicated by the black square (male) or circle (female). Homozygote mutations within each family are indicated by MM; heterozygotes by MN; normal on both alleles by NN; and not assessed as NA. A slash through the symbol indicates that the subject is deceased, and a double line between the parents indicates a consanguineous union.
Table 2.
Mutations of PCSK1 by Kindred
| Mutation Name | Nucleotide change | coord | dbsnp137 | Exon | Nucleotide | Allele freq | Protein product | Genotype | Kindred |
|---|---|---|---|---|---|---|---|---|---|
| MISSENSE: | |||||||||
| p.G209R | c.625G>A | 5:95751821 | – | 6 | GGG -> AGG @625 | – | Conserved residue | Homozygous | #2 |
| p.P258T | c.772C>A | 5:95748132 | – | 7 | CCT-> ACT@772 | – | Conserved residue | Homozygous | #2 |
| p.N423K | c.1269C>A | 5:95735818 | – | 10 | AAC-> AAA @1269 | – | Conserved residue | Homozygous | #11 |
| p.F548S | c.1643T>C | 5:95733119 | – | 12 | TTT->TCT@1643 | – | Conserved residue | Homozygous | #9 |
| p.G593R | c.1777G>A | 5:95730675 | – | 13 | GGG->AGG@1777 | – | Conserved residue | Homozygous | #1 |
| NONSENSE: | |||||||||
| p.M1X | c.2T>C | 5:95768745 | – | 1 | ATG->ACG@2 | – | Truncated | Homozygous | #6a/6b |
| p.Y231X | c.693C>G | 5:95751753 | – | 6 | TAC->TAG@693 | – | Truncated | Homozygous | #10 |
| p.Q337X | c.1009C>T | 5:95746564 | – | 8 | CAA->TAA@1009 | – | Truncated | Homozygous | #4 |
| p.R405X | c.1213C>T | 5:95735874 | – | 10 | CGA->TGA@1213 | – | Truncated | Homozygous | #5a/#5b |
| DELETION: | |||||||||
| p.V450fsX1 | c.1349_1352delTGGA | 5:95735735-38 | – | 10 | GTGGAT->GTTTAG@1349 | – | Deletion | Homozygous | #8 |
| SPLICE SITE: | |||||||||
| IVS8+1G>T | c.1095+1T | 5:95746477 | – | I-8 | |GTA->TTA | – | Intron donor site loss | Homozygous | #3 |
| IVS8+1G>A | c.1095+1A | 5:95746477 | – | I-8 | |GTA->ATA | – | Intron donor site loss | Homozygous | #7 |
Enzyme Assay
Clones containing cDNAs encoding the various PC1/3 mutant variants were transfected into HEK293 cells as described in more detail in Supplement 1. Enzymatic activity of secreted recombinant PC1/3 proteins present in conditioned medium was measured as previously described 13. Maximum rates were obtained from the later portion of the kinetic measurement curves and compared to WT PC1/3 wells. All experiments were independently repeated at least three times.
Results
CLINICAL PHENOTYPE
General Clinical Characteristics
Ten subjects were the offspring of known consanguineous relationships. Eighty-five percent of the subjects analyzed (11 of 13) were males (Table 1). Two of the subjects (#4, #7) died at 8 and 15 months of life, respectively, during prolonged hospitalizations secondary to presumed central venous line sepsis. In three families (#6, #8, #10), three other children died as either neonates or during the late childhood period with a similar clinical course of chronic diarrhea prior to the diagnosis of the index case. One case (#2) was lost to clinical follow-up after late infancy. The oldest proband of this cohort is at present 17 years old (#11), and 6 of the subjects are older than 6 years of age.
Table 1.
Summary of Clinical Phenotype of 13 Subjects with PCSK1 Mutations
| ID # | 1 | 2 | 3 | 4 | 5a | 5b | 6a | 6b | 7 | 8 | 9 | 10 | 11 | Summary |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ethnicity | Hispanic | Indian | Turkish | Turkish | African | African | Arab | Arab | Turkish | Arab | Turkish | Turkish | Canadian | Diverse |
| Sex | M | M | F | M | M | M | M | M | M | M | M | F | M | 11 of 13 male |
| Outcome; Age | Alive; 7.5 yo | Alive; 6 yo | Alive; 15 yo | Dead @ 8 mo; sepsis | Alive; 3.7 yo | Alive; 9.3 yo | Alive; 12.8 yo | Alive; 2.9 yo | Dead @ 15 mo; sepsis | Alive; 5.5 yo | Alive; 3.8 yo | Alive; 3.0 yo | Alive; 17 yo | 11 of 13 alive χ 7.7 +/−4.8 |
| Consanguinity | Yes | Yes | Yes | Yes | No | No | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes, 12 of 13 |
| Family history | No | No | No | No | Yes; #87b | Yes; #87a | Yes, 2; 1 died & #113 | Yes, 2; 1 died & #88 | No | Yes; 1 died @ 5 yo | No | Yes; died | No | Yes, 4 of 12 |
| Birth Wt Kg | 3.85 | 3.3 | 3.3 | 3.1 | 3.55 | 3.7 | 3.13 | 2.95 | 3.2 | 3.1 | 3.8 | 3.6 | 3.5 | χ3.4+/−0.3 |
| Age presented | 3 wk | 1 wk | 1 wk | 1 wk | 1 wk | 1 wk | 2 wk | 1 wk | 4 wk | 8 wks | 3 wk | 2 wk | 2 wk | χ 2.3 +/−1.9 |
| Generalize malabsoprtion | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes, 13 of 13 |
| Intest. Biopsy; Normal | Yes | Yes | No; mild villus atropy | Yes | No; villus atropy; eos | Not done | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes, 0 of 12 normal |
| Chronic IV Nut (age start; end) | Yes (1 to 16 mo) | Yes (3 to 12 mo) | Yes (10 to 34 mo) | Yes (4 to 8 mo) | Yes (1 to 20 mo) | Never treated w/ IV Nut | Yes (1.5 to 11 mo) | Yes (1 to 12 mo) | Yes (3 to 15 mo) | Yes (6 to 14 mo) | Yes (10 to 23 mo) | Yes (1 to 12 mo) | Yes (1 to 12 mo) | Yes, 12 of 13 |
|
Early growth: FTT/age; WT Z score |
Yes/2mo; WT Z −1.3 | Yes/2mo; WT Z −2.1 | Yes/10mo; WT Z −6.56 | Yes/4 mo; WT Z −5.63 | Yes/1 wk; WT Z−1.7 | Yes/6 mo; WT Z−6.6 | Yes/2 mo; WT Z−2.66 | Yes/2 wk; WT Z−2.5 | Yes/3 mo; WT Z−3.24 | Yes/2 mo; WT Z −4.0 | Yes/4 mo; WT Z−1.9 | Yes/2.5 mo; WT Z−2.24 | Yes/1 mo; WT Z−3.15 | Yes, 13 of 13 χ −3.35+/−1.8 |
|
Late growth: Obese/age WT Z score HT Z score |
Yes/7.7 yo; BMI Z+2.4 WT Z+1.81 HT Z−1.4 |
N/A | Yes/15yo; BMI Z+1.7 WT Z+1.0 HT Z−1.78 |
N/A | Yes/3.7yo; BMI Z+2.7 WT Z+1.4 HT Z−0.7 |
Yes/9.3 yo; BMI Z+2.33 WT Z+2.15 HT Z−0.21 |
Yes/12.8yo; BMI Z+2.6 WT Z +2.5 HT Z−0.6 |
Yes/2.9yo; BMI Z+2.55 WT Z+0.0 HT Z−2.8 |
N/A | No/5.5yo; BMI Z+0.2 WTZ −1.21 HT Z −1.26 |
Yes/3.5yo; BMI Z+3.62 WT Z +2.18 HT Z −0.68 |
Yes/2.4 yo; BMI Z+3.43 WT Z +2.7 HT Z−0.1 |
Yes/17 yo; BMI Z+2.36 WTZ+2.03 HT Z−1.12 |
Yes, 9 of 10 BMI +2.5+/− 0.9 WT +1.5+/− 1.2 HT−1.0+/− |
| Proinsulin | ND | ND | ND | ND | Elevated | Elevated | Elevated | Elevated | ND | Elevated | Elevated | ND | Elevated | 7 of 13 |
| Postprandial hypoglycemia | Yes | Yes | Yes | No | Yes | Yes | No | No | Yes | No | Yes | Yes | No | 8 of 13 |
| Polydipsia& Polyuria | Yes | Yes | Yes | No | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | 11 of 13 |
| Diabetes Insipidus confirmed; Age, treated | Yes, @ 18 mo; DDAVP | N/A | Yes, @3.5 yo; DDAVP | N/A | N/A | Yes, @ 13.5 mo; DDAVP | Yes, @ 1 mo; DDAVP | N/A | N/A | No | Yes, @ 13 mo; DDAVP | Yes, @ 19 mo; DDAVP | Yes, @3 yo; not responsive to DDAVP | 7 of 13 |
| Hypogonadism | Micropenis | N/A | Yes, delay puberty | N/A | No | Micropenis; hypospadias | No | No | No | No | No | N/A | Micropenis; delay puberty | 4 of 13 |
| Hypoadrenalism confirmed; Age, treated | Yes, @6.5 yo | N/A | Yes, @5.5 yo | No | Yes, @12mo | Yes, @6mo | Yes, @1mo | Yes, @ 3 mo | N/A | No | Yes, @ 14 mo; hydrocortisone with stress | Yes, @ 19 mo; hydrocortisone | No | 8 of 13 |
| GH deficiency confirmed; Age, treated | N/A | N/A | Yes, @5.5 yo | N/A | No | No | Yes, @9.5yo | Yes, @2.5 yo | N/A | No | No | No | Yes, @ 14 yo | 4 of 13 |
| Hypothyroid confirmed; Age, treated | Yes, @6.5 yo | N/A | Yes, @5.5 yo | No | No | No | Yes, @ 1 mo | Yes, @ 3 wk | Yes, @ 1 mo | Yes, @ 4 mo | No | Yes, 29 mo | Yes, @ 17 yo | 8 of 13 |
N/A, not assessed; Yes, confirm symptom or test; No, does not have symptom, or the test was negative; yo (years-of-age); mo (months-of-age).
Diarrhea/Nutrition and Growth Characteristics
All subjects were born at full term gestation with normal weights (χ, 3.4±0.3 Kg). All children presented with evidence of dehydration, metabolic acidosis, and diarrhea during the first two months of life (χ, 2.3±2.0 weeks; range 1 to 8 weeks), with slightly less than half (6/13) presenting during the first week of life. The diarrheal symptoms failed to resolve upon selective elimination of carbohydrates (glucose, lactose or sucrose), amino acids, and fats during various dietary challenges. In all cases, the diarrhea was malabsorptive type, based on assessment of stool electrolytes and the resolution of diarrhea during states of fasting. Eleven of the 13 subjects had intestinal biopsies, and all but two were reported as normal. The two cases (# 3, #5a) with abnormal biopsies had evidence of mild villous atrophy without overt inflammation.
Most cases (8 of 13) began parenteral nutrition within the first three months of life secondary to severe diarrhea and failure to thrive, and all but one case (#5b) required prolonged intravenous nutrition. While two cases required parenteral nutrition beyond 17 months of age (#3, #9), the remaining children were on exclusive enteral feeds by 1.5 years of age (χ, 15.8±7.8 months of age). Although the dependency of parenteral nutrition diminishes with time, these subjects continue to experience significant malabsorptive diarrhea and loose stools throughout childhood. Many of these children were hospitalized for prolonged periods of time for extensive clinical and laboratory evaluations, including numerous endoscopy, dietary challenges, and nutritional rehabilitation; some were hospitalized chronically because of the unavailability of home parenteral nutritional support (# 4, #7).
All of the infants that initially received prolonged parenteral nutrition had severe failure to thrive prior to starting nutritional support, and had a weight standard deviation score (z-score) of less than −3 (χ, −3.35±1.8). Interestingly, as the subjects aged beyond early infancy, their weight increased significantly, and out of proportion to height. Specifically, of the five cases that have reached mid-childhood (6 years of age; case #1, #3, #5b, #6a, #11), the weight z score was more than +2 (χ, 1.9±0.5), and height z-score was −1 (χ, −1.0±0.6). All of these children were moderately obese and had a mean body-mass-index (BMI) z-score of +2.3±0.3. Representative growth charts of cases #1, #9 and #11 illustrate that despite poor growth in early infancy, significant increases in weight and BMI are characteristic of this disorder years after parenteral nutrition as been discontinued (Supplement 2).
Endocrine Characteristics
Only 7 of the 13 subjects were confirmed to have elevated serum proinsulin levels, as this assay was not available at all parent institutions, or because the diagnosis was established post-mortem by genetic testing. Of those cases where proinsulin levels were assessed, the values were significantly elevated for the various reference laboratories. Episodes of postprandial hypoglycemia were documented on multiple occasions in 8 of the 13 cases.
Polydipsia and polyuria were common symptoms identified in at least 11 of the 13 cases. Irritability and aggressive water-seeking behavior were common complaints, especially during occasions when access to water was impaired. Diabetes insipidus was established in eight cases, and the average age of this diagnosis was ~18 months old, with a range of 1 to 42 months. Seven of these children were managed with intranasal desmopressin (DDAVP), and the other with water restriction. Of all of the endocrinopathies other than malabsorptive diarrhea, DI or partial DI was identified most consistently. In one case (#11), despite laboratory and clinical evidence of partial DI, the subject failed to respond as expected to appropriate doses of DDAVP.
At least three of the males had hypogonadism with small testis and micropenis, and at least two responded to testosterone therapy. One child (#11) was documented to have central hypogonadism with low stimulated serum levels of LH and FSH, and testosterone, and responded appropriately to testosterone administration. One of the two females (#3) has reached pubertal age and experienced delayed puberty with feminization on estradiol therapy.
Adrenal function was assessed in at least 11 cases and central adrenal insufficiency was observed in 8 children at 1 month to 5.5 years of age. Diagnosis was based on low basal cortisol and ACTH levels, or insufficient response to ACTH testing. Seven subjects receive daily hydrocortisone therapy, and one (#9) receives only stress coverage.
Similarly, central hypothyroidism was identified in 8 cases and was either not assessed or not observed in 5 cases. The hypothyroidism was hypothalamic in origin with normal or low serum TSH levels and low T4. The age range of detection was also wide, from 1 month to 17 years of age.
Growth hormone deficiency was identified in at least 4 cases as assessed by stimulation tests and/or abnormalities of serum IGFBP3 and IGF-1 levels. All of the children diagnosed with growth hormone deficiency were treated with growth hormone with good response.
SEQUENCING OF PCSK1
We sequenced each of PCSK1’s 14 exons in DNA from saliva samples from every index patient and in a subset of parents and siblings. We observed one homozygote mutation in 12 of the samples and two homozygote mutations in the remaining sample (#2). None of the variants were previously identified in dbSNP 137 or in 1092 or 6500 individuals in the 1000 Genomes and NHLBI datasets, respectively; and are very rare (MAF<0.001%) (Table 2, Figure 1, Supplement 1). The p.G593R variant was previously described as a compound heterozygote 3, 15.
Two severe nonsense mutations, p.M1X and p.R405X, were identified in four subjects (#6A/6B and #5A/#5B) from two families. The p.M1X mutation deletes the gene’s usual initiation codon, and the next in-frame alternative initiation codon is M125, located within exon 3, while the closest out-of-frame ATG is 78 nucleotides downstream of the initiation codon. The p.R405X mutation results in an entire deletion of the protein’s P, and CT domains (Table 2, Figure 2).
Figure 2. Domain structure and mutation locations within prepro-PC1/3.
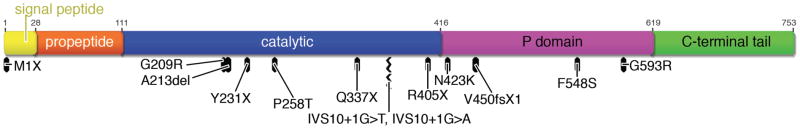
Overview of PC1/3 protein regions with locations of mutations presented in this study and previously published mutations.
Two other nonsense mutations, p.Y231X and p.Q337X, were identified in two other subjects (#10, #4). A frame shift mutation (p.V450fsX1) in the index case from another family (#8) would be predicted, if stable, to result in a severely truncated protein that would be expected to lack the catalytic, P and CT domains (Table 2, Figures 1–2). Two essential splice site mutations (IVS8+1G>T and IVS8+1G>A), located at the identical obligate acceptor nucleotide in intron 8, were identified in two unrelated subjects (#3, #7), and are predicted to severely alter the gene’s correct splicing pattern.
All of the missense variants were identified in the homozygote state in the index cases, and these altered amino acid residues are evolutionarily conserved from H. sapiens to D. rerio (Figure 3), suggesting their importance. Two mutations (p.G209R and p.P258T) were identified in the same subject (#2); these alter residues within the PC1/3 catalytic domain, substituting a large basic residue for a small polar residue, and a hydroxylated polar residue for a rigid nonpolar residue, respectively (Figures 1–3). Two missense variants, p.N423K and p.G593R, in patients #11 and #1 respectively, are significant polar to charged basic residue changes within the P domain. Finally, also within the P domain, in case #9, the p.F548S variant substitutes a hydroxylated polar residue for a highly hydrophobic amino acid.
Figure 3. Location of 5 missense mutations in the PC1/3 gene family in conserved domains.
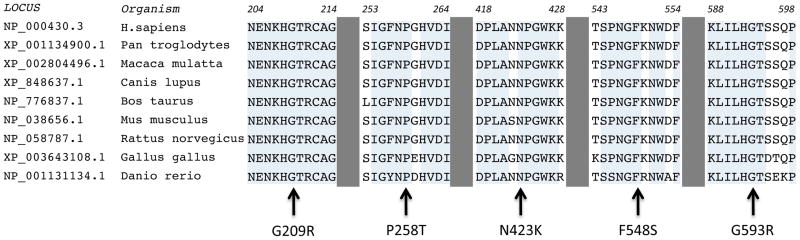
Alignment of missense variants to members of PC1/3 gene family.
FUNCTIONAL ANALYSIS AND IN VITRO ASSESSMENT OF MUTANT PC1/3s
Expression of recombinant wild-type PC1/3 and various mutant PC1/3s
The PC1/3 patient mutations were placed into a human PC1/3 expression vector, expressed in HEK cells, and the conditioned supernatant assayed for PC1/3 activity; cell lysate and medium were also assessed for protein content by Western blotting. As can be seen in Figure 4, none of the truncation mutants resulted in a cellular product (panel A); however, the frameshift variant, p.V450fsX1, generated a secreted 55 kDa truncated PC1/3 product. In contrast, all of the PC1/3 proteins containing point mutations were synthesized as evidenced by their presence in cell lysates (panel B), though only three were secreted into the medium (p.P258T, p.N423K, p.F548S). The p.N423K and the p.F548S proteins were both secreted somewhat inefficiently compared to WT. Interestingly, neither of these two 87 kDa PC1/3 mutant proteins exhibited the lower molecular mass proteins in the medium typical of C-terminal truncation, in contrast to the p.P258T point mutant. The p.P258T PC1/3 variant appeared to be more efficiently secreted than the other two secreted point mutants, and was also able to mature to smaller forms similarly to WT.
Figure 4. Western blot of PC1/3 wild-type and mutant protein expression.

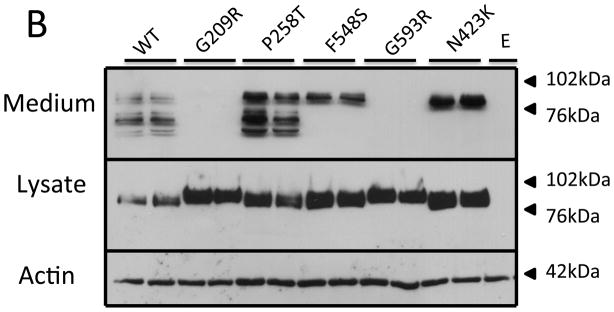
HEK293 cells were transfected with empty vector (E), wild-type PC1/3 (WT), or PC1/3s containing novel mutations. Media and cells were subjected to Western blotting using amino-terminally directed PC1/3 primary antiserum. Truncation mutations are shown in Panel A, and missense mutations are shown in Panel B. β-actin was used as a loading control.
Enzymatic activity of recombinant wild-type PC1/3 and various mutant PC1/3s
Most of the mutations resulted in PC1/3 forms lacking any secreted enzymatic activity (Figure 5); this includes all of the truncation mutants, as well as the p.V450fsX1 frame shift variant. While the F548S protein was secreted, it was completely inactive. Two other missense mutations exhibited partial activity, P258T and N432K; the former exhibited 60% of wild-type activity, while the latter exhibited very little activity (11% of wild-type).
Figure 5. Enzymatic activity of wild-type PC1/3 and novel PC1/3 variants.

The enzymatic activities of secreted PC1/3 proteins in conditioned medium were assayed using a fluorogenic assay. Four replicates per transfection condition were assayed in triplicate, and maximum rates were normalized to WT PC1/3. Maximum activity rates are shown as the mean ±S.D., n=4 wells. Data represent one of 3 independent experiments. Bars represent mean ± standard deviation, and unpaired Student’s t-test was used to assess difference between mutant and WT activities with *:P-value <0.001, **:P-value <0.05. All experiments were independently repeated in triplicate wells at least three times, with similar results.
Discussion
This study describes the clinical outcome and molecular basis of PC1/3 deficiency in 13 patients followed through the ages of 3 to 17. All 13 subjects had homozygous mutations, whereas in three previously reported cases, one was a homozygote and two were compound heterozygotes 4, 6, 8. We confirm that this disorder is characterized in the early years by a significant risk of mortality and failure to thrive secondary to severe generalized malabsorptive diarrhea. However, the children studied here had considerable improvement in mortality beyond 18 months of age, despite persistent morbidity concomitant with the development of an array of major endocrinopathies.
Generalized malabsorptive diarrhea is the endocrinopathy that dominates the early clinical picture. Similar to children with enteric anendocrinosis-associated mutations of NEUROG3, the subjects’ diarrheal symptoms failed to abate with the elimination of selective nutrients 2. Also like children with NEUROG3 mutations, the majority of the cases had a normal crypt-villus-axis and an absence of a dominant inflammatory component.
The assortment of endocrinopathies associated with PC1/3 deficiency distinguishes this disorder from enteric anendocrinosis-associated mutations of NEUROG3 who develop insulin-dependent diabetes mellitus in early childhood, but do not appear to develop other endocrine abnormalities 2. Nevertheless, the enteric endocrinopathy associated with each of these disorders appear to be indistinguishable. It should be noted that NEUROG3 is a helix-loop-helix transcription factor that is required and sufficient to drive the development of enteroendocrine and islet cells, and is therefore essential to produce all components of endocrine cells, including hormone and processing enzymes such as PC1/3.
All but one (#5b) of the children required a prolonged course of parenteral nutrition therapy; however, the untreated child’s failure to thrive was the severest of all those encountered (weight, z score −6.6). Unlike most forms of congenital enteropathy, which require life-long parenteral nutrition, the PC1/3-deficient children were weaned off intravenous nutrition by 18-months of age. We speculate that the hormones processed by PC1/3 and secreted by enteroendocrine cells are important to support the particularly high caloric intake necessary for growth during early infancy, but that this requirement diminishes thereafter.
It is remarkable how closely the Pcsk1 null mouse model mimics the clinical course seen in the human subjects described here. The Pcsk1 null mouse has a high postnatal mortality rate, with only one-third surviving beyond seven days of life, and most succumbing by day two 10. The null pups appear similar to controls until the third day, when considerable differences in weight become apparent, and those that do survive beyond the first week exhibit significant growth retardation that was attributed to defective processing of GHRH10. Interestingly, mild diarrhea is discernible in the older mice, despite normal intestinal architecture.
Multiple prohormones secreted from enteroendocrine cells are processed by PC1/3 (Supplement 3) 7. However, we have been unable to identify a murine model where deletion of an enteric hormone, and/or its corresponding putative receptor, is associated with early postnatal mortality as seen in the Pcsk1- and Neurog3-null models 10, 14. These findings suggest either that there might be another novel peptide processed by PC1/3 that enhances assimilation of a broad group of nutrients; or that redundant hormones have this role, and selective depletion does not recapitulate the early mortality seen in humans and mice with null PCSK1 and NEUROG3 mutations 2, 3, 10, 14. This raises the possibility that exogenous administration of the hormone(s) might have beneficial effects in attenuating the severity of the diarrhea in this unique group of patients.
Nearly all of the PCSK1 mutations studied here destroyed the enzymatic activity of PC1/3 when examined in an HEK cell expression system. The majority of truncation mutations likely underwent mRNA decay and intracellular degradation since PC1/3t was undetectable in the lysate. In contrast, the missense point mutations resulted in a variety of biochemical phenotypes. The missense mutants p.G593R and p.G209R, and the nonsense mutant p.V450fsX1, apparently failed to traverse the secretory pathway; while intracellular proteins were observed, they were not secreted. These highly conserved glycine residues (Figure 4b) may be essential to protein folding. The p.G593R mutation was previously identified in a compound heterozygote state in the initial index case, and impaired secretion of this variant was recently reported by others 15. The p.P258T substitution, identified in kindred #2, resulted in protein able to traverse the secretory pathway efficiently; the secreted protein exhibited robust enzymatic activity against the fluorogenic substrate (Figure 5). In the PC1/3 model this residue, is located in an exterior beta turn of the catalytic domain, a position that may not be important for protein folding 16. Proband #2 also is a homozygote for the p.G209R mutation which exhibits no detectable enzymatic activity; this second variant likely accounts for the subject’s clinical phenotype. The p.N423K substitution also resulted in a secreted but very weakly active enzyme that was apparently unable to mature to smaller species. This residue is located in a loop in close proximity to the P domain and the catalytic triad, a location that is apparently integral to C-terminal cleavage. Lack of C-terminal cleavage is predicted to result in severe loss of activity, consistent with the observed results 7. Further analysis of mutant processing should be tested in a model cell system that contains regulated secretory granules, in which C-terminal processing should be enhanced.
Our biochemical results are consistent with others who sequenced PC1/3 from 845 obese patients and found eight other novel missense mutations identified in eight different heterozygote carriers 15. Seven of these mutated PC1/3s exhibited moderate impaired synthesis or activity, and mutations likely altered enzyme folding and stability and folding of the enzyme. In a larger cohort of obese European patients, these missense mutations were associated with a 8.7-fold higher risk of obesity15.
Indeed, PC1/3 has frequently been implicated in the polygenic and monogenic forms of obesity and has an essential role in POMC processing; POMC-derived peptides represent a key component of the leptin-signaling pathway 3, 4, 17. PC1/3 also processes the central orexigenic hormones NPY and agouti-related protein (AgRP) that compete with α-melanocyte-stimulating hormone for the melanocortin receptor 4 (MC4R), expressed in the hypothalamus 18. Paradoxically, despite the anticipated loss of PC1/3 processing of both central (NPY, AgRP and orexin) and peripheral (ghrelin) orexigenic hormones 18–20 in these subjects, our probands exhibited polyphagia throughout childhood. Attenuation of a PC1/3-dependent anorexigenic signal such as PYY should enhance appetite 21, 22. Given PC1/3’s extensive role in processing many of the peptidergic components of pathways regulating energy balance and appetite, we might have anticipated more profound obesity in our probands. While it is conceivable that other proprotein convertases such as PC2 might compensate for the loss of PC1/3 activity, we hypothesize that the milder form of obesity in this cohort is due in part to the persistent malabsorption that distinguishes homozygote PCSK1 deficiency from these other disorders.
Several severe monogenic obesity disorders have been described along this leptin-MC4R pathway, and all are typified by obesity that is noticeable within the first several months of life, and persisting throughout adulthood as class III morbid obesity 23. For instance, cohorts with severe MC4R heterozygote mutations have a mean BMI index z-score of +3.9 23. This is in stark contrast to our PCSK1 probands that experience profound failure to thrive during early infancy, and only moderate obesity during the late childhood and adolescence period (Table 1). Indeed, the first reported PCSK 1 proband ever described, a middle-aged woman, 4 had class I obesity (BMI 34.4, z-score +1.9), with a height (z-score −0.35), and weight (z-score +1.9) that resemble many of the index cases described here. However, she reportedly weighed 36 Kg (z-score +5.3) at 3 years of age 3; this is significantly more than any of the children included in this study (Table 1).
Four subjects in this cohort exhibited linear growth deficiencies and received therapy for growth hormone deficiency. Stunted growth was not described in the two earlier cases of PC1/3 deficiency who reached childhood ages 3, 4, 6. Reduced linear growth in the later ages clearly appeared to contribute to the high BMI values, as the mean height z-score was −1. These observations raise the possibility that the more common mild PCSK1 mutations in heterozygote carriers may be particularly prone to an elevated BMI because of a concomitant diminution of linear growth 9. It would certainly be interesting to assess these and other clinical and laboratory parameters in the obligate heterozygotes from our cohort, since the defective PCSK1 allele is severely affected.
In mice, PC1/3 processes proGHRH, and in Pcsk1 null mice hypothalamic GHRH and circulating and hepatic IGF1 levels are low, contributing to their growth disparity 10. Species differences in the cleavage site sequence of proGHRH suggest that humans might be less prone to GH deficiency; however, an inherent species difference in the specificity of the convertases towards the human and mouse precursors has not been directly assessed. The proGHRH cleavage site is also processed efficiently by furin 10. In addition to GHRH, peripheral and hypothalamic control of GH secretion also involves somatostatin and ghrelin, which are also processed by PC1/3 24.
Reproductive systems were also strongly affected by PC1/3 mutations. The convertase(s) that processes hypothalamic proGnRH has not been defined in humans, but PC2 has been implicated in rats 25. In our cohort, two males were formally diagnosed with laboratory evidence of central hypogonadism with evidence of micropenis and small soft testis that were treated successfully with testosterone, and one female is on estradiol treatment for primary hypogonadism. Leptin deficiency is also associated with hypothalamic hypogonadism via several mechanisms including kisspeptin, the natural ligand for GPR54, which modifies GnRH release 25. The convertase that processes the kisspeptin precursor has not been elucidated, but it is conceivable that PC1/3 deficiency may impair GnRH synthesis by this alternative route.
Hypoadrenalism was identified in each of the three previously described patients with PCSK1 deficiency 3, 6, 8. Similarly, eight subjects from our cohort had evidence of central adrenal insufficiency with low basal cortisol levels and low to normal ACTH levels. In the hypothalamus, the CRH precursor is processed by PC2, and mature CRH can modulate the endoproteolytic processing of POMC by enhancing PC1/3 expression 26. However, POMC processing does not appear to be entirely dependent on PC1/3, as low normal ACTH levels were detected in the previously described PC1/3-deficient subjects despite low cortisol and elevated POMC levels in serum 4, 8.
Seven children had laboratory evidence of at least partial central diabetes insipidus, with serum hyper-osmolarity, urine hypo-osmolarity, and low serum arginine vasopressin (AVP) levels; they were treated with DDAVP. Since adrenal insufficiency may diminish the excretion of free water, it is conceivable that the evidence of DI might only be revealed in subjects with normal cortisol levels. While infants with DI are generally managed with extra free water and not with DDAVP to maintain normal hydration, it is possible that the parenteral nutrition may have also masked the vasopressin deficiency. Interestingly, the antidiuretic response to DDAVP administration was poor in at least one case (#11), suggesting a potential nephrogenic component. It is conceivable that an abundance of the inactive vasopressin precursor may compete with DDAVP for binding to the vasopressin (V2) receptor, and impair activation of the aquaporin-2 water channels. While direct in vitro data assessing the efficacy of PC1/3 and PC2 in processing of the human vasopressin precursor is also lacking, it is conceivable that differential age-dependent expression of PC1/3 and PC2 in the hypothalamus might explain the development of DI in our cohort beyond early infancy.
We also identified central hypothyroidism (low serum levels of TSH and T4) in seven of the subjects, similar to a previously described case of PC1/3 deficiency 8, In the hypothalamus, prothyrotropin-releasing hormone (pro-TRH) is processed by both PC1/3 and PC2 to a hypothalamic tripeptide that stimulates TSH synthesis and release, in a leptin-dependent mechanism 27.
Interestingly, females are significantly underrepresented in all known cases of PC1/3 deficiency, suggesting potential embryonic lethality. In this study, we found a predominance of males (11 of 13 patients). Including the three previously reported subjects (1 of 3), and two other subjects not reported here (2 of 2), 14 of 18 cases with PC1/3 deficiency are males (p=0.031, exact binomial two-tailed) 3, 6, 8. We failed to identify any asymptomatic homozygote females in our various kindreds. Although a degree of prenatal mortality was detected in the mouse model, differences in male/female ratio in those few Pcsk1 null mice that survived the early postnatal mortality were not described 10.
In conclusion, this study provides a broad understanding of the full clinical phenotype associated with the autosomal recessive form of PC1/3 deficiency. While enteroendocrine cell dysfunction (e.g. enteric dysendocrinosis) dominates the early clinical phenotype, continued malabsorption likely attenuates the severity of the obesity seen at later ages. Specific hormones with profound effects in enhancing nutrient assimilation have not been definitively elucidated, but this study strongly implies that such hormones must be generated by PC1/3 processing. In our cohort, we found clinical and laboratory evidence of GH deficiency, central DI and hypogonadism in males, three clinically important features not previously identified. Homozygote females were also significantly underrepresented in our cohort, suggesting that PC1/3 activity in utero may be particularly required for females. We also raise the possibility that the elevated BMIs that are associated with the more common autosomal dominant form of PC1/3 deficiency may be related in part to an impairment of linear growth (GH deficiency). Finally, our study illustrates the complexity of an evolving phenotype, and highlights the importance of establishing the correct underlying diagnosis to guide treatment and physicians’ and parents’ expectations.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (#DK083762), and California Institute of Regenerative Medicine (CIRM), RT2-01985 to MGM, and DA05084 to IL.
Abbreviations
- DDAVP
intranasal desmopressin
- PCSK1
proprotein convertase subtilisin/kexin type 1
- DI
diabetes insipidus
- GHRH
growth hormone-releasing hormone
- POMC
opiomelanocortin
- α-MSH
α-melanocyte-stimulating hormone
- NPY
neuropeptide Y
- AgRP
agouti-related protein
- MC4R
melanocortin receptor 4
- GH
growth hormone
- Peptide YY
(PYY)
- GnRH
gonadotropin releasing hormone
- TRH
thyrotropin-releasing hormone
- CRH
corticotropin-releasing hormone
- z-score
standard-deviation score
- BMI
body-mass-index
- WT
wild-type
- MAF
minor allele frequency
Footnotes
Disclosures: None of the authors have potential financial, professional, or personal conflicts to disclose.
Martín G. Martín = SCD, AID, DM, CR, OF, SS
Iris Lindberg = SCD, AD, AID, DM, CR, SS
R. Sergio Solorzano-Vargas = SCD, AD, AID, TMS
Jiafang Wang = AD, TMS
Yaron Avitzur = SCD, AD, DM, CR
Robert Bandsma = SCD, AD, DM, CR
Christiane Sokollik = AD, DM
Sarah Lawrence = AD, DM, CR
Lindsay A. Pickett = AD, AID, DM, TMS
Zijun Chen = AD, TMS
Odul Egritas = AD, CR
Buket Dalgic = AD, CR
Valeria Albornoz = AD, TMS
Lissy de Ridder = AD, DM, CR
Jessie Hulst = AD, DM, CR
Faysal Gok = AD, CR
Ayşen Aydoğan = AD, CR
Abdulrahman Al-Hussaini = AD, CR
Deniz Engin Gok = AD, CR
Michael Yourshaw = AID, DM, CR, SA
S. Vincent Wu = SCD, DM, CR
Galen Cortina = SCD, AID, DM
Sara Stanford = AD, DM, TMS
Senta Georgia = AD, AID, DM
Authors’ contribution:
study concept and design = SCD
acquisition of data = AD
analysis and interpretation of data = AID
drafting of the manuscript = DM
critical revision of the manuscript for important intellectual content = CR
statistical analysis = SA
obtained funding = OF
technical or material support = TMS
study supervision = SS
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Berni Canani R, Terrin G, Cardillo G, et al. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J Pediatr Gastroenterol Nutr. 2010;50:360–366. doi: 10.1097/MPG.0b013e3181d135ef. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Cortina G, Wu SV, et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. New England Journal of Medicine. 2006;355:270–280. doi: 10.1056/NEJMoa054288. [DOI] [PubMed] [Google Scholar]
- 3.Jackson RS, Creemers JW, Ohagi S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–306. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 4.O’Rahilly S, Gray H, Humphreys PJ, et al. Brief report: impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N Engl J Med. 1995;333:1386–90. doi: 10.1056/NEJM199511233332104. [DOI] [PubMed] [Google Scholar]
- 5.Hogenauer C, Meyer RL, Netto GJ, et al. Malabsorption due to cholecystokinin deficiency in a patient with autoimmune polyglandular syndrome type I. N Engl J Med. 2001;344:270–274. doi: 10.1056/NEJM200101253440405. [DOI] [PubMed] [Google Scholar]
- 6.Jackson RS. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. Journal of Clinical Investigation. 2003;112:1550–1560. doi: 10.1172/JCI18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoshino A, Lindberg I. Peptide Biosynthesis: Prohormone Convertases 1/3 and 2. In: Fricker LD aDL., editor. Colloquium Series on Neuropeptides. 1. Morgan and Claypool Life Sciences Publishers; 2012. [Google Scholar]
- 8.Farooqi IS, Volders K, Stanhope R, et al. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab. 2007;92:3369–73. doi: 10.1210/jc.2007-0687. [DOI] [PubMed] [Google Scholar]
- 9.Benzinou M, Creemers JW, Choquet H, et al. Common nonsynonymous variants in PCSK1 confer risk of obesity. Nature Genetics. 2008;40:943–5. doi: 10.1038/ng.177. [DOI] [PubMed] [Google Scholar]
- 10.Zhu X, Zhou A, Dey A, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci U S A. 2002;99:10293–8. doi: 10.1073/pnas.162352599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lloyd DJ, Bohan S, Gekakis N. Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Hum Mol Genet. 2006;15:1884–93. doi: 10.1093/hmg/ddl111. [DOI] [PubMed] [Google Scholar]
- 12.Vindrola O, Lindberg I. Biosynthesis of the prohormone convertase mPC1 in AtT-20 cells. Mol Endocrinol. 1992;6:1088–94. doi: 10.1210/mend.6.7.1508222. [DOI] [PubMed] [Google Scholar]
- 13.Hoshino A, Kowalska D, Jean F, et al. Modulation of PC1/3 activity by self-interaction and substrate binding. Endocrinology. 2011;152:1402–11. doi: 10.1210/en.2010-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gradwohl G, Dierich A, LeMeur M, et al. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Creemers JWM, Choquet H, Stijnen P, et al. Heterozygous Mutations Causing Partial Prohormone Convertase 1 Deficiency Contribute To Human Obesity. Diabetes. 2012;61:383–390. doi: 10.2337/db11-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henrich S, Lindberg I, Bode W, et al. Proprotein convertase models based on the crystal structures of furin and kexin: explanation of their specificity. J Mol Biol. 2005;345:211–27. doi: 10.1016/j.jmb.2004.10.050. [DOI] [PubMed] [Google Scholar]
- 17.Zhou A, Bloomquist BT, Mains RE. The prohormone convertases PC1 and PC2 mediate distinct endoproteolytic cleavages in a strict temporal order during proopiomelanocortin biosynthetic processing. J Biol Chem. 1993;268:1763–9. [PubMed] [Google Scholar]
- 18.Creemers JW, Pritchard LE, Gyte A, et al. Agouti-related protein is posttranslationally cleaved by proprotein convertase 1 to generate agouti-related protein (AGRP)83-132: interaction between AGRP83-132 and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology. 2006;147:1621–31. doi: 10.1210/en.2005-1373. [DOI] [PubMed] [Google Scholar]
- 19.Brakch N, Rist B, Beck-Sickinger AG, et al. Role of prohormone convertases in pro-neuropeptide Y processing: coexpression and in vitro kinetic investigations. Biochemistry. 1997;36:16309–20. doi: 10.1021/bi9714767. [DOI] [PubMed] [Google Scholar]
- 20.Zhu X, Cao Y, Voogd K, et al. On the processing of proghrelin to ghrelin. J Biol Chem. 2006;281:38867–70. doi: 10.1074/jbc.M607955200. [DOI] [PubMed] [Google Scholar]
- 21.Batterham RL, Cowley MA, Small CJ, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418:650–4. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 22.Boey D, Lin S, Karl T, et al. Peptide YY ablation in mice leads to the development of hyperinsulinaemia and obesity. Diabetologia. 2006;49:1360–70. doi: 10.1007/s00125-006-0237-0. [DOI] [PubMed] [Google Scholar]
- 23.Farooqi IS, Keogh JM, Yeo GS, et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 24.Galanopoulou AS, Kent G, Rabbani SN, et al. Heterologous processing of prosomatostatin in constitutive and regulated secretory pathways. Putative role of the endoproteases furin, PC1, and PC2. J Biol Chem. 1993;268:6041–9. [PubMed] [Google Scholar]
- 25.Elias CF. Leptin action in pubertal development: recent advances and unanswered questions. Trends Endocrinol Metab. 2012;23:9–15. doi: 10.1016/j.tem.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong W, Seidel B, Marcinkiewicz M, et al. Cellular localization of the prohormone convertases in the hypothalamic paraventricular and supraoptic nuclei. J Neurosci. 1997;17:563–75. doi: 10.1523/JNEUROSCI.17-02-00563.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez VC, Goldstein J, Stuart RC, et al. Regulation of hypothalamic prohormone convertases 1 and 2 and effects on processing of prothyrotropin-releasing hormone. J Clin Invest. 2004;114:357–69. doi: 10.1172/JCI21620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.