

Abstract
Heat shock protein 90 promotes tumor progression and survival and has emerged as a vital therapeutic target. Previously we reported that the combinatorial treatment of 17AAG/sihsp90α significantly downregulated Hsp90α mRNA and protein levels in Glioblastoma Multiforme (GBM). Here we investigated the ability of cell penetrating peptide (Tat48–60 CPP)-mediated siRNA-induced hsp90α knockdown as a single agent and in combination with 17-allylamino-17-demethoxygeldanamycin (17-AAG) to induce tumor growth inhibition in GBM and whether it possessed therapeutic implications. GBM and non-tumorigenic cells exposed to siRNA and/or 17-AAG were subsequently assessed by qRT-PCR, immunofluorescence, FACS analysis, quantitative Akt, LDH leakage and cell viability assays. PAGE was performed for serum stability assessment. A combination of siRNA/17-AAG treatment significantly induced Hsp90α gene and protein knockdown by 95% and 98%, respectively, concomitant to 84% Akt kinase activity attenuation, induced cell cycle arrest and tumor-specific cytotoxicity by 88%. Efficient complex formation between CPP and siRNA exhibited improved serum stability of the siRNA with minimal intrinsic toxicity in vitro. The preliminary in vivo results showed that combination therapy induced hsp90α knockdown and attenuated Akt kinase activity in intracranial glioblastoma mouse models. The results imply that RNAi-mediated hsp90α knockdown increases 17-AAG treatment efficacy in GBM. In addition, the cytotoxic response observed was the consequence of downregulation of hsp90α gene expression, reduced Akt kinase activity and S-G2/M cell cycle arrest. These results are novel and highlight the ability of Tat to efficiently deliver siRNA in GBM and suggest that the dual inhibition of Hsp90 has therapeutic potentials.
Keywords: Hsp90α, Tat, sihsp90α/CPP complex, Cell penetrating peptides, Glioblastoma, GBM, Hsp90 inhibitor, Akt, Cell-cycle arrest
Highlights
-
•
17-AAG–siRNA dual treatment exhibits significant anti-cancer activity in GBM.
-
•
Combination therapy induced Hsp90α gene/protein knockdown causing Akt inactivation.
-
•
Hsp90α inhibition causes S-G2/M cell cycle arrest and GBM-specific cytotoxicity.
-
•
Efficient siRNA/CPP interaction improves serum stability of siRNA.
-
•
RNAi-mediated hsp90α knockdown increases GBM sensitivity to 17-AAG.
1. Introduction
Heat shock protein 90 is a vital molecular chaperone involved in regulating cellular processes by chaperoning its oncogenic clientele including EGFR, PDGFR, Cdks, Raf, Akt and p53 [1,2]. Many of these signalling proteins are overexpressed or mutated in GBM, indicating Hsp90-dependent tolerance to genetic alterations that would otherwise be fatal. 17-Allylamino-17-demethoxy-geldanamycin (17-AAG), a benzoquinone antibiotic and binds to the N-terminal domain of Hsp90 protein with higher binding affinity than ATP and has been subsequently shown to attenuate tumor growth, induce cell cycle arrest and apoptosis [1].
Gene therapy has received major impetus in recent years as RNAi reaches clinical trials [3]. Low serum stability, renal clearance, non-specific immune stimulation and poor cellular uptake are key factors that have restricted the use of RNAi in systemic applications [4]. We have previously combined the application of RNAi using small interfering RNA (siRNA) and the current chemotherapeutic drug temozolomide (TMZ) resulting in significant gene knockdown and induced chemosensitivity in GBM [5]. Furthermore, a combination of RNAi and 17-AAG manifested significant gene and protein knockdown, concomitant to suppressed Akt levels and reduced GBM growth [6]. The effects of combination treatment were tumor-specific as non-tumorigenic cells exhibited no significant cytotoxicity. Although successful RNAi response is relatively straight forward in vitro, achieving high transfection is challenging in vivo [7].
Cell penetrating peptides (CPP) are short cationic peptides ranging up to 30 amino acids in length. These peptides, together with siRNA, bind to the anionic cell membrane and prompts cellular internalization through a endocytosis-mediated process [8]. The therapeutic applications of non-covalently linked siRNA/CPP complexes (SCC) have been recently reported in vitro and in vivo [9,10]. The Tat48–60 is a short cationic peptide derived from the human immunodeficiency virus type 1 (HIV-1) Tat transcriptional activator protein has been extensively employed for intracellular delivery of ONs including siRNA in vitro and in vivo [11].
In this study, we determined the effects of hsp90α-specific siRNA with and without 17-AAG on GBM cell growth and survival. Dual treatment significantly reduced GBM cell growth and the cytotoxicity was tumor-specific. This induced growth inhibition was a consequence of reduced Hsp90α gene expression concomitant with repressed Akt kinase activity and cell cycle arrest in GBM.
2. Results
2.1. RNAi and 17-AAG induce hsp90α knockdown in GBM
To examine the gene silencing potential of the combination treatment, hsp90α expression was quantitated by qRT-PCR. U87-MG cells treated with SCC showed significant hsp90α downregulation after 48 h, exhibiting a CPP concentration-dependent knockdown of up to 30% (Fig. 1A). After 72 h, although significant gene knockdown was achieved at a lower CPP concentration, however, increasing the CPP concentration weakened the RNAi response. The combination treatment produced a significant magnitude of hsp90α knockdown that increased with increasing CPP concentration, yielding up to 95% after 48 h (p ≤ 0.001). A similar but slightly weakened RNAi response was observed after 72 h. All the qRT-PCR fragments were also resolved by agarose gel electrophoresis (2%) (data not shown).
Fig. 1.
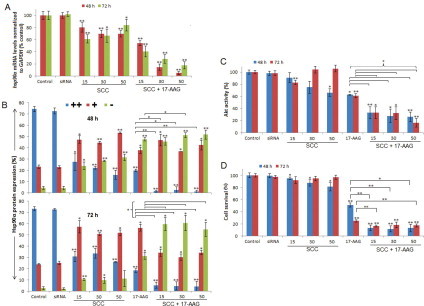
A combination of SCC and 17-AAG induces Hsp90α gene and protein knockdown, loss of Akt kinase activity and growth inhibition in GBM. (A) hsp90α knockdown quantitated by qRT-PCR following treatment with SCC (CPP/siRNA ratio of 15–50) and 17-AAG as single agents or as a dual combination. (B) Hsp90α protein levels determined by immunofluorescence utilizing laser scanning confocal microscopy. For each sample, 150 cells were counted and the protein levels were categorized into three groups namely, strong (++), weak (+) and no expression (−). (C) The combination treatment attenuated Akt kinase activity. (D) Cell viability assessment using CellTiter-Glo luminescent cell viability assay. Cells administered with DMSO (0.0001% v/v) were used as negative control for 17-AAG treated cells and cells exposed to CPP alone (refer Fig. 4C) served as a negative control for all the treated samples. siRNA represents negative control where cells were exposed to neat siRNA. siRNA corresponds to sihsp90α. Data values are mean ± SD, n = 3. *p ≤ 0.05 and **p ≤ 0.001 were considered statistically significant.
2.2. Combination treatment with RNAi and 17-AAG depletes Hsp90α protein levels in GBM
To examine whether RNAi and 17-AAG administration resulted in Hsp90α protein repression in glioblastoma, control and treated cells were categorized into three groups depicting the extent of expression, namely, strong (++), weak (+) and no expression (−), taking into consideration the intensity, contrast and sharpness of the image. SCC exhibited an RNAi response that increased with CPP concentration resulting in 84% protein reduction was achieved after 48 h with 32% exhibiting complete inhibition (Fig. 1B). After 72 h, however, the response declined as 74% protein repression was achieved and only 10% showed no protein expression. Additionally, the combination treatment with siRNA and 17-AAG resulted in 98% and 95% protein downregulation with 52% and 61% resulting in no protein expression after 48 and 72 h, respectively. Administration of 17-AAG resulted in 80% protein repression including 48% that exhibited complete protein downregulation after 48 h. The benefit of concomitant gene and protein inhibition was evident with up to 52% and 61% displaying complete protein downregulation (p ≤ 0.001). A statistical comparison revealed a significant benefit for the combination therapy against 17-AAG alone after 48 and 72 h, thus suggesting a potential therapeutic value of this treatment.
2.3. RNAi and 17-AAG promotes downregulation of Hsp90 clientele Akt kinase activity
Since Hsp90 is essential for client protein stability and activity, we examined the effects of the treatment on Akt kinase activity in GBM. RNAi response mediated by SCC reduced Akt activity by 34% and 17% after 48 and 72 h exposure, respectively (Fig. 1C). The combination treatment significantly reduced Akt kinase activity by 74% and 84% after 48 and 72 h, respectively (p ≤ 0.001). U87-MG cells exposed to 17-AAG alone exhibited a 37% and 39% reduction in Akt activity after 48 and 72 h. The effects of the combination treatment were statistically more significant than 17-AAG alone (p ≤ 0.05).
2.4. RNAi synergizes with 17-AAG to enhance treatment efficacy in glioblastoma
To examine whether siRNA-mediated gene knockdown exerts an additional efficacy on growth inhibition of GBM, U87-MG cells were treated with increasing concentrations of SCC ranging from 15- to 50-fold CPP molar excess to siRNA and 17-AAG. The SCC alone resulted in significant reduction in tumor cell viability yielding up to 19% growth inhibition after 48 h (Fig. 1D). On the other hand, SCC showed no significant benefit on growth inhibition of GBM after 72 h. U87-MG cells exposed to 17-AAG alone yielded 49% and 75% growth inhibition after 48 and 72 h, respectively. Furthermore, the combination of SCC and 17-AAG demonstrated a significant cytotoxicity in GBM at all CPP concentrations resulting in 88% and 84% tumor growth inhibition (p ≤ 0.001). Statistical analysis revealed a significant benefit of adding hsp90α-specific siRNA to 17-AAG on growth inhibition of GBM compared to 17-AAG alone (p ≤ 0.001). A clear correlation between the Hsp90α mRNA and protein expression profile, Akt activity and cell viability was observed in GBM.
2.5. The effects of SCC and 17-AAG in non-tumorigenic cells
To determine whether the combination of SCC and 17-AAG possesses therapeutic value in the treatment of GBM, SVGp12 cells were treated with SCC and 17-AAG and its effects on hsp90α gene expression and cell viability was assessed. qRT-PCR analysis revealed negligible levels of hsp90α expression after 48 and 72 h (Fig. 2A). Although, treatments with SCC and 17-AAG were observed to slightly induce hsp90α levels, its effects on SVGp12 cell growth was somewhat unaltered after 48 and 72 h displaying no more than 7% growth inhibition (Fig. 2B).
Fig. 2.
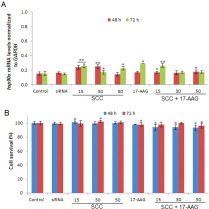
Cytotoxic effects of the combination treatment with SCC and 17-AAG in non-tumorigenic cells of neural origin. (A) An assessment of hsp90α gene expression was performed by qRT-PCR. (B) Hsp90 inhibition using siRNA and 17-AAG exhibits minimal cytotoxicity in non-tumorigenic cells. SCC represents CPP/siRNA ratio of 15–50). Data values are mean ± SD, n = 3. *p ≤ 0.05 and **p ≤ 0.001 were considered statistically significant.
2.6. SCC and 17-AAG causes cell cycle arrest in GBM
As cell cycle progression is a critical element of growth, FACS analysis was performed to determine whether Hsp90α inhibition results in cell cycle arrest in GBM. We examined the effects of SCC, 17-AAG and a combination of SCC and 17-AAG after 48 and 72 h. Compared to the untreated control, U87-MG cells displayed G1 and S phase arrest following 17-AAG exposure and an S phase arrest post SCC treatment after 48 and 72 h, respectively (Fig. 3). Furthermore, the combination treatment induced accumulation of cells in the G2/M and S phase after 48 and 72 h, respectively (p ≤ 0.001). However, the only apparent effect of 17-AAG was an S phase arrest after 72 h in SVGp12 cells.
Fig. 3.

SCC and 17-AAG causes cell cycle arrest in GBM. The cell cycle distribution in treated and control samples was quantitated by FACS analysis using a flow cytometer and recording 10,000 events per sample. 1 represents control, 2: siRNA, 3–5: SCC 15–50, 6: 17-AAG, and 7–9: SCC + 17-AAG. Data values are mean ± SD, n = 3. *p ≤ 0.05 and **p ≤ 0.001 were considered statistically significant.
2.7. CPP does not exhibit acute or long-term toxicity
To determine whether siRNA efficiently binds to the CPP, a gel shift assay was performed. The peptide bound to the siRNA in a dose-dependent manner was observed to completely retard the siRNA at 15-fold peptide molar excess (Fig. 4A).
Fig. 4.

Cytotoxic effect of the CPP interaction with siRNA. (A) A gel shift assay to confirm that CPP effectively binds to the siRNA. M represents siRNA marker, 0: free siRNA and 1–20: CPP/siRNA molar ratios. (B) LDH leakage assay to assess membrane integrity of cells treated with increasing concentrations of CPP and different SCC ratios ranging from 3–20 μM and 15–100 (peptide/siRNA ratio), respectively. Control cells were lysed with LDH assay lysis solution representing 100% LDH leakage was used as a control. (C) Long-term toxicity analysis of CPP. (D) CPP induces hsp90α expression in GBM. (E) The effects of CPP and SCC on Hsp90α protein expression in GBM (refer Fig. 1B for legend key). (F) CPP exhibits no regulatory effect on Akt kinase activity in GBM. Data values are mean ± SD, n = 3. *p ≤ 0.05 and **p ≤ 0.001 were considered statistically significant.
To determine whether CPP alone and SCC exhibit intrinsic toxicity that could influence the observed gene silencing effects, membrane disturbance and viability were assessed. An LDH leakage assay was used to measure the acute membrane disturbance caused by peptides at high concentrations. Peptide alone showed no more than 6% LDH leakage at 50-fold peptide molar excess to siRNA and at CPP concentration of 10 μM (Fig. 4B). However, SCC induced ∼15% membrane leakage at a very high CPP/siRNA molar ratio of 100 or 20 μM in GBM, consequently, CPP concentrations used in other experiments did not exceed 10 μM. In SVGp12 cells, Tat induced no more than 3% LDH leakage at concentrations of 20 μM or 100-fold peptide molar excess. In addition, long-term toxicity assessment using CellTiter-Glo assay showed that CPP induced no more than 2% growth inhibition in U87-MG or SVGp12 cells (Fig. 4C).
Moreover, qRT-PCR analysis revealed a regulatory role of the CPP on transcriptional regulation of hsp90α in GBM (Fig. 4D), but not in non-tumorigenic cells (data not shown). Increasing concentrations of CPP induced hsp90α levels after 48 and 72 h. On the contrary, CPP administration causes marginal protein knockdown at 48 and 72 h time points (Fig. 4E). Akt kinase activity assessment presented no significant effect of CPP in GBM at all concentrations used in this study (Fig. 4F). Finally, CPP showed accumulation of GBM cells in the S phase at various concentrations after 48 and 72 h but exhibited no effect on cell cycle distribution in SVGp12 cells.
2.8. Complex formation with CPP improves serum stability of siRNA
Stability studies were performed to characterize the susceptibility of aqueous siRNA and SCC in 50% human serum as examined by polyacrylamide gel electrophoresis. Neat siRNA was unstable in serum after 1 h and exhibited complete degradation after 6 h (Fig. 5). In contrast, SCC demonstrated improved stability in serum at higher peptide concentrations. At 30-fold peptide molar excess, siRNA integrity was maintained after 24 h whereas at 50-fold molar excess, intact siRNA was detected after 36 h.
Fig. 5.

SCC improves serum stability of siRNA. M represents siRNA marker and 0 indicates free siRNA. This is representative of three such independent experiments.
2.9. GBM growth in vivo
Following intracranial implantation, stained sections of tumor specimen clearly showed the presence of a tumor resulting in tissue distortion (Fig. 6A). Furthermore, tumor tissue exhibited high luciferase expression compared to normal tissue (Fig. 6B). The luciferase activity in treated samples in vitro manifested a dose-dependent reduction with up to 73% luciferase knockdown achieved after 24 h (data not shown).
Fig. 6.

SCC and 17-AAG induce hsp90α knockdown and attenuate Akt kinase activity in vivo. (A) H & E stained tissue sections (n = 2) and (B) luciferase quantitation indicate presence of tumor. Values are mean, n = 2. Treated specimens (n = 3) show reduced hsp90α expression (C) and Akt kinase activity (D) compared to control (n = 1).
2.10. Combination treatment downregulates hsp90α and Akt activity in vivo
SCC combined with 17-AAG exhibited 73% hsp90α knockdown after 24 h (Fig. 6C). The hsp90α expression in tumor tissue was normalized to GAPDH expression as well as normal brain tissue within each sample to accurately quantitate gene expression. In xenograft models, combination treatment exhibited 75% reduction in Akt kinase activity after 24 h (Fig. 6D). The Akt activity in tumor specimens was normalized to normal brain tissue within each sample to accurately quantitate the Akt expression. The qRT-PCR and Akt kinase activity results demonstrate a strong correlation.
3. Discussion
In GBM, Hsp90 levels are highly induced to support a range of activated oncoproteins and signalling pathways that glioblastomas are dependent on [12]. Hsp90α, the inducible isoforms of Hsp90, is highly expressed at both mRNA and protein levels in glioma cell lines and tissues but not in normal cell lines and tissues [13]. This corroborates Hsp90α as an anti-glioma therapeutic target. We have previously demonstrated significant siRNA-mediated hsp90α knockdown concomitant to induced chemosensitivity in GBM in vitro [5]. However, the major issue in gene therapy is the poor bioavailability of the negatively charged siRNA molecules. Commonly used transfection systems such as cationic lipids or viruses have several disadvantages; the use of cationic lipids is restricted to in vitro and viruses have the tendency to stimulate immunogenic responses in vivo [14]. Likewise, the efficacy of many delivery systems is compromised by the presence of serum proteins. In this study, the ability of CPP to promote siRNA-mediated RNAi and subsequently induce growth inhibition when combined with 17-AAG was investigated in GBM.
The SCC significantly reduced cell viability after 48 h and induced hsp90α knockdown after 48 and 72 h that corroborate the ability of the CPP to deliver functional siRNA into the cells. The hsp90α knockdown failed to impose cytotoxic effects after 72 h which could be attributed to the lack of siRNA durability [15], and the protein half-life of 36 h [6] rationalizing the necessity of a protein inhibitor to achieve additional efficacy on growth inhibition. Localization studies of SCC revealed a predominant nuclear accumulation along with diffused cytoplasmic staining. Increasing the peptide concentration resulted in higher siRNA yields in the cytoplasm, an observation that correlated well with RNAi response in vitro implying that the cytoplasmic localization of siRNA is critical for maximum gene knockdown. Interestingly, SCC repressed Akt activity after 48 and 72 h, however, the Akt activity levels recovered at higher CPP concentrations. A plausible explanation could be that vital components of the survival signalling pathway such as Akt may be regulated by other chaperones following Hsp90α inhibition.
A combination of SCC and 17-AAG demonstrated synergistic growth inhibition to enhance treatment efficacy in GBM as compared to 17-AAG after 48 and 72 h. While the combination treatment induced hsp90α knockdown comparable to that of 17-AAG, Hsp90α protein expression analysis reveal a benefit of adding SCC as protein levels were significantly depleted compared to 17-AAG alone after 48 and 72 h. The Akt activity was assessed to quantitate the extent of Hsp90 inhibition [16]. Similar effects were recorded for Akt activity where the combination treatment significantly downregulated its activity. Moreover, results reveal a malign addiction of GBM to Hsp90α mainly ascribed to the induced cytotoxicity as a result of SCC and 17-AAG administration. These observations confirmed that the dual approach to target Hsp90α yields better anti-tumor effects in GBM compared to either treatment independently. Similar results were obtained in a preliminary in vivo study whereby nude mice were utilized as carriers of exogenously introduced human tumor that closely simulates human disease. Due to the limited numbers of animals available for this investigation [a control group (n = 1) and treatment group (n = 3)], in vivo data presented are preliminary and requires additional repeats in order to validate the results. Nonetheless, the data is encouraging.
CPP alone did not display a significant regulatory effect on the Akt activity at concentrations of 10 μM that corresponds with its effects on GBM viability where no long-term toxicity was detected. Similarly, treatment with CPP or SCC exhibited no cytotoxic effects in non-tumorigenic cells even when associated with 17-AAG. These observations indicate that Hsp90α in SVGp12 cells bears a latent/uncomplexed conformation which corroborates the lack of the requirement for functional/active chaperones in non-tumor cells demarcated by the negligible transcriptional expression. This not only establishes the dependence of tumors to molecular chaperones for stabilizing oncogenic clientele responsible for malignant transformation but also the combination treatment may be critical in a clinical setting whereby a window of dosing can be established to primarily target glioblastoma cells.
Cyclin-dependent kinase 4 (cdk4), a serine/threonine kinase functions when bound to active Hsp90 and regulates the G1 cell cycle progression once activated by Akt [17]. Similarly, Hsp90 client cdk2 kinase plays an essential role in DNA replication and S/G2 transition [18]. Cell division cycle 2 kinase (cdc2) is responsible for G2/M transition and requires functional Hsp90 for its activity [1]. The combination treatment induced a G2/M arrest and an S phase arrest after 48 and 72 h, respectively. Results suggest that SCC and 17-AAG-induced cell cycle arrest was the consequence of Hsp90α gene expression downregulation and Akt activity attenuation. Moreover, the disparity in cell cycle distribution could be due to a change in conformation of the chaperone complexes associated with these kinases at different time points. CPP exhibited an S phase arrest in GBM but not in non-tumorigenic cells which may indicate non-specific interference with the transcriptional regulation of S/G2 transition proteins such as cyclin A and cdk2.
CPP exhibited no long-term toxicity at concentrations used in this study which contradicts its effect on hsp90α transcription levels in GBM. These could be uncharacterized effects of the CPP on the transcriptional regulation of hsp90α, on par with previous observations [19]. Additionally, since the CPP is derived from the DNA binding domain of HIV-1 trans-activator protein, it is conceivable that it may directly interferes with the gene transcription [11].
Presently, 17-AAG is in phase II/III clinical trials in adults with solid tumor malignancies [20]. These results collectively indicate that the dual targeting of Hsp90 using RNAi and protein inhibitors possess anti-tumor activity in GBM. The combination treatment provided additional efficacy on growth inhibition of glioblastoma cells compared to 17-AAG alone. In addition, the cytotoxic response observed was the consequence of downregulation of Hsp90α gene and protein expression, reduced Akt kinase activity and S-G2/M cell cycle arrest. The tumor-specific response described herein suggests that the dual inhibition of Hsp90 may have therapeutic potential in GBM.
4. Materials and methods
4.1. Cell culture
Human Glioblastoma Multiforme (U87-MG) and human non-tumorigenic (SVGp12) cell lines were purchased from the European Collection of Cell Cultures and the American Type Culture Collection, respectively. The cells were propagated in EMEM (Lonza) containing FBS (10%), l-glutamine (2 mM), non-essential amino acids (1% v/v), and sodium pyruvate (1 mM) and maintained in a humidified incubator at 37 °C with 5% CO2. Cultures were restricted to 5–7 passages for all the experiments.
4.2. Peptide preparation
The Tat48–60 CPP was prepared using a standard strategy as described previously [21]. The amino acid sequence of the CPP was H-GRKKRRQRRRPPQ-NH2 (MW: 1718.06).
4.3. Treatment with siRNA and 17-AAG
The siRNA specific to Hsp90AA1 gene with the sequence: sense, (5′) CGUGAUAAAGAAGUAAGCGtt (3′); and antisense, (5′) CGCUUACUUCUUUAUCACGtt (3′) was resuspended in nuclease-free water to achieve a stock concentration of 100 μM (Ambion, UK).
U87-MG and SVGp12 cells were seeded in 96-well plates or 25 cm2 culture flasks 24 h prior to treatment. The siRNA/CPP complexes (SCC) were prepared by mixing siRNA (200 nM) with increasing CPP concentrations giving rise to siRNA:peptide molar ratios of 1:15–1:50 in serum-free medium at 37 °C for 30 min. Fresh serum-supplemented medium was added to the cells after 4 h and incubated for a further 48 and 72 h. For combination treatment, 17-AAG was added following the initial 4 h incubation to achieve a final concentration of 0.225 μM. For the in vivo experiments, SCC (5 mg/kg) were formed in nuclease-free water at 50-fold peptide molar excess in half the injection volume (100 μl) using 0.5 mM siRNA and 10 mM CPP stock solutions. After the 30 min incubation, 100 μl of 10.8% mannitol solution was added to SCC and injected into the tail-vein of the mice. Subsequently, the mice were injected with 17-AAG (i.p., 80 mg/kg). Mice treated with luciferase siRNA (sense: 5′ACGCCAAAAACAUAAAGAAAG3′ and antisense: 5′UUCUUUAUGUUUUUGGCGUCU3′) and 17-AAG were used as negative controls and the left striatums were used as normal brain tissues.
4.4. Cell viability assay
The percent of viable cells was assessed using CellTiter-Glo® luminescent cell viability assay (Promega) as described previously [6]. Briefly, U87-MG and SVGp12 cells were seeded at a density of 1000 cells/well in 96-well plates in triplicates 24 h prior to treatment with SCC and 17-AAG and the luminescent signal was recorded using Tecan GENios Pro® (Tecan, Austria).
4.5. qRT-PCR analysis
Isolation of mature mRNA and cDNA synthesis was performed as described previously [6]. Gene specific cDNA primers (hsp90α sense: 5′ tctggaagatccccagacac 3′, antisense: 5′ agtcatccctcagccagaga 3′; and GAPDH sense: 5′ gagtcaacggatttggtcgt 3′, anti-sense: 5′ ttgattttggagggatctcg 3′) were obtained from TIB MOLBIOL (Germany) designed using the Primer3 software (http://primer3.wi.mit.edu/) and using the gene sequence (NM_002046.4) obtained from NCBI. The standard nucleotide BLAST was performed using the GAPDH primer sequence and the human genomic + transcript nucleotide database available from BLAST that showed 1 hit for transcripts which was Homo sapiens GAPDH transcript variant 1 (NM_002046.4) showing 100% identity. It is unlikely that pseudogene interference would occur since the mRNA Isolation Kit (Roche Life Sciences, UK) ensures high purity of mRNA (free of DNA and other RNAs) which was subsequently used for cDNA synthesis and amplification. qRT-PCR was performed using FastStart DNA MasterPLUS SYBR Green 1 as described previously [13]. The hsp90α expression profiles were normalized to the GAPDH expression and plotted as a percentage.
4.6. Immunofluorescence
A standard protocol was followed for immunofluorescence described in depth in a previous publication [6]. The primary monoclonal antibody and FITC-conjugated secondary antibody were obtained from Cambridge Bioscience (UK). The images were taken using Axiovert 200M LSM 510 laser scanning confocal microscope (Carl Zeiss Ltd.) and analyzed using the Zeiss LSM Image Browser software (Carl Zeiss Jena GmbH).
4.7. Akt/PKB kinase activity assay
Protein extraction, quantitation and Akt kinase activity assessment was performed as described previously [6].
4.8. Cell cycle analysis
U87-MG and SVGp12 cells were seeded at a density of 1 × 105 cells/T25 flask 24 h prior to treatment with SCC and 17-AAG. Cells were harvested, washed in PBS and fixed in ice-cold ethanol (70%). After 30 min, cells were incubated in PBS supplemented with propidium iodide (50 μg/ml; Sigma, UK) and RNase A (100 μg/ml; Sigma, UK) for 30 min at room temperature. The DNA content was analyzed by FACSAria flow cytometer (Becton Dickinson, UK) and the cell cycle profiles were obtained using the FACSDiva software (Becton Dickinson).
4.9. Gel mobility assay
SCC was prepared as described earlier at molar ratios ranging from 1:1 to 1:20 (siRNA:peptide) and were subjected to electrophoresis on 15% TBE poly-acrylamide gel (Bio-Rad, UK) in TBE buffer at 100 V under non-denaturing conditions and stained with ethidium bromide (0.5 μg/ml) and analyzed by UV-transilluminator GENE GENIUS Bioimaging system (Syngene).
4.10. Membrane disturbance measurements
The membrane integrity was measured using the In Vitro Toxicology Assay Kit according to manufacturer's instructions (TOX7; Sigma, UK). U87-MG and SVGp12 cells (1 × 104/well) were seeded in 96-well plates 24 h prior to treatment with increasing concentrations CPP or SCC in serum-free medium for 30 min at 37 °C. The samples were centrifuged and transferred to a clean 96-well flat bottom plate. LDH assay mixture was added and the absorbance measured at 490 nm using Tecan GENios Pro®. Serum-free medium without cells was used as a blank and untreated cells lysed with LDH Assay Lysis Solution (1/10 total volume) was used as a control.
4.11. Serum stability of siRNA
Neat siRNA (1 μg) and SCC were incubated in EMEM with human serum (50%) at 37 °C. Aliquots (20 μl) were taken at 1, 3, 6, 24, and 36 h intervals and supplemented with Proteinase K (200 μg/ml; Sigma, UK) and SDS (0.5% w/v). After 1 h incubation at 37 °C that facilitates segregation and digestion of the CPP, the samples were stored at −80 °C. The siRNA was extracted from the SCC using phenol/chloroform/isoamyl alcohol (25:24:1; Sigma, UK) and analyzed on 15% TBE polyacrylamide gel in TBE buffer (1×) at 100 V under non-denaturing conditions. The gels were stained with ethidium bromide and images were developed by UV-transilluminator GENE GENIUS Bioimaging system.
4.12. RNAi experiments in vivo
A luciferase expressing cell line (U87-MG-luc2; Caliper Life Sciences) was cultured and propagated as stated above. The intracranial implantation of U87-MG-luc2 cells into the right striatums was performed utilizing U87-MG-luc2 cells (1 × 106 cells/5 μl of fresh media without serum) were injected into the right striatums of homozygous female nude mice using a stereotaxic frame at coordinates A = +1, L = +2, V = +3.5 from Bregma. Once tumor growth was detected, the mice were injected with SCC and 17-AAG and the tissues were harvested after 24 h and stored at −80 °C immediately. For luciferase quantitation, protein was extracted using Reporter Lysis Buffer (Promega, Sweden) and quantitated using DC Protein Assay according to manufacturer's instructions (Bio-Rad). qRT-PCR and Akt kinase activity was performed as described previously. For H&E staining, tissue specimens collected were frozen immediately in a isobutanol bath on dry ice. The tissue sections were mounted on glass slides and stained with haematoxylin and eosin. All the procedures for animal care and tumor cell implantation followed approved animal protocols and guidelines of the Estonian Laboratory Animal Ethics Committee (approval number 19 dated 25th September 2009, number 69 and 70 dated 9th February 2011).
4.13. Statistical analysis
The data presented are the mean of three independent experiments ± SD (unless stated otherwise) and was analyzed using the PASW Statistics 18 package using the One-Sample Student's T-test and Paired-Sample T-test. A p value of *p ≤ 0.05 and **p ≤ 0.001 was considered as statistically significant.
5. Funding sources
This study was supported by a Grant from the Sydney Driscoll Neuroscience Foundation (SDNF) and the School of Pharmacy and Biomedical Sciences (UCLan). The work was also supported by the Estonian Government through the targeted financing SF0180027s08 and the European Regional Development Fund [project Tumor-Tech (3.2.1001.11–0008)].
6. Conflict of interest disclosures
No competing financial interests exist.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Garcia-Morales P., Carrasco-Garcia E., Ruiz-Rico P., Martinez-Mira R., Menendez-Gutierrez M.P., Ferragut J.A., Saceda M., Martinez-Lacaci I. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G(2)/M arrest in glioblastoma cell lines. Oncogene. 2007;26:7185–7193. doi: 10.1038/sj.onc.1210534. [DOI] [PubMed] [Google Scholar]
- 2.Karkoulis P.K., Stravopodis D.J., Margaritis L.H., Voutsinas G.E. 17-Allylamino-17-demethoxygeldanamycin induces downregulation of critical Hsp90 protein clients and results in cell cycle arrest and apoptosis of human urinary bladder cancer cells. BMC Cancer. 2010;10:481. doi: 10.1186/1471-2407-10-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daka A., Peer D. RNAi-based nanomedicines for targeted personalized therapy. Adv. Drug Delivery Rev. 2012;64:1508–1521. doi: 10.1016/j.addr.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 4.Lundberg P., El-Andaloussi S., Sutlu T., Johansson H., Langel U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007;21:2664–2671. doi: 10.1096/fj.06-6502com. [DOI] [PubMed] [Google Scholar]
- 5.Cruickshanks N., Shervington L., Patel R., Munje C., Thakkar D., Shervington A. Can hsp90alpha-targeted siRNA combined with TMZ be a future therapy for glioma? Cancer Invest. 2010;28:608–614. doi: 10.3109/07357901003630967. [DOI] [PubMed] [Google Scholar]
- 6.Mehta A., Shervington L., Munje C., Shervington A. A novel therapeutic strategy for the treatment of glioma, combining chemical and molecular targeting of Hsp90α. Cancers. 2011;3:4228–4244. doi: 10.3390/cancers3044228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng S., Wei B., Xu R., Zhang K., Wang L., Zhang R., Li J. TAT peptides mediated small interfering RNA delivery to Huh-7 cells and efficiently inhibited hepatitis C virus RNA replication. Intervirology. 2009;52:135–140. doi: 10.1159/000220597. [DOI] [PubMed] [Google Scholar]
- 8.Meade B.R., Dowdy S.F. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv. Drug Delivery Rev. 2008;60:530–536. doi: 10.1016/j.addr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han L., Zhang A., Wang H., Pu P., Jiang X., Kang C., Chang J. Tat-BMPs-PAMAM conjugates enhance therapeutic effect of small interference RNA on U251 glioma cells in vitro and in vivo. Hum. Gene Ther. 2010;21:417–426. doi: 10.1089/hum.2009.087. [DOI] [PubMed] [Google Scholar]
- 10.Kim S.W., Kim N.Y., Choi Y.B., Park S.H., Yang J.M., Shin S. RNA interference in vitro and in vivo using an arginine peptide/siRNA complex system. J. Controlled Release. 2010;143:335–343. doi: 10.1016/j.jconrel.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Moschos S.A., Jones S.W., Perry M.M., Williams A.E., Erjefalt J.S., Turner J.J., Barnes P.J., Sproat B.S., Gait M.J., Lindsay M.A. Lung delivery studies using siRNA conjugated to TAT(48–60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjugate Chem. 2007;18:1450–1459. doi: 10.1021/bc070077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins V.P. Brain tumours: classification and genes. J. Neurol. Neurosurg. Psychiatry. 2004;75(Suppl. 2):ii2–ii11. doi: 10.1136/jnnp.2004.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shervington A., Cruickshanks N., Lea R., Roberts G., Dawson T., Shervington L. Can the lack of HSP90 protein in brain normal tissue and cell lines, rationalise it as a possible therapeutic target for gliomas? Cancer Invest. 2008;26:900–904. doi: 10.1080/07357900802087259. [DOI] [PubMed] [Google Scholar]
- 14.Mae M., Langel U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr. Opin. Pharmacol. 2006;6:509–514. doi: 10.1016/j.coph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 15.McAnuff M.A., Rettig G.R., Rice K.G. Potency of siRNA versus shRNA mediated knockdown in vivo. J. Pharm. Sci. 2007;96:2922–2930. doi: 10.1002/jps.20968. [DOI] [PubMed] [Google Scholar]
- 16.Liao Y., Hung M.C. Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2010;2:19–42. [PMC free article] [PubMed] [Google Scholar]
- 17.Shervington A., Cruickshanks N., Wright H., Atkinson-Dell R., Lea R., Roberts G., Shervington L. Glioma: what is the role of c-Myc, hsp90 and telomerase? Mol. Cell Biochem. 2006;283:1–9. doi: 10.1007/s11010-006-2495-z. [DOI] [PubMed] [Google Scholar]
- 18.Xu W., Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin. Cancer Res. 2007;13:1625–1629. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 19.Fotin-Mleczek M., Welte S., Mader O., Duchardt F., Fischer R., Hufnagel H., Scheurich P., Brock R. Cationic cell-penetrating peptides interfere with TNF signalling by induction of TNF receptor internalization. J. Cell Sci. 2005;118:3339–3351. doi: 10.1242/jcs.02460. [DOI] [PubMed] [Google Scholar]
- 20.Siegel D., Yan C., Ross D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharmacol. 2012;83:1033–1040. doi: 10.1016/j.bcp.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones S., Holm T., Mager I., Langel U., Howl J. Characterization of bioactive cell penetrating peptides from human cytochrome c: protein mimicry and the development of a novel apoptogenic agent. Chem. Biol. 2010;17:735–744. doi: 10.1016/j.chembiol.2010.05.018. [DOI] [PubMed] [Google Scholar]